Introduction
Colorectal cancer (CRC) is one of the most prevalent
malignancies worldwide and is usually not diagnosed until it is at
the advanced or metastatic stage (1). Surgical resection is the primary
treatment option for CRC, followed by chemotherapy for patients who
cannot undergo surgery (2).
First-line drugs, such as irinotecan, oxaliplatin, fluorouracil,
capecitabine and calcium folinate, and more recently developed
targeted drugs, such as bevacizumab, cetuximab and gefitinib, as
well as combinations of these drugs have been used against CRC
(3). However, patients frequently
develop chemoresistance, which is the major cause of treatment
failure (4). Some new drug
combinations (5,6) and genetic interventions (7,8) have
achieved tumor cell chemo-sensitization. For example, the
combination therapy of oxaliplatin and coxsackievirus A11 increases
the oncolytic activity in oxaliplatin-resistant CRC cells (5). Guanine nucleotide-binding protein
subunit β-5 knockdown enhances cetuximab cytotoxicity in
KRAS-mutant CRC cells (8).
Nevertheless, drug resistance is still a major challenge that needs
to be managed in order to improve therapeutic efficacy.
Sirtuins (SIRTs) are NAD+-dependent
protein deacetylases that are localized to specific cellular
compartments, including the nucleus (SIRT1, SIRT6 and SIRT7),
cytoplasm (SIRT2 and SIRT5) and mitochondria (SIRT3 and SIRT4)
(9). SIRTs act as tumor activators
or suppressors through regulating metabolism, genomic stability or
cancer stem cell proliferation (10,11).
SIRT expression levels in CRC cells are correlated with
chemosensitivity (12). Prolonged
exposure to drugs can promote SIRT1-induced mitochondrial oxidative
phosphorylation, resulting in chemoresistance and tumor survival
(13), while deletion of SIRT2
confers resistance to MEK inhibitors in KRAS mutant (mut) CRC cells
(14). Resveratrol-mediated
inhibition of CRC cells is accompanied by DNA damage and SIRT6
upregulation (15). SIRT inhibitors,
including EX527 (an inhibitor of SIRT1), AGK2 (an inhibitor of
SIRT2) and sirtinol (an inhibitor of SIRT1 and SIRT2) have shown
anti-neoplastic effects in CRC cells (16-18).
However, it is unclear whether different SIRT inhibitors act
synergistically or antagonistically when combined with other
chemotherapeutic drugs against CRC.
In the present study the effect of multiple SIRT
inhibitors and other drugs on TP53 in wild-type (wt) and mut
CRC cell lines was analyzed. Bioinformatics analysis was
additionally used to indicate the status of SIRT1 and protein
deacetylation regulatory genes in TP53wt CRC
cells compared to the TP53mut cells. The likely
mechanism underlying the antagonistic effect of SIRT inhibitors
with other agents was explored in TP53wt CRC
cells. These data suggested that the sensitivity of CRC cells to
multiple drug combinations is governed by the p53 mutation
status.
Materials and methods
Cell lines and culture
conditions.
CRC cell lines HCT116 (ATCC® CCL-247™,
KRASmut and TP53wt) and SW620
(ATCC® CCL-227TM, KRASmut and
TP53mut R273H) were obtained from American Type
Culture Collection and tested for mycoplasma contamination and STRs
were confirmed. The characteristics of SW620 cells have been
previously defined in relevant studies (19,20) and
on the ATCC website (https://www.atcc.org/products/all/CCL-227.aspx#characteristics).
Cells were cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.)
and 1% penicillin/streptomycin at 37˚C under 5% CO2.
Chemotherapeutic agents
All chemotherapeutic agents were purchased from
Selleck Chemicals LLC and the following stock solutions were
prepared in dimethyl sulfoxide (DMSO) or PBS: 1 M nicotinamide
(NAM), 50 mM EX527, 10 mM AGK2, 2 mg/ml cisplatin, 25 mg/ml
5-fluorouracil (5-FU), 10 mM irinotecan, 10 mg/ml oxaliplatin, 10
mg/l paclitaxel, 10 µM gefitinib, 5 mg/ml LY294002, 2 M
dichloroacetate (DCA) and 1.5 M metformin. All drugs were freshly
added to the medium for the experiments.
Cytotoxicity assay
HCT116 and SW620 cells were seeded at a density of
1x104 cells/well in 96-well plates and allowed to adhere
for 24 h. The cells were then treated with drugs in triplicate at
the indicated concentrations for 72 h. After the medium was
discarded, fresh medium containing 10 µl CCK-8 solution (Dojindo
Molecular Technologies, Inc.) was added to each well. The
absorbance values at 450 nm were measured and cell viability was
calculated as the ratio of the absorbance values between the
drug-treated and equal dose vehicle (up to 0.5% DMSO in
PBS)-treated cells. IC50 values of the different drugs
were determined using inhibition dose-response curves with variable
slopes, as previously described (21).
Drug screening
The synergistic or antagonistic effects of various
drugs were analyzed according to the Chou-Talalay method (22) using the CompuSyn software program
(Version 1.0.1; ComboSyn, Inc.). The combination index (CI) was
calculated as D1/Dx1 +
D2/Dx2, wherein D1 or
D2 are the inhibitory concentrations of the individual
drugs and Dx1 or Dx2 the inhibitory
concentration of the drugs when used in combination. A CI<1 and
>1 indicate synergistic and antagonistic effects, respectively
(22). The -log10 of the
CI value was used to define chemo-sensitization (positive value) or
antagonism (negative value). At least three independent experiments
were performed.
Cell cycle assay
CRC cell lines were treated with vehicle (0.1% DMSO
in PBS), cisplatin (2 and 0.2 µg/ml), NAM (3 and 5 mM) or a
combination of these drugs for 72 h at 37˚C. The treated cells were
fixed in 70% ethanol for at least 12 h at -20˚C, followed by
incubation with 500 µl propidium iodide/RNase Staining Buffer
Solution (BD Pharmingen; BD Biosciences) for 15 min at room
temperature. The stained cells were assessed by flow cytometry
using a FACSMelody Flow Cytometer (BD Biosciences) and the
proportion of cells in the different cell cycle stages were
analyzed using the Modfit LT software (version 3.1; Verity Software
House).
