Introduction
Chronic progressive external ophthalmoplegia (CPEO)
is a mitochondrial disease characterized by chronic progressive
blepharoptosis, ocular dyskinesia, ptosis, limited of eye movement
and bulbar muscle weakness of varied severity (1,2). The
condition typically appears in adults aged 18-40 years and slowly
worsens over time. Limb involvement is rarely observed with CPEO,
and patients may only develop mild limb weakness. Therefore, the
disease is easily misdiagnosed as ocular myasthenia gravis,
oculopharyngeal muscular dystrophy, Fisher syndrome or other
ophthalmic diseases (3).
Mitochondria are cellular organelles found in all nucleated human
cells. CPEO can be caused by mitochondrial DNA (mtDNA) point
mutations, a single large scale mtDNA deletions, duplications or
multiple mtDNA deletions, secondary to nuclear mutations to genes
including ANT1, POLG1, POLG2, OPA1,
C10orf2 and SLC25A4 (3,4). In
total, ~70% of patients with CPEO were found to carry single
large-scale mtDNA deletions, whilst a small number of patients have
also been found to carry mtDNA point mutations (3-5).
Skeletal muscle biopsies are used to confirm the diagnosis of CPEO,
where >2% of the ragged red fibres (RRF) in skeletal muscle
biopsies are strongly suggestive of mitochondrial cytopathy
(6).
CPEO is a rare disorder with a low prevalence. In
the 20 years between January 1997 and January 2018, only 12
patients with CPEO were encountered among a total of 1,060 muscle
biopsies that were performed at the People's Hospital of Jiaozuo
City. In the present study, the clinical data of the 12 patients
with CEPO were retrospectively analyzed, with the aim of
investigating the clinical manifestations, neuroelectrophysiology
and muscle pathology of patients with CEPO, while improving the
current understanding of CPEO and promoting early diagnosis.
Materials and methods
Ethics statement
The present study was approved by the ethics
committee of the People's Hospital of Jiaozuo City (Jiaozuo,
China). All patients have provided written informed consent for
publication.
Subjects
This retrospective study summarized and analyzed the
clinical data of 12 patients who had been diagnosed with CPEO from
a total of 1,060 muscle biopsies performed in the People's Hospital
of Jiaozuo City between January 1997 and January 2018. There were 6
males and 6 females, aged between 9 and 56 years, with an average
age of 32 years. The disease duration was 2-25 years, with an
average of 13 years. The onset age was 7-35 years, with an average
of 17.2 years.
Electrophysiological examination
Concentric needle electromyography as well as
sensory and motor nerve conduction velocities in all the patients
were measured using an Evoked Potential/Electromyography Measuring
System (MEB-9200K; Nihon Kohden Corporation). Deltoid, biceps
brachii, iliopsoas, gluteus maximus, vastus medialis, tibialis
anterior and gastrocnemius muscles were selected for
electromyography. Data were recorded and analyzed, including: i)
spontaneous potential during the quiet period (occurrence in more
than two sites was considered an abnormality); ii) time limit,
amplitude and percentage of polyphasic waves of 20 motor unit
action potentials during contractions at small forces; and iii)
waveform and peak-to-peak amplitude of the recruitment potential
during contractions at large forces. Conduction velocities of the
median nerve, ulnar nerve, common peroneal nerve, posterior tibial
nerve and sural nerve were determined. The ulnar nerve and axillary
nerve were selected for repetitive nerve stimulation.
Pathological examination
All the patients underwent open biopsy after local
anesthesia, including biceps brachii in 8 cases, deltoid in 1 case
and quadriceps femoris in 3 cases. A portion of each muscle
specimen was frozen in liquid nitrogen, and sliced into 8-µm frozen
sections. Sections were subjected to conventional histological,
enzyme and histochemical staining, including hematoxylin and eosin
(H&E), modified Gomori trichrome, reduced nicotinamide adenine
dinucleotide tetrazolium reductase (NADH-TR), oil red O, periodic
acid-Schiff reaction and ATPase (pH 4.2, 4.3, 10.6 and 10.8).
Results were visualized using a light microscope (magnifications,
x200 and x400).
H&E staining
Frozen sections were first stained with hematoxylin
solution for 5-10 min, rinsed under running water for 5 min and
stained with eosin solution for 3 min at room temperature. The
sections were then dehydrated with an alcohol gradient, cleared in
xylene and mounted in neutral gum.
Modified Gomori trichrome
staining
Frozen sections were immersed in hematoxylin
solution for 5-10 min at room temperature, rinsed under running
water for 5 min, and then immersed in Gomori trichrome solution for
30 min at room temperature. After rinsing with distilled water, the
sections were dehydrated with an alcohol gradient, cleared in
xylene and mounted in neutral gum.
NADH-TR staining
Frozen sections were incubated with NADH-TR staining
solution for 30 min in a thermostat at 37˚C, rinsed with distilled
water, air-dried and finally sealed with glycerin gelatin.
Oil Red O staining
Frozen sections were stained in Oil Red O staining
solution for 30 min at room temperature. Following rinsing with
running water for 5 min, sections were counterstained with
hematoxylin solution for 1 min at room temperature, rinsed with
running water for 2 min, and sealed with glycerin gelatin.
Periodic acid-Schiff reaction
staining
Frozen sections were fixed in Carnoy's solution for
10 min at room temperature. Following rinsing with running water
for 5 min, the sections were immersed in 1% periodic acid solution
for 5 min, rinsed with distilled water for 2 min, and then immersed
in Schiff solution for 10 min at room temperature. After washing
with running water for 5 min, sections were counterstained with
hematoxylin solution for 2-3 min at room temperature, and rinsed
with running water for 5 min. Then the sections were dehydrated
with an alcohol gradient, cleared in xylene and mounted in neutral
gum.
ATPase staining
Two 8-µm frozen sections per patient were incubated
with sodium acetate solution (pH 4.2 and pH 4.3) for 10 min at room
temperature. Another two, 8-µm frozen sections were incubated with
calcium barbital solution (pH 10.6 and pH 10.8) at 37˚C for 10 min.
The four sections were then rinsed with running water for 5 min and
incubated with ATP/calcium barbital solution (pH 9.4) at 37˚C for
30 min. They were then incubated in 2% cobalt chloride for 3 min at
room temperature, rinsed with distilled water, immersed in 1%
ammonium sulfide for 3 min at room temperature, and rinsed with
running water for 5 min.
Electron microscopic examination
Another portion of each muscle specimen was fixed in
2.5% glutaraldehyde for 24 h at room temperature; washed three
times with sodium dimethylarsenate buffer; post-fixed in 1% osmic
acid (in sodium dimethylarsenate buffer) for 2 h at room
temperature; dehydrated with either an alcohol or acetone series;
and then embedded in epoxy resin 618 at 37˚C for 24 h and at 60˚C
for 48 h. The specimens were sliced into ultrathin sections (60 nm)
using a glass knife. The sections were stained with uranyl acetate
at room temperature for 20 min, rinsed thoroughly with water and
stained with lead citrate at room temperature for 20 min, following
which these sections were dried and observed using a H-7500
transmission electron microscope (magnification, x10,000; Hitachi,
Ltd.).