Western blotting
HCT116 and SW620 cells were treated with 5 mM NAM or
vehicle (PBS) and lysed on ice with RIPA buffer (Nanjing KeyGen
Biotech Co., Ltd.) containing protease inhibitors. The lysates were
centrifuged at 15,000 x g, 4˚C for 20 min to remove the cell debris
and the concentration of total protein was determined using the BCA
Protein Quantification kit [Yeasen Biotechnology (Shanghai) Co.,
Ltd.]. 20 µg (5-20 µl volume) protein in each lane were separated
by SDS-PAGE (7.5% separating gel) and transferred to PVDF membranes
(GE Healthcare). The membranes were sequentially incubated with the
primary antibodies overnight at 4˚C and secondary antibodies for 1
h at room temperature and the bands were detected using ECL HRP
substrate (Sigma-Aldrich; Merck KGaA) in the bioanalytical imaging
system c300 (Azure Biosystems, Inc.). The following primary
antibodies were used: anti-p53 (cat. no. 10442-1-AP; ProteinTech
Group, Inc.), anti-histone H3K9 acetylation (cat. no. A7255;
ABclonal, Inc.), anti-histone H3 (cat. no. 17168-1-AP; ProteinTech
Group, Inc.), anti-phospho-p53 (cat. no. 9284; Cell Signaling
Technology, Inc.), anti-p21 (cat. no. 10355-1-AP; ProteinTech
Group, Inc.), anti-SIRT1 (cat. no. 60303-1-Ig; ProteinTech Group,
Inc.) and anti-GAPDH (cat. no. G9545; Sigma-Aldrich; Merck KGaA).
The anti-GAPDH antibody was used as the reference antibody.
HRP-labeled goat anti-rabbit (cat. no. KGAA35; Nanjing KeyGen
Biotech Co., Ltd.) and goat anti-mouse (cat. no. KGAA37; Nanjing
KeyGen Biotech Co., Ltd.) secondary antibodies were used.
Microarray datasets and gene set
enrichment analysis (GSEA)
Gene expression profiles of TP53wt
and TP53mut CRC and other tumor cell lines
(GSE41258, GSE57343) were downloaded from the Gene Expression
Omnibus (GEO) database (accessed on April 22nd, 2019) (23,24). The
GSE41258 dataset included the expression data of the
TP53wt lines HTB39 (GSM1012660), LNCaP (prostate
cancer cell line, GSM1012661) and LOVO (GSM1012662). The datasets
also contained the TP53mut lines DLD1 (S241F,
GSM1012656), HCT15 (S241F P153A, GSM1012657), HT29 (R273H,
GSM1012659), SW1116 (A159D, GSM1012665), SW620 (R273H, GSM1012666)
and WiDr (R273H, GSM1012667). The GSE57343 dataset included SW620
(GSM1380254-GSM1380259) and HCT116 (GSM1380296-GSM1380301) cell
lines. GSEA (version 4.0.3; Broad Institute, Inc.) was performed
using the above datasets to explore potential Kyoto Encyclopedia of
Genes and Genomes (KEGG) pathways and protein acetylation or
deacetylation-related Gene Ontology (GO) gene sets using the
Molecular Signatures Database (MsigDB, version 7.0; Broad
Institute, Inc.). Heat maps of differentially expressed genes
(DEGs) were drawn using GraphPad Prism 7 (GraphPad Software, Inc.),
as demonstrated in a previous study (25).
Statistical analysis
GraphPad Prism 7 was used for statistical analysis
of cell cycle data. One-way analysis of variance (ANOVA) followed
by Bonferroni post-hoc test was used to analyze the differences
between each two groups in the cell cycle assays. Data are
expressed as the mean ± SD of three independent experiments and
P<0.05 was considered statistically significant.
Results
SIRT inhibitors show synergistic
effects with chemotherapeutic agents in TP53 mut CRC cells
The HCT116 (KRASmut and
TP53wt) and SW620 (KRASmut and
TP53mut) cells were treated with NAM (a broad
spectrum SIRT inhibitor), EX527 (a SIRT1 inhibitor) or AGK2 (a
SIRT2 inhibitor) and the respective IC50 values were
calculated (Fig. 1). Similarly, the
IC50 values of cisplatin, 5-FU, irinotecan, oxaliplatin,
paclitaxel, EGFR inhibitor gefitinib, PI3K inhibitor LY294002,
pyruvate dehydrogenase kinase inhibitor DCA and the gluconeogenesis
inhibitor metformin were determined (Fig. 2). These findings suggested that, SIRT
inhibitors and chemotherapeutic agents had tumor inhibitory effects
on CRCs.