Results
Clinical characteristics
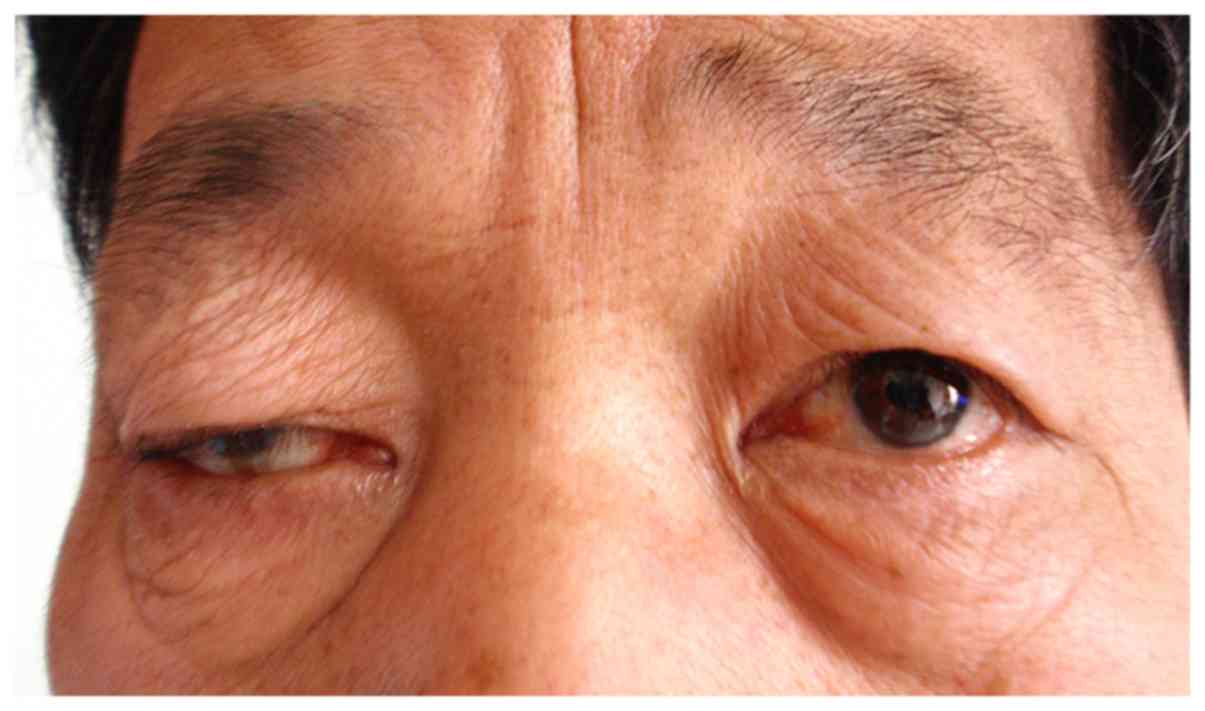
All 12 patients had varying degrees of chronic
progressive blepharoptosis. The eyelids covered half of the pupil
in 6 patients, two-thirds of the pupil in 4 patients and the whole
pupil in 2 patients (Fig. 1). Of the
12 patients, 11 patients suffered from obvious ocular dyskinesia
(including 8 patients with binocular fixation in the central
position) and 3 patients had obvious binocular horizontal and
vertical eye movement disorders. All patients had a normal pupil
size and a light reflex without diplopia. The disease duration was
between 2-25 years. The onset age was 7-35 years, with an average
of 17.2 years. Four patients developed mild limb weakness several
years after onset, and 1 patient displayed swallowing
difficulties.
Neuroelectrophysiological changes
Needle electromyography in 12 patients revealed
abnormal changes in 6 patients, including myogenic lesions in 4
patients, a neurogenic lesion in 1 patient and mixed
myogenic/neurogenic lesions in 1 patient. A decreased sensory nerve
conduction velocity was observed in 2 patients. One of these 2
patients had a decreased sensory conduction velocity of the median
nerve and posterior tibial nerve, and the other had a decreased
sensory conduction velocity of the sural nerve. Negative symptoms
were determined in 12 patients after repetitive stimulation.
Pathological changes in muscles
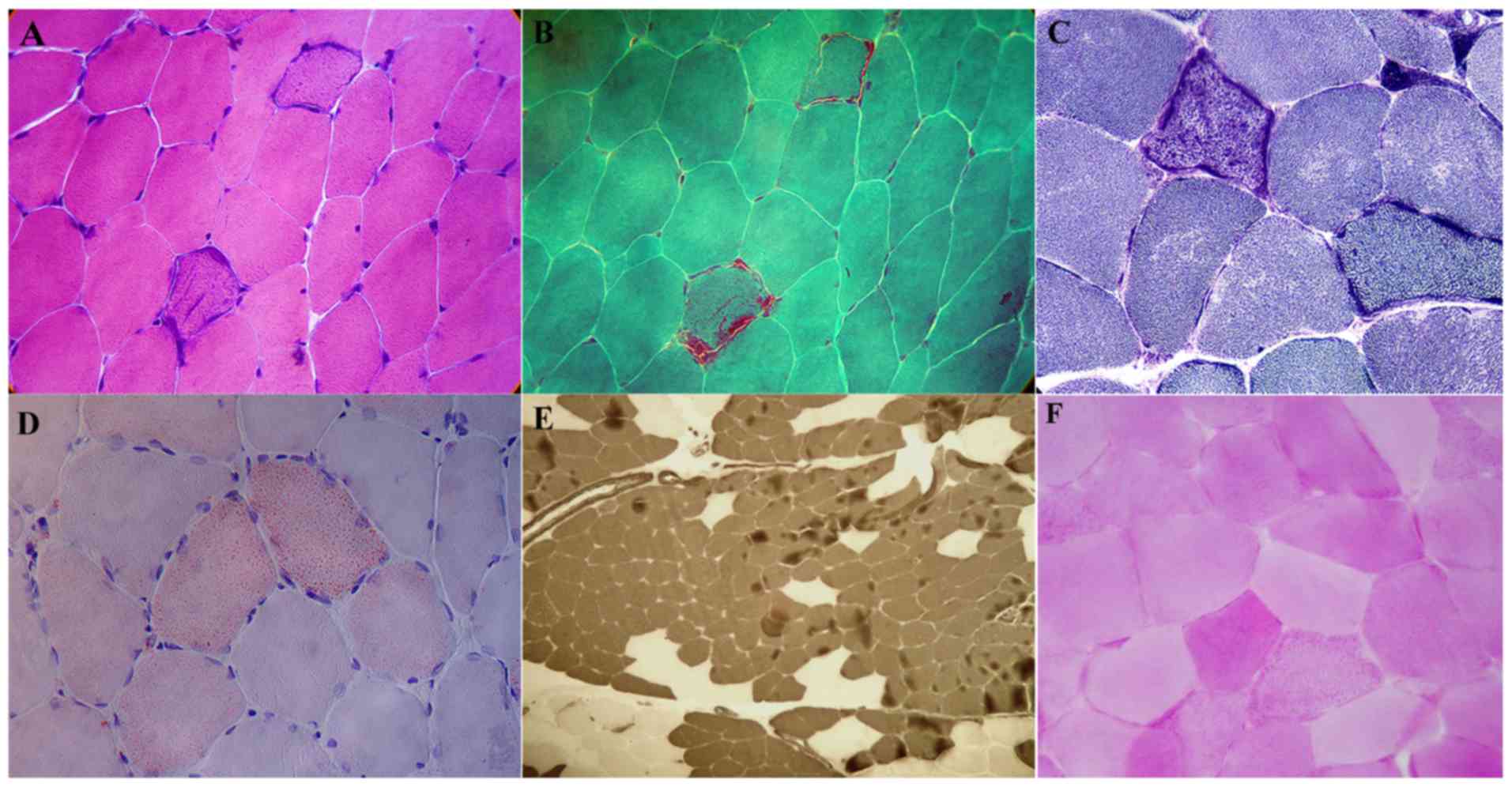
H&E staining demonstrated that among the 12
patients, muscle fiber size was normal in 10 patients and a few
angular atrophic muscle fibers were visible in 2 patients. In 11
patients, basophilic muscle fibers were irregularly scattered in
the sarcoplasm and some fibers were broken (Fig. 2A). No degeneration or necrosis of
muscle fibers and no infiltration of inflammatory cells were
evident in any of the 12 patients. Modified Gomori trichrome
staining revealed several typical ragged-red fibers (Fig. 2B). NADH-TR staining showed that the
edges of the muscle fibers were deeply stained in 11 of 12 patients
(Fig. 2C). The Oil Red O staining
demonstrated that among 12 patients, there was a marked increase in
lipids in some of the muscle fibers from 4 patients, predominantly
in type I muscle fibers (Fig. 2D).