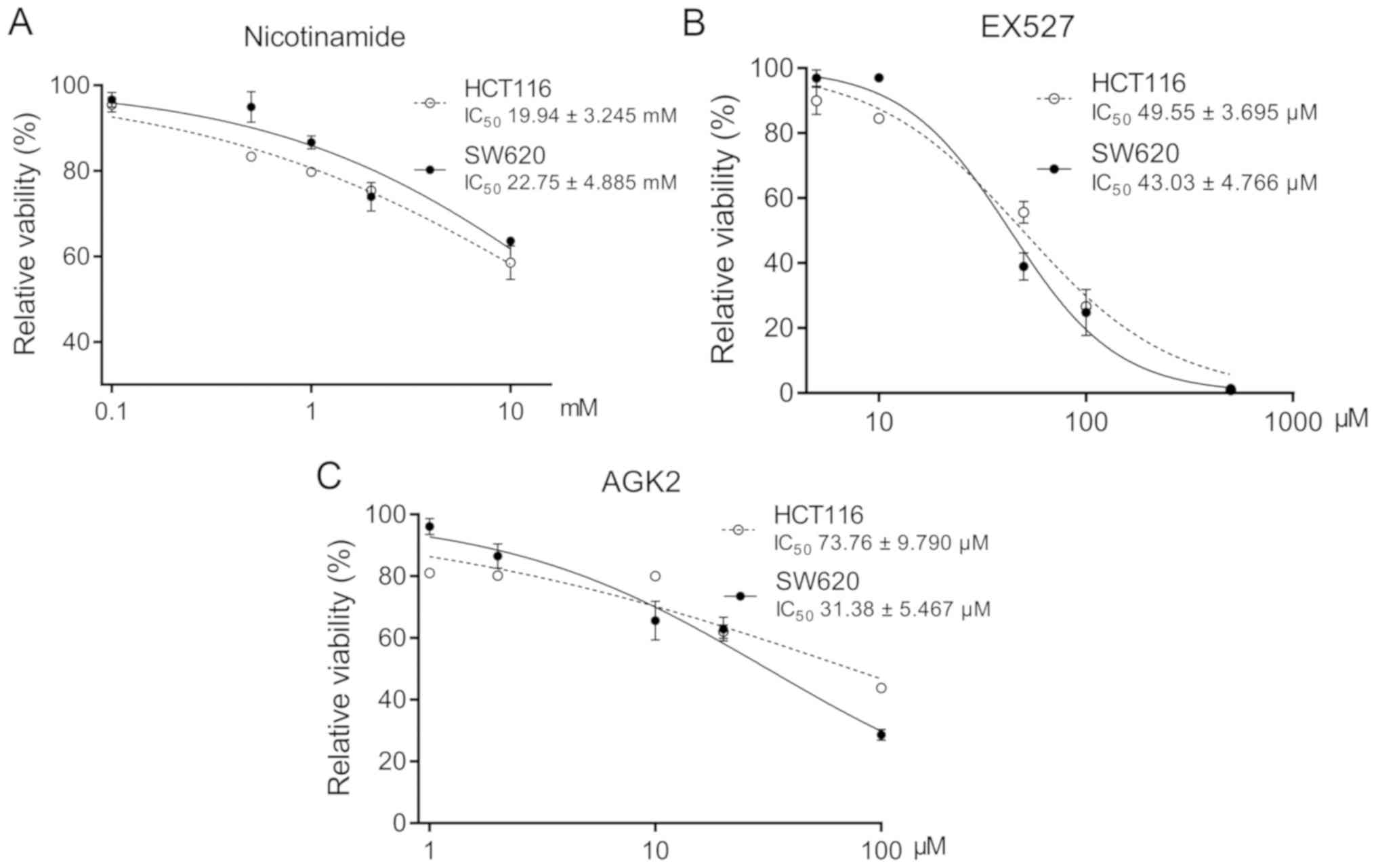 | Figure 1Cytotoxicity of SIRT inhibitors on
TP53wt and TP53mut CRC cells.
The viability of HCT116 (TP53wt) and SW620
(TP53mut) cells treated with (A) 0.1, 0.5, 1, 2
and 10 mM nicotinamide; (B) 5, 10, 50, 100 and 500 µM EX527; or (C)
1, 2, 10, 20 and 100 µM AGK2 for 72 h. The IC50 values
for each agent in both cell lines are presented as the mean ± SD of
three independent experiments. CRC, colorectal cancer; SIRT,
sirtuin; wt, wild-type; mut, mutant. |
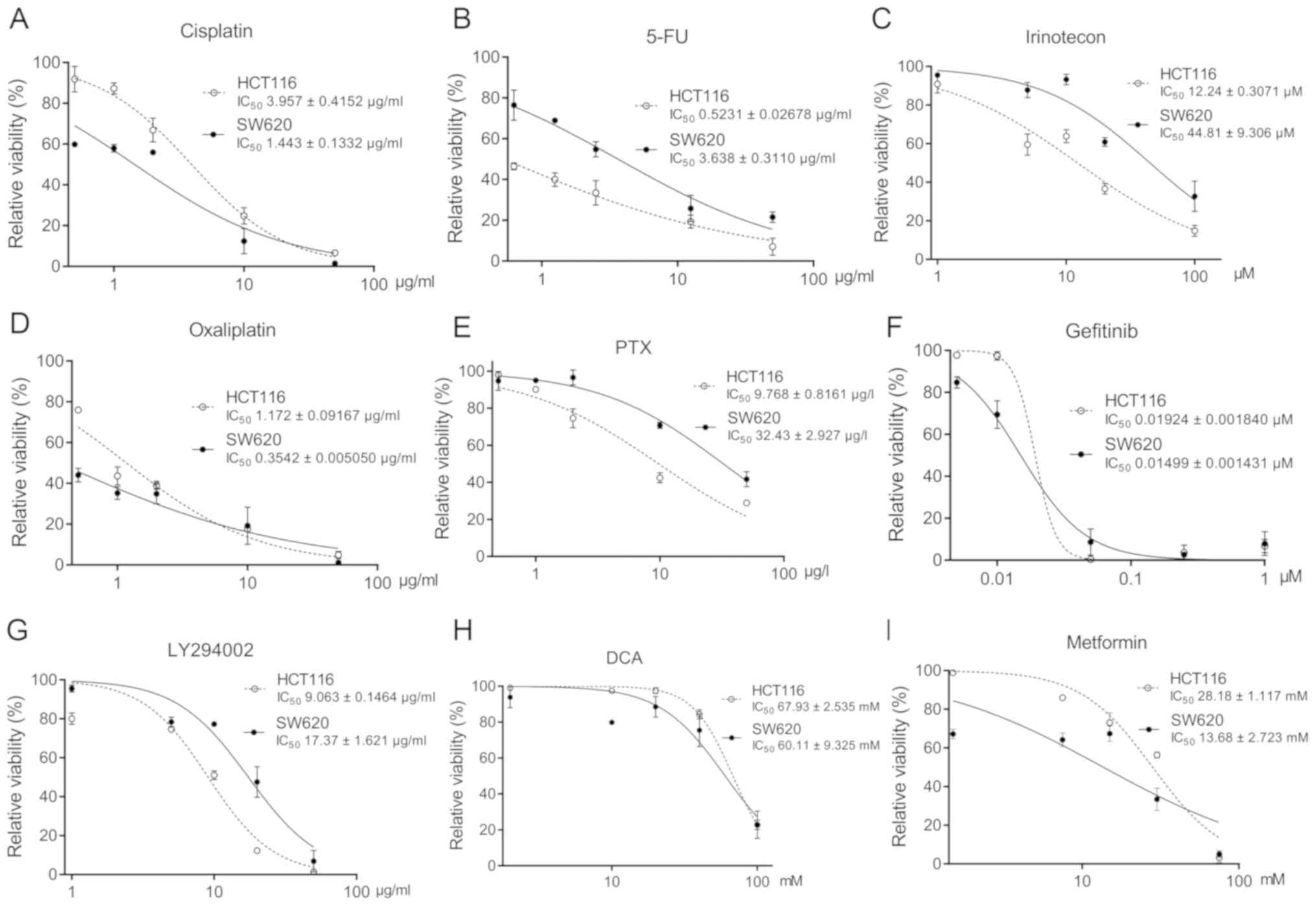 | Figure 2Cytotoxicity of chemotherapeutic
agents on TP53wt or TP53mut CRC
cells. Viability of HCT116 and SW620 cells treated with (A) 0.5, 1,
2, 10 and 50 µg/ml cisplatin; (B) 0.625, 1.25, 2.5, 12.5 and 50
µg/ml 5-FU; (C) 1, 5, 10, 20 and 100 µM irinotecan; (D) 0.5, 1, 2,
10 and 50 µg/ml oxaliplatin; (E) 0.5, 1, 2, 10 and 50 µg/l PTX; (F)
0.005, 0.01, 0.05, 0.25 and 1 µM gefitinib; (G) 1, 5, 10, 20 and 50
µg/ml LY294002; (H) 2, 10, 20, 40 and 100 mM DCA; and (I) 1.5, 7.5,
15, 30 and 75 mM metformin for 72 h. The IC50 values are
presented as the mean ± SD of three independent experiments. 5-FU,
5-fluorouracil; CRC, colorectal cancer; SIRT, sirtuin; wt,
wild-type; mut, mutant. |
Inhibition curves were used to predict the 30%