ATPase staining demonstrated that type I and II muscle fibers were
distributed normally in 9 patients. Both types of muscle fiber were
abnormally distributed in 3 patients, which showed fiber-type
grouping, where the type I fibers were more prevalent (Fig. 2E). Periodic acid-Schiff reaction
staining did not reveal any abnormalities in the 12 patients
(Fig. 2F).
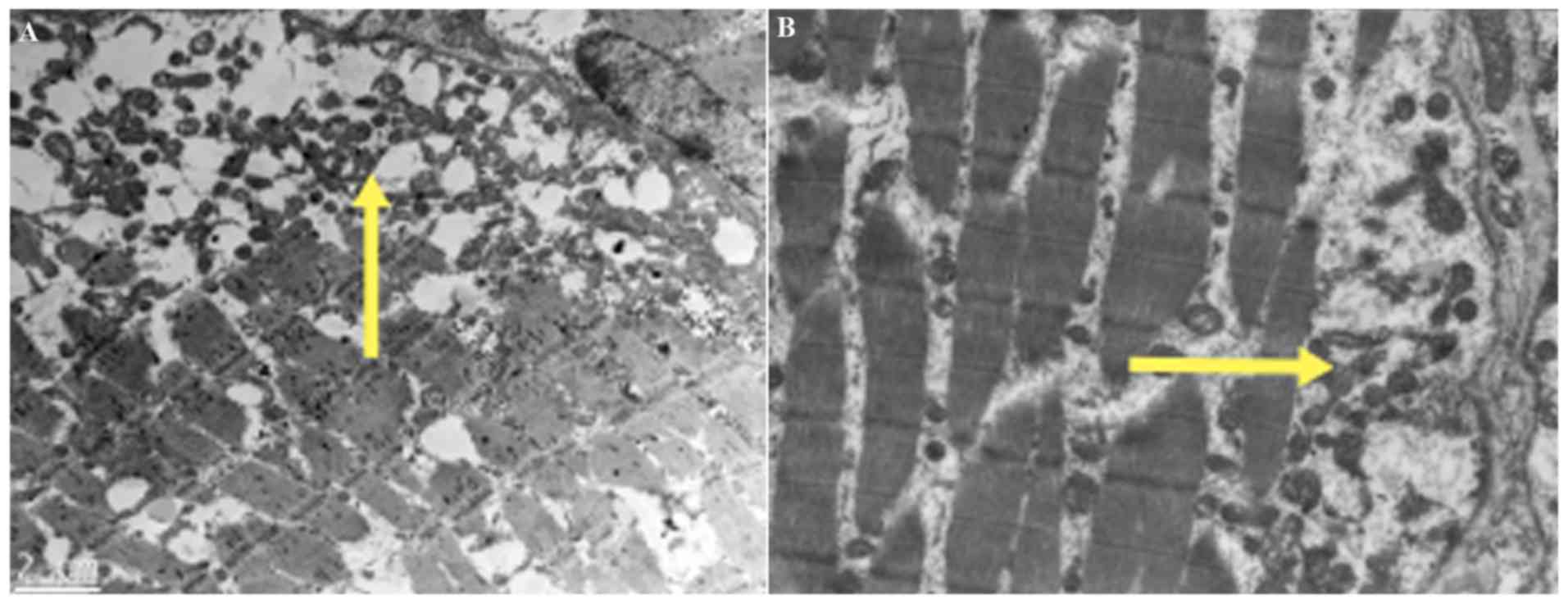
Transmission electron microscopy showed that among
the 5 patients tested, there were increased mitochondria and
abnormal mitochondrial aggregation with varied shapes and sizes
observed between the subsarcolemma and myofibrils in 4 of the 5
patients (Fig. 3).
Discussion
CPEO is a rare mitochondrial myopathy that can occur
at any age, but is commonly found in children and young people
(7). Most of the cases are sporadic
and familial cases are rare (8). In
the present study, the 12 patients were sporadic cases, with an
onset age of 7-35 years and an average age of onset of 17.2 years.