inhibitory concentration of each agent in both the HCT116 and SW620
cells (Table I), followed by the
calculation of the CI and the -log10 CI. In the
TP53wt HCT116 cells, cisplatin, 5-FU,
oxaliplatin, gefitinib, LY294002 and metformin were antagonistic to
the SIRT inhibitors, whereas irinotecan and paclitaxel acted
synergistically (Fig. 3A-C). In the
TP53mut SW620 cells, the majority of the
chemotherapeutic agents showed a weak synergism with the SIRT
inhibitors. Cell cycle analysis also confirmed that when used in
combination, cisplatin and NAM were not more effective for
preventing the G1-S transition of HCT116 cells compared with
cisplatin alone (Fig. 3D). However,
the G1-S transition was significantly reduced with a combination of
cisplatin and NAM when compared to the use of each drug
individually in the SW620 cells (Fig.
3E). These results suggested that SIRT inhibitors may
antagonize chemotherapeutic drugs in TP53wt CRC
cells, while inhibition of SIRTs may make TP53mut
cells more sensitive to other chemotherapeutic agents in this
assay.
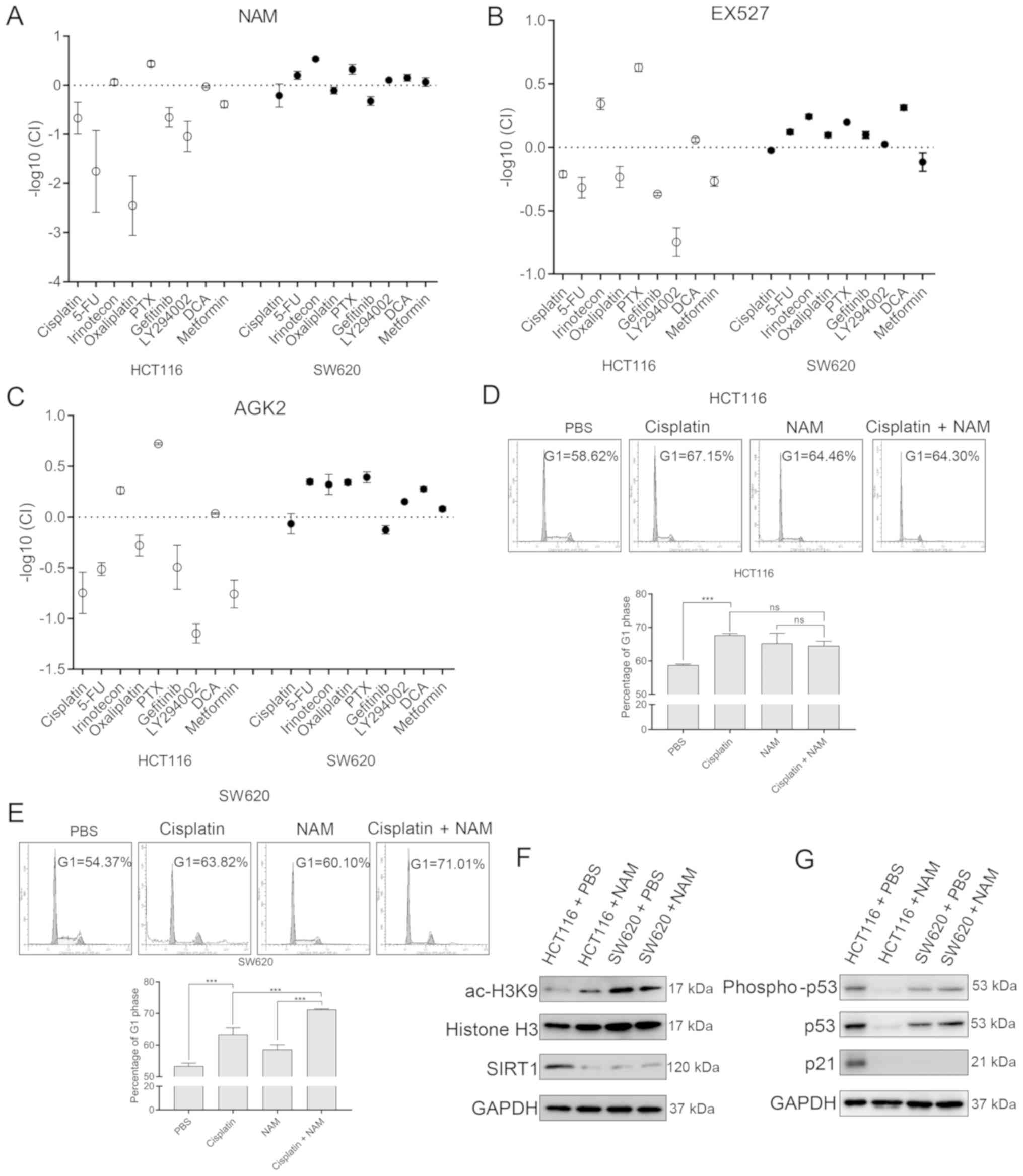 | Figure 3SIRT inhibitors antagonize
chemotherapeutic agents in TP53wt CRC cells by
reducing wild-type 53 protein levels. The log CI values between (A)
NAM, (B) EX527 or (C) AGK2 and various chemotherapeutic agents.
Cell cycle profile of (D) HCT116 and (E) SW620 cells treated with
cisplatin and/or NAM for 72 h. Data are presented as the mean± SD
of three independent experiments. (F and G) Immunoblots showing
levels of p53, phospho-p53, p21, SIRT1 and histone H3K9 acetylation
in HCT116 and SW620 cells treated with 5mM NAM or vehicle for 72 h.
***P<0.001; ns, no significance. CRC, colorectal
cancer; CI, combination index; NAM, nicotinamide; SIRT, sirtuin;
wt, wild-type; mut, mutant. |
 | Table IEstimated concentration of each agent
that led to 30% inhibition (70% survival) of the two CRC cell
lines. |
Table I
Estimated concentration of each agent
that led to 30% inhibition (70% survival) of the two CRC cell
lines.
| | Concentration in
each cell type |
|---|
| Drug | HCT116 | SW620 |
|---|
| Nicotinamide | 3 mM | 5 mM |
| EX527 | 20 µM | 35 µM |
| AGK2 | 15 µM | 10 µM |
| Cisplatin | 2 µg/ml | 0.2 µg/ml |
| 5-Fluorouracil | 0.2 µg/ml | 0.9 µg/ml |
| Irinotecon | 4.6 µM | 15 µM |
| Oxaliplatin | 0.45 µg/ml | 0.05 µg/ml |
| Paclitaxel | 4 µg/l | 5.7 µg/l |
| Gefitinib | 0.02 µM | 0.01 µM |
| LY294002 | 3.6 µg/ml | 10 µg/ml |
|
Dichloroacetate | 50 mM | 26 mM |
| Metformin | 17 mM | 3.4 mM |
P53 status and its level in CRC cells
determines the combined effects of SIRT inhibitor and
chemotherapeutic agents
TP53 is frequently mutated into a
proto-oncogene in tumor cells (26),
which encodes a highly acetylated protein that is unstable and
degrades easily (27). Therefore,
the present study aimed to determine whether similar p53 protein
levels and acetylation status existed in HCT116 and SW620 cells. In
the present study the level of the deacetylase SIRT1 was
significantly higher in HCT116 cells when compared to SW620 cells,
with high levels of wt p53. In SW620 cells, the level of the
deacetylase SIRT1 was lower than that in HCT116 cells, which may
lead to increased levels of acylated and easily-degraded p53 mut
(Fig. 3F). Treatment with NAM
reduced the levels of the wt p53 protein, activated p-p53Ser15 and
its downstream target p21 in HCT116 cells. This effect was not
observed in the SW620 cells with a mutated p53 protein (Fig. 3G).
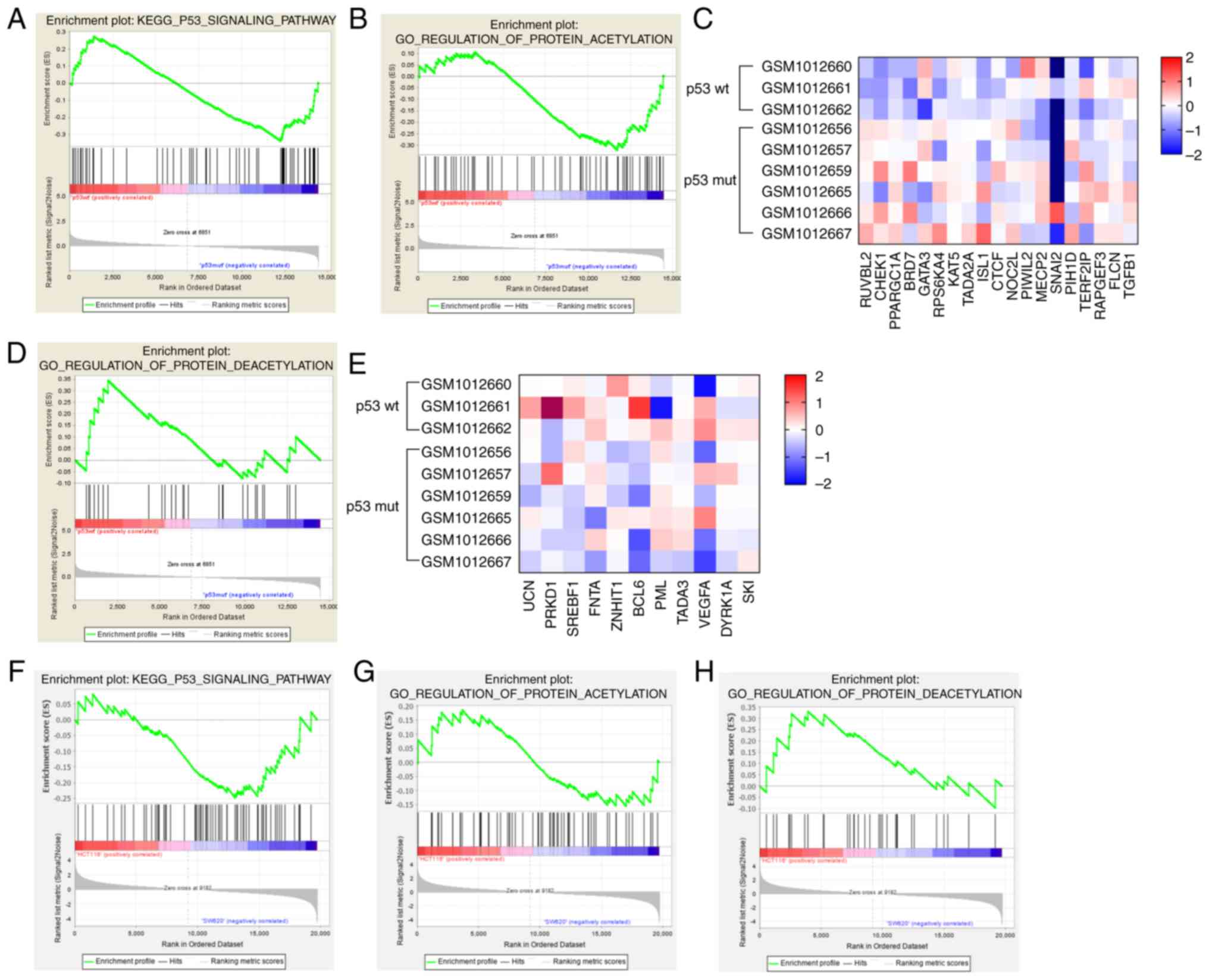
Enrichment of genes associated with
the GO-term protein deacetylation in CRC cells with wt p53
expression
The transcriptome data of TP53wt
and TP53mut cancer cell lines was extracted from
the RNA-Seq GSE41258 dataset and submitted to GSEA for enrichment
analysis. KEGG pathway analysis did not reveal any significant
differences in the enrichment of genes related to the term ‘p53
signaling pathway’ between the cell lines (Fig. 4A). However, TP53mut
cells were enriched in genes related to the GO-term ‘protein
acetylation’ (Fig. 4B), which were
also differentially expressed compared to that in the
TP53wt cells (Fig.