The average onset age was older compared with that of previous
studies (9,10). Patients with CPEO were mainly
characterized by progressive blepharoptosis and ocular dyskinesia
(7). In the present study, the 12
patients who were analyzed suffered from unilateral or bilateral
blepharoptosis, then gradually developed ocular dyskinesia followed
by binocular fixation in the central position. Diplopia rarely
occurs during the clinical progression of CPEO (11). In the present study, only one case
presented short-term and mild diplopia. Patients may develop severe
extraocular muscle paralysis without diplopia since the bilateral
extraocular muscles are involved and paralysis of the extraocular
muscles progresses slowly as extraocular muscles can compensate for
each other (11). The main clinical
symptoms of CPEO are blepharoptosis and paralysis of the
extraocular muscles in the early stages. Limb movement disorders
rarely occur, so patients are often first seen by an
ophthalmologist (7,12). Among the 12 patients included in the
present study, 9 were first treated by an ophthalmologist and 5
patients were diagnosed with weakness of the levator palpebrae
superioris muscle. Three patients were treated with two eyelid
lifting surgeries. CPEO is easily misdiagnosed as ocular myasthenia
gravis in the clinic (12). In the
present study, 8 patients were initially diagnosed with ocular
myasthenia gravis and 6 patients received short-term treatment with
pyridostigmine bromide, but their clinical symptoms did not
improve. The causes of clinical misdiagnosis are potentially due to
a lack of awareness of CPEO by clinicians and the failure to
consider CPEO as the diagnosis. Clinicians may only pay attention
to ptosis in the first diagnosis, and not notice or consider the
signs of binocular eye movement disorders. In myasthenia gravis,
blepharoptosis is more serious than ocular dyskinesia, and diplopia
is obvious. Moreover, the symptoms of diplopia tend to fluctuate,
with the presence of fatigue. However, paralysis of the extraocular
muscles in patients with CPEO is often more prominent than
blepharoptosis, and eventually bilateral eyeballs are fixed in the
central position, while diplopia symptoms are not clear. Taken
together, a diagnosis of CPEO should be considered first in
patients with extraocular muscle paralysis which presents as more
serious than blepharoptosis, if the neostigmine test shows a
negative result.
Neuroelectrophysiological testing is a conventional
examination for the diagnosis of neuromuscular diseases. Repetitive
nerve stimulation is an effective method for differentiating CPEO
from other neuromuscular diseases (12,13).
Repetitive nerve stimulation demonstrated negative results in all
12 patients in the present study, which did not support
neuromuscular junction lesions. Needle electromyography revealed
abnormal changes in 6 patients, including myogenic lesion in 4
patients, a neurogenic lesion in 1 patient and mixed
myogenic/neurogenic lesions in 1 patient. These results indicated
that although no obvious limb weakness symptoms were observed in
the clinic, some abnormal changes were evident from the
electrophysiological examination. Furthermore, besides myogenic
lesions, a neurogenic lesion or mixed lesions were also be seen in
individual patients. In nerve conduction measurements, 2 patients
demonstrated decreased sensory nerve conduction velocity. This
suggests that patients with CPEO may also have peripheral nerve
damage, which has rarely been reported in previous studies
(11,12).
Muscle biopsies are an important method for the
diagnosis of mitochondrial myopathy in CPEO. In 1963, Engel and
Cunningham (14) first discovered
ragged-red fibers using modified Gomori trichrome staining. In
1977, Shapira et al (15)
proposed mitochondrial encephalomyopathies. In 1988, Yamamoto and
Nonaka (16) described CPEO, which
provided an in-depth understanding of the muscle pathology of
mitochondrial diseases. When ragged-red fibers are found in muscle
fibers, it indicates that there are a large number of abnormally
proliferating and aggregated mitochondria in the muscle fibers
(6). It is generally considered that
>2% of the total fibers presenting as ragged-red fibers is an
important basis for the diagnosis of mitochondrial myopathy
(6). In the present muscle biopsies,
the proportion of ragged-red fibers was >2%. Thus, CPEO could be
diagnosed by this in combination with the clinical manifestations.