4C). Consistent with this, genes associated with the GO term
‘protein deacetylation’ were enriched in the
TP53wt cells (Fig.
4D and E). GSEA of the HCT116 and SW620 transcriptomes (from
the GSE7343 dataset) similarly showed a downregulation of genes
associated with the KEGG term ‘p53 signaling pathway’ and the
GO-term ‘protein deacetylation’ in SW620 cells (Fig. 4F-H). Taken together, these results
suggested that the protein deacetylation machinery may be more
activated in the TP53wt compared to
TP53mut CRC cells. With the blockage of the SIRTs
inhibitors, the stable wt p53 was significantly reduced, which may
antagonize the action of SIRT inhibitors in combination with a
chemotherapeutic drug.
Discussion
Studies show that ~60% of CRC cases harbor
TP53 mutations, which correlate with greater malignancy
(28). The tumor suppressor
TP53 is a ‘master regulator’ of cellular processes including
the cell cycle, apoptosis and DNA damage repair (29). Gain-of-function mutations in
TP53, such as V143A, R248Q, R273H and R280K, confer a
malignant phenotype on tumor cells by promoting proliferation,
invasion, metastasis and chemo-resistance (30). In CRC cells, mut p53 protein binds to
STAT3 and activates the pro-tumorigenic Jak2/STAT3 signaling
pathway, which increases tumor invasiveness, leading to a worse
prognosis (31). In addition, mut
p53 also drives CRC progression and chemo-resistance by increasing
cancer stem cell renewal and reprogramming tumor-associated
macrophages (32,33). Distinct therapeutic strategies are
needed against TP53wt and
TP53mut CRCs, as they are likely to differ in
their chemo-sensitivities. For example, the therapeutic potential
of ascorbic acid is higher when used in combination with first-line
drugs such as 5-FU or oxaliplatin in TP53mut CRC
cells (34). Furthermore, the Wee1
inhibitor MK1775 induces apoptosis in the TP53mut
HT29 and SW480 cells and sensitizes them to irinotecan (35). In contrast, the histone deacetylase
inhibitors, valproic acid and capecitabine, are antagonistic in
p53-deficient CRC cells, but act synergistically in cells
expressing normal or mut p53(36).
In the present study, SIRT inhibitors sensitized
TP53mut CRC cells to chemotherapeutic agents and
TP53wt CRC cells to only irinotecan or
paclitaxel, while antagonizing the other drugs. These findings
suggested that SIRT inhibitors are promising for
TP53mut refractory or drug-resistant CRC but not
suitable for TP53wt CRC.
The role of SIRTs in tumorigenesis, tumor
progression and metastasis is controversial. SIRT1 acts as tumor
suppressor in TP53mut hepatocellular carcinoma
and its high levels predict a favorable prognosis (37). In esophageal squamous cell carcinoma;
however, miR-34a-mediated inhibition of SIRT1 and induction of p53
exerted an anti-tumor effect (38).
Some SIRT inhibitors retard tumor growth by attenuating the
deacetylase activity of SIRTs and downregulating tumorigenic
signaling pathways. For example, the antipsychotic drug
chlorpromazine induces apoptosis in CRC cells by downregulating
SIRT1(39) and the SIRT inhibitor
benzimidazole also inhibits growth of CRC cells (40). SIRT inhibitors used in the present
cytotoxicity and cell cycle experiments appeared to show an
inhibitory effect on two CRC cell lines. However, cell cycle
analysis only determines the proportion of viable dividing cell
populations, which presents limitations in determining the
proportion of apoptotic cells (41).
In addition, SIRT levels were not the decisive reason for the
different chemosensitivity in HCT116 and SW620 cells in the present
study. This can be concluded because after NAM treatment SIRT1 was
induced to be the same level in the two cell lines, which suggested
that the baseline levels of SIRT1 were the same for the combination
usage of the SIRT inhibitors and those chemotherapeutic agents.
From these data, it was concluded that the baseline difference of
SIRT1 in these two tested cell lines could not be a driving or
effective factor that leads to the efficacy-divergency of the
combination treatment.
p53 protein binds to HDAC6 and HSP90 to form a
complex, which protects it from ubiquitin protease-mediated
degradation (42). HDAC6 inhibitors
interfere with the formation of this complex and degrade p53 to an
unstable state (43). In the present
study, NAM suppressed SIRT activity and reduced the stability of
both wt and mut p53. However, the overall levels of acetylated p53
were low in the TP53wt HCT116 cells when compared
with TP53mut SW620 cells, which corresponded with
high levels of the stable p53 protein. Therefore, a reduced pool of
stable p53 was underlying the antagonism between NAM and multiple
chemotherapeutic agents.
In conclusion, the experimental data and
bioinformatics analysis in the present study suggested that
TP53 status may be responsible for the divergence in CRC
cell chemosensitivity profiles. The findings also suggested that a
combination of SIRT inhibitors and first-line drugs may be
beneficial for patients with TP53mut CRC.
Acknowledgements
Not applicable.