In addition to Gomori trichrome staining, H&E, Oil Red O and
ATPase staining were also performed. Scattered, abnormal muscle
fibers with deeply stained coarse granules were visible in the
H&E-stained sections. The 11 patients who had ragged-red fibers
on Gomori trichrome staining also had abnormal deeply basophilic
staining of muscle fibers as observed by H&E staining. Oil Red
O staining was positive in 4 patients, suggesting that CPEO could
be combined with abnormal lipid metabolism, resulting in lipid
deposition in muscle fibers. ATPase staining demonstrated the
abnormal distribution of type I and II muscle fibers in 3 patients,
where the fibers grouped together with predominantly type 1 fibers.
This abnormal distribution of muscle fibers suggests that there may
be peripheral nerve damage in patients with CPEO. In one of the
patients with abnormally distributed muscle fibers,
electromyography tests showed neurogenic damage to some of the
muscles examined and there was a slightly decreased conduction
velocity of the peripheral nerves. These data also support that
CPEO could be accompanied by peripheral nerve damage.
With the rapid development of molecular tests, many
mtDNA fragment losses and point mutations have been identified in
patients with CPEO (17-19).
Sundaram et al (20)
performed muscle biopsies and gene detection in 45 patients with
CPEO, and found neurogenic atrophy in seven biopsies. Mitochondrial
gene mutations were found in 10 of the 11 patients who presented
with a normal muscle biopsy. Lehmann et al (21) found that peripheral nerve involvement
was associated with multiple deletions in mtDNA fragments. Gene
detection has become a new diagnostic basis for CPEO, and should be
conducted in future studies to identify the loss of mtDNA fragments
and mutation sites.
It should be noted that the present study has
limitations. Firstly, the sample size is small. Since CPEO is a
rare disorder, inclusion of more patients with CPEO is very
difficult, but the inclusion of more patients in future
multi-center studies would be useful to clarify the clinical
feature of different types of CPEO. Secondly, a number of possible
factors were not analyzed, for example the disease duration and age
of the patients could have affected the results and needs to be
further investigated. Finally, given the importance of gene
detection in the diagnosis of CPEO, further studies are needed to
investigate the genetic abnormalities associated with patients with
CPEO, to further improve the molecular-pathological diagnosis of
CPEO.
In conclusion, the present study suggests that if
patients present with obvious extraocular muscle paralysis and do
not have symptoms of diplopia, the possibility of CPEO should be
considered. Ragged-red fibers identified by Gomori trichrome
staining and the H&E staining of muscle biopsies from patients
is essential for confirming the diagnosis of CPEO.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HL contributed to the study conception and design.
QQQ, HL, QQ, XZ, YZ contributed to the acquisition, analysis and
interpretation of data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the People's Hospital of Jiaozuo City. All patients
have given written informed consent.
Patient consent for publication
All patients have given written informed consent for
publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
López-Gallardo E, López-Pérez MJ, Montoya
J and Ruiz-Pesini E: CPEO and KSS differ in the percentage and
location of the mtDNA deletion. Mitochondrion. 9:314–317.
2009.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Negro R, Zoccolella S, Dell’aglio R, Amati
A, Artuso L, Bisceglia L, Lavolpe V, Papa S, Serlenga L and
Petruzzella V: Molecular analysis in a family presenting with a
mild form of late-onset autosomal dominant chronic progressive
external ophthalmoplegia. Neuromuscul Disord. 19:423–426.
2009.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Murdock J, Thyparampil PJ and Yen MT:
Late-onset development of eyelid ptosis in chronic progressive
external ophthalmoplegia: A 30-year follow-up. Neuroophthalmology.
40:44–46. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Filosto M, Mancuso M, Nishigaki Y,
Pancrudo J, Harati Y, Gooch C, Mankodi A, Bayne L, Bonilla E,
Shanske S, et al: Clinical and genetic heterogeneity in progressive
external ophthalmoplegia due to mutations in polymerase γ. Arch
Neurol. 60:1279–1284. 2003.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Brandon BR, Diederich NJ, Soni M, Witte K,
Weinhold M, Krause M and Jackson S: Autosomal dominant mutations in
POLG and C10orf2: Association with late onset chronic progressive
external ophthalmoplegia and Parkinsonism in two patients. J
Neurol. 260:1931–1933. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Walker UA, Collins S and Byrne E:
Respiratory chain encephalomyopathies: A diagnostic classification.
Eur Neurol. 36:260–267. 1996.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Chaturvedi S, Bala K, Thakur R and Suri V:
Mitochondrial encephalomyopathies: Advances in understanding. Med
Sci Monit. 11:RA238–RA246. 2005.PubMed/NCBI
|
|
8
|
Biousse V and Newman NJ:
Neuro-ophthalmology of mitochondrial diseases. Curr Opin Neurol.
16:35–43. 2003.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Bing Q, Hu J, Li N, Zhao Z, Shen HR and
Yuan HH: Clinical and pathological features of chronic progressive
external ophthalmoplegia. Linchuang Shenjingbingxue Zazhi.
22:175–177. 2009.(In Chinese).
|
|
10
|
Sun L, Lu JH and Lu ZZ: Clinical
manifestations, pathological changes and diagnosis of chronic
progressive external ophthalmoplegia (twelve cases attached). Fudan
Daxue Yixueban. 36:212–215. 2009.(In Chinese).
|
|
11
|
Wallace DK, Sprunger DT, Helveston EM and
Ellis FD: Surgical management of strabismus associated with chronic
progressive external ophthalmoplegia. Ophthalmology. 104:695–700.
1997.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wu JL, Yan CZ, Wang QZ, Liu SP, Gao SQ,
Zhang YQ and Li DN: Chronic progressive external ophthalmophegia:
Clinical and pathological analysis of 22 cases. Zhonghua Shenjinke
Zazhi. 38:737–740. 2005.(In Chinese).
|
|
13
|
Fournier E and Tabti N: Clinical
electrophysiology of muscle diseases and episodic muscle disorders.
Handb Clin Neurol. 161:269–280. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Engel WK and Cunningham GG: Rapid
examination of muscle tissue. An improved trichrome method for
fresh-frozen biopsy sections. Neurology. 13:919–923.
1963.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Shapira Y, Harel S and Russell A:
Mitochondrial encephalomyopathies: A group of neuromuscular
disorders with defects in oxidative metabolism. Isr J Med Sci.
13:161–164. 1977.PubMed/NCBI
|
|
16
|
Yamamoto M and Nonaka I: Skeletal muscle
pathology in chronic progressive external ophthalmoplegia with
ragged-red fibers. Acta Neuropathol. 76:558–563. 1988.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Lee SJ, Na JH, Han J and Lee YM:
Ophthalmoplegia in mitochondrial disease. Yonsei Med J.
59:1190–1196. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Sachdev A, Fratter C and McMullan TF:
Novel mutation in the RNASEH1 gene in a chronic progressive
external ophthalmoplegia patient. Can J Ophthalmol. 53:e203–e205.
2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Viscomi C and Zeviani M: MtDNA-maintenance
defects: Syndromes and genes. J Inherit Metab Dis. 40:587–599.
2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Sundaram C, Meena AK, Uppin MS, Govindaraj
P, Vanniarajan A, Thangaraj K, Kaul S, Kekunnaya R and Murthy JM:
Contribution of muscle biopsy and genetics to the diagnosis of
chronic progressive external opthalmoplegia of mitochondrial
origin. J Clin Neurosci. 18:535–538. 2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Lehmann D, Kornhuber ME, Clajus C, Alston
CL, Wienke A, Deschauer M, Taylor RW and Zierz S: Peripheral
neuropathy in patients with CPEO associated with single and
multiple mtDNA deletions. Neurol Genet. 2(e113)2016.PubMed/NCBI View Article : Google Scholar
|