Funding
This study was supported by research grants from
National Natural Science Foundation of China (grant nos. 81903065
and 81803581), Shanghai Key Laboratory of Molecular Imaging (grant
no. 18DZ2260400), The Collaborative Innovation Key Project of
Shanghai University of Medical and Health Sciences (grant no.
SPCI-18-18-003) and the Program for Professor of Special
Appointment (grant no. Eastern Scholar TP2018080) at Shanghai
Institutions of Higher Learning.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HY performed experiments and wrote the manuscript.
JY and ZZ designed the study and revised the manuscript. HY, JY and
ZZ contributed to the cell cycle assay and GSEA data analysis. HY,
DW and YC performed the cytotoxicity assay and drug sensitivity
analysis. YC and YJ performed statistical and bioinformatics
analysis. All authors read and approved the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Usher-Smith JA, Walter FM, Emery JD, Win
AK and Griffin SJ: Risk prediction models for colorectal cancer: A
Systematic Review. Cancer Prev Res (Phila). 9:13–26.
2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Matsuda T, Yamashita K, Hasegawa H,
Oshikiri T, Hosono M, Higashino N, Yamamoto M, Matsuda Y, Kanaji S,
Nakamura T, et al: Recent updates in the surgical treatment of
colorectal cancer. Ann Gastroenterol Surg. 2:129–136.
2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Redondo-Blanco S, Fernández J,
Gutiérrez-Del-Río I, Villar CJ and Lombó F: New insights toward
colorectal cancer chemotherapy using natural bioactive compounds.
Front Pharmacol. 8(109)2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wu T, Wang Z, Liu Y, Mei Z, Wang G, Liang
Z, Cui A, Hu X, Cui L, Yang Y, et al: Interleukin 22 protects
colorectal cancer cells from chemotherapy by activating the STAT3
pathway and inducing autocrine expression of interleukin 8. Clin
Immunol. 154:116–126. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wang B, Ogata H, Takishima Y, Miyamoto S,
Inoue H, Kuroda M, Yamada K, Hijikata Y, Murahashi M, Shimizu H, et
al: A novel combination therapy for human oxaliplatin-resistant
colorectal cancer using oxaliplatin and coxsackievirus A11.
Anticancer Res. 38:6121–6126. 2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Chen M, Liang X, Gao C, Zhao R, Zhang N,
Wang S, Chen W, Zhao B, Wang J and Dai Z: Ultrasound triggered
conversion of porphyrin/camptothecin-fluoroxyuridine triad
microbubbles into nanoparticles overcomes multidrug resistance in
colorectal cancer. ACS Nano. 12:7312–7326. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Ou J, Peng Y, Yang W, Zhang Y, Hao J, Li
F, Chen Y, Zhao Y, Xie X, Wu S, et al: ABHD5 blunts the sensitivity
of colorectal cancer to fluorouracil via promoting autophagic
uracil yield. Nat Commun. 10(1078)2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Park SM, Hwang CY, Cho SH, Lee D, Gong JR,
Lee S, Nam S and Cho KH: Systems analysis identifies potential
target genes to overcome cetuximab resistance in colorectal cancer
cells. FEBS J. 286:1305–1318. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Zhu S, Dong Z, Ke X, Hou J, Zhao E, Zhang
K, Wang F, Yang L, Xiang Z and Cui H: The roles of sirtuins family
in cell metabolism during tumor development. Semin Cancer Biol.
57:59–71. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Carafa V, Altucci L and Nebbioso A: Dual
tumor suppressor and tumor promoter action of sirtuins in
determining malignant phenotype. Front Pharmacol.
10(38)2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Mei Z, Zhang X, Yi J, Huang J, He J and
Tao Y: Sirtuins in metabolism, DNA repair and cancer. J Exp Clin
Cancer Res. 35(182)2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhu Y, Wang G, Li X, Wang T, Weng M and
Zhang Y: Knockout of SIRT4 decreases chemosensitivity to 5-FU in
colorectal cancer cells. Oncol Lett. 16:1675–1681. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Vellinga TT, Borovski T, de Boer VC,
Fatrai S, van Schelven S, Trumpi K, Verheem A, Snoeren N, Emmink
BL, Koster J, et al: SIRT1/PGC1α-dependent increase in oxidative
phosphorylation supports chemotherapy resistance of colon cancer.
Clin Cancer Res. 21:2870–2879. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Bajpe PK, Prahallad A, Horlings H,
Nagtegaal I, Beijersbergen R and Bernards R: A chromatin modifier
genetic screen identifies SIRT2 as a modulator of response to
targeted therapies through the regulation of MEK kinase activity.
Oncogene. 34:531–536. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
San Hipólito-Luengo Á, Alcaide A,
Ramos-González M, Cercas E, Vallejo S, Romero A, Talero E,
Sánchez-Ferrer CF, Motilva V and Peiró C: Dual effects of
resveratrol on cell death and proliferation of colon cancer cells.
Nutr Cancer. 69:1019–1027. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Oon CE, Strell C, Yeong KY, Östman A and
Prakash J: SIRT1 inhibition in pancreatic cancer models:
Contrasting effects in vitro and in vivo. Eur J Pharmacol.
757:59–67. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ma W, Zhao X, Wang K, Liu J and Huang G:
Dichloroacetic acid (DCA) synergizes with the SIRT2 inhibitor
Sirtinol and AGK2 to enhance anti-tumor efficacy in non-small cell
lung cancer. Cancer Biol Ther. 19:835–846. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Hsu YF, Sheu JR, Lin CH, Yang DS, Hsiao G,
Ou G, Chiu PT, Huang YH, Kuo WH and Hsu MJ: Trichostatin A and
sirtinol suppressed survivin expression through AMPK and p38MAPK in
HT29 colon cancer cells. Biochim Biophys Acta. 1820:104–115.
2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wang Y, Yang L, Zhang J, Zhou M, Shen L,
Deng W, Liang L, Hu R, Yang W, Yao Y, et al: Radiosensitization by
irinotecan is attributed to G2/M phase arrest, followed by enhanced
apoptosis, probably through the ATM/Chk/Cdc25C/Cdc2 pathway in
p53-mutant colorectal cancer cells. Int J Oncol. 53:1667–1680.
2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Sedic M, Poznic M, Gehrig P, Scott M,
Schlapbach R, Hranjec M, Karminski-Zamola G, Pavelic K and
Kraljevic Pavelic S: Differential antiproliferative mechanisms of
novel derivative of benzimidazo[1,2-alpha]quinoline in colon cancer
cells depending on their p53 status. Mol Cancer Ther. 7:2121–2132.
2008.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Sen Z, Zhan XK, Jing J, Yi Z and Wanqi Z:
Chemosensitizing activities of cyclotides from Clitoria
ternatea in paclitaxel-resistant lung cancer cells. Oncol Lett.
5:641–644. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Sheffer M, Bacolod MD, Zuk O, Giardina SF,
Pincas H, Barany F, Paty PB, Gerald WL, Notterman DA and Domany E:
Association of survival and disease progression with chromosomal
instability: A genomic exploration of colorectal cancer. Proc Natl
Acad Sci USA. 106:7131–7136. 2009.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Li H, Chiappinelli KB, Guzzetta AA,
Easwaran H, Yen RW, Vatapalli R, Topper MJ, Luo J, Connolly RM,
Azad NS, et al: Immune regulation by low doses of the DNA
methyltransferase inhibitor 5-azacitidine in common human
epithelial cancers. Oncotarget. 5:587–598. 2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhang Y, He W and Zhang S: Seeking for
correlative genes and signaling pathways with bone metastasis from
breast cancer by integrated analysis. Front Oncol.
9(138)2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Leroy B, Fournier JL, Ishioka C, Monti P,
Inga A, Fronza G and Soussi T: The TP53 website: An integrative
resource centre for the TP53 mutation database and TP53 mutant
analysis. Nucleic Acids Res. 41:D962–D969. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Blagosklonny MV, Trostel S, Kayastha G,
Demidenko ZN, Vassilev LT, Romanova LY, Bates S and Fojo T:
Depletion of mutant p53 and cytotoxicity of histone deacetylase
inhibitors. Cancer Res. 65:7386–7392. 2005.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Nakayama M and Oshima M: Mutant p53 in
colon cancer. J Mol Cell Biol. 11:267–276. 2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Aubrey BJ, Strasser A and Kelly GL:
Tumor-suppressor functions of the TP53 pathway. Cold Spring Harb
Perspect Med. 6(6)2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Parrales A and Iwakuma T: Targeting
oncogenic mutant p53 for cancer therapy. Front Oncol.
5(288)2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Schulz-Heddergott R, Stark N, Edmunds SJ,
Li J, Conradi LC, Bohnenberger H, Ceteci F, Greten FR, Dobbelstein
M and Moll UM: Therapeutic ablation of gain-of-function mutant p53
in colorectal cancer inhibits Stat3-mediated tumor growth and
invasion. Cancer Cell. 34:298–314.e7. 2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Cooks T, Pateras IS, Jenkins LM, Patel KM,
Robles AI, Morris J, Forshew T, Appella E, Gorgoulis VG and Harris
CC: Mutant p53 cancers reprogram macrophages to tumor supporting
macrophages via exosomal miR-1246. Nat Commun.
9(771)2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Solomon H, Dinowitz N, Pateras IS, Cooks
T, Shetzer Y, Molchadsky A, Charni M, Rabani S, Koifman G, Tarcic
O, et al: Mutant p53 gain of function underlies high expression
levels of colorectal cancer stem cells markers. Oncogene.
37:1669–1684. 2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Pires AS, Marques CR, Encarnação JC,
Abrantes AM, Marques IA, Laranjo M, Oliveira R, Casalta-Lopes JE,
Gonçalves AC, Sarmento-Ribeiro AB, et al: Ascorbic acid
chemosensitizes colorectal cancer cells and synergistically
inhibits tumor growth. Front Physiol. 9(911)2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Yin Y, Shen Q, Tao R, Chang W, Li R, Xie
G, Liu W, Zhang P and Tao K: Wee1 inhibition can suppress tumor
proliferation and sensitize p53 mutant colonic cancer cells to the
anticancer effect of irinotecan. Mol Med Rep. 17:3344–3349.
2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Terranova-Barberio M, Pecori B, Roca MS,
Imbimbo S, Bruzzese F, Leone A, Muto P, Delrio P, Avallone A,
Budillon A, et al: Synergistic antitumor interaction between
valproic acid, capecitabine and radiotherapy in colorectal cancer:
Critical role of p53. J Exp Clin Cancer Res. 36(177)2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Zhang ZY, Hong D, Nam SH, Kim JM, Paik YH,
Joh JW, Kwon CH, Park JB, Choi GS, Jang KY, et al: SIRT1 regulates
oncogenesis via a mutant p53-dependent pathway in hepatocellular
carcinoma. J Hepatol. 62:121–130. 2015.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Ye Z, Fang J, Dai S, Wang Y, Fu Z, Feng W,
Wei Q and Huang P: MicroRNA-34a induces a senescence-like change
via the down-regulation of SIRT1 and up-regulation of p53 protein
in human esophageal squamous cancer cells with a wild-type p53 gene
background. Cancer Lett. 370:216–221. 2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Lee WY, Lee WT, Cheng CH, Chen KC, Chou
CM, Chung CH, Sun MS, Cheng HW, Ho MN and Lin CW: Repositioning
antipsychotic chlorpromazine for treating colorectal cancer by
inhibiting sirtuin 1. Oncotarget. 6:27580–27595. 2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Tan YJ, Lee YT, Yeong KY, Petersen SH,
Kono K, Tan SC and Oon CE: Anticancer activities of a benzimidazole
compound through sirtuin inhibition in colorectal cancer. Future
Med Chem. 10:2039–2057. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Fraker PJ, King LE, Lill-Elghanian D and
Telford WG: Quantification of apoptotic events in pure and
heterogeneous populations of cells using the flow cytometer.
Methods Cell Biol. 46:57–76. 1995.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Li D, Marchenko ND and Moll UM: SAHA shows
preferential cytotoxicity in mutant p53 cancer cells by
destabilizing mutant p53 through inhibition of the HDAC6-Hsp90
chaperone axis. Cell Death Differ. 18:1904–1913. 2011.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Ryu HW, Shin DH, Lee DH, Choi J, Han G,
Lee KY and Kwon SH: HDAC6 deacetylates p53 at lysines 381/382 and
differentially coordinates p53-induced apoptosis. Cancer Lett.
391:162–171. 2017.PubMed/NCBI View Article : Google Scholar
|