Introduction
Diabetes mellitus is a life-threatening and complex
metabolic disorder affecting multiple systems in the body. The
morbidity rate of diabetes in China is 9.7% and continues to grow
(1). Diabetes is considered a major
risk factor for coronary heart disease, with the mortality rate of
cardiovascular disease in diabetic patients estimated at 65%
(2). Diabetic cardiomyopathy (DCM)
is a type of diabetic heart disease first discovered in
1972(3) and appears as myocardial
dysfunction in diabetic patients without valvular heart disease,
hypertension, or coronary artery disease (CAD) (3,4).
Epidemiologic studies indicated that DCM was closely associated
with progressive increase of relative wall thickness, left atrium
and left ventricular mass and impaired glucose tolerance (4). Research on the etiopathogenesis of DCM
revealed that hyperglycemia played a decisive role in the
development of DCM (5).
Diabetes mellitus is characterized by several
important changes in micro-vascular architecture, including
subendothelial matrix deposition, abnormal capillary permeability,
fibrosis surrounding arterioles and micro-aneurysm formation
(6). Hyperglycemia can activate
protein kinase C and stimulate vascular endothelial cells to
enhance the production of vasoconstrictor prostanoids, which can
promote endothelial dysfunction, as well as ventricular and
myocardial hypertrophy (7,8). On the other hand, hyperglycemia also
can aggravate intracellular oxidative stress, which then induces
myocardial injury (9). Moreover,
oxidative stress is exacerbated by reactive oxygen species (ROS)
production in the mitochondria of diabetic cardiac tissues
(10). Aberrant ROS production also
increases pro-inflammatory response and myocardial apoptosis
(11).
Peroxisome proliferator-activated receptors (PPARs)
are important nuclear hormone receptors (12) consisting of 3 types (PPAR-α, PPAR-γ
and PPAR-δ), each of which is encoded by separate genes and
distinguished by specific ligands, functions, and distributions
(12). A previous study described
that transgenic mice with PPAR-α overexpression develop
cardiomyopathy similar to the diabetes mellitus condition (13).
Acetylation is a critical mechanism for regulating
the activity of widespread enzymes associated with mitochondrial
metabolism, such as the fatty acid oxidation, citric acid cycle,
antioxidant defense and the electron transport system (14-16).
Sirtuin 3 (SIRT3) is the primary deacetylase in the mitochondria
(17). Intriguingly, SIRT3 activity
and expression were found to be decreased in an obesity animal
model, possibly leading to obesity-related reduction of antioxidant
defense and oxidative metabolism (18). However, very little is known about
the function and mechanism of SIRT3 in DCM. In this study, the AC16
cell line was used as a cardiomyocyte model coupled with SIRT3
overexpression or inhibition (by 3-TYP), as well as treatment with
PPAR-α agonist (Wy14643) or antagonist (GW6471) under high
glucose/euglycemia conditions. The interaction between SIRT3 and
PPAR-α was investigated.
Materials and methods
Chemicals and reagents
Glucose was from Sigma-Aldrich (Shanghai, China).
Annexin V-FITC apoptosis detection kit, CCK8 assay kit and cellular
reactive oxygen species detection kit were obtained from Beyotime
Biotechnology. 3-TYP and Wy14643 were from Selleck Chemicals, and
GW6471 from R&D Systems China. TRIzol reagent and reverse
transcription kits were obtained from Thermo Fisher Scientific,
Inc. PPAR-α antibody was from Abcam. SIRT3, cleaved caspase-3, Bax,
Bcl2, JNK1/2, phosphorylated JNK1/2 (p-JNK1/2) and glyceraldehyde
3-phosphate dehydrogenase (GAPDH) antibodies were from Cell
Signaling Technology.
The study was approved by the Ethics Committee of
Central Hospital of Minhang District (Shanghai, China).
AC16 cells
AC16 human cardiomyocyte cell line was obtained from
the Chinese Academy of Sciences Cell Bank (http://www.cellbank.org.cn/, Shanghai, China). Cells
were incubated with DMEM media (HyClone) supplemented with 100 U/ml
penicillin and 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.) at 37˚C (5% CO2). AC16 cells at
logarithmic phase were used for follow-up experiments.
Experimental groups
Effects of glucose treatment on SIRT3 and PPAR-α
expression in AC16 cells. AC16 cells were treated with
different concentrations of glucose (5.5, 10, 30 and 50 mM, added
into glucose-free DMEM media) for 48 h. Relative mRNA expression
and protein levels of SIRT3 and PPAR-α were detected at 24 and 48 h
after treatment.
Effects of SIRT3 overexpression and PPAR-α
activation/inhibition on AC16 cells under high glucose
condition. AC16 cells were divided into 5 groups: Control group
(5.5 mM glucose); G 30 mM + EPC group (30 mM glucose + empty
plasmid control); G 30 mM + SIRT3 OE group (30 mM glucose + SIRT3
overexpression); G 30 mM + SIRT3 OE + GW6471 group (30 mM glucose +
SIRT3 overexpression + PPAR-α antagonist, GW6471); and G 30 mM +
Wy14643 group (30 mM glucose + PPAR-α agonist, Wy14643). First,
glucose (5.5 or 30 mM) was added into glucose-free DMEM media in
respective groups for 24 h. Then, SIRT3 OE or EPC lentivirus (JRDun
Biotech) (Table I), Wy14643 (100 µM,
dissolved in DMSO) and GW6471 (10 µM, dissolved in DMSO) were added
into AC16 cells, respectively for 72 h. Proliferation of AC16 cells
was examined at 0, 24, 48 and 72 h after lentivirus treatment. Cell
apoptosis and reactive oxygen species levels were measured at the
end of the experiment, along with SIRT3 mRNA expression and SIRT3,
cleaved caspase-3, Bax, Bcl2, JNK1/2 and p-JNK1/2 protein
levels.
 | Table ISIRT3 (NM_001017524)/SIRT3 promoter
sequence and primers. |
Table I
SIRT3 (NM_001017524)/SIRT3 promoter
sequence and primers.
| Gene name | Primer sequence
(5'-3') |
|---|
| SIRT3 | F:
CGGAATTCATGGCGTTCTGGGGTTG |
| CDS | R:
CGGGATCCCTATTTGTCTGGTCCATCAAGC |
| Semi- | F:
CCCCTCCGTCTCCCTCTATC |
| qRT-PCR (ChIP) | R:
CAACCCCAGAACGCCATG |
| SIRT3 | F:
CCCTCGAGACGGCGGAAGTGGTTG |
| promoter | R:
CCAAGCTTTCCCTGCCGCCAAG |
Effects of SIRT3 inhibitor (3-TYP) and PPAR-α
agonist (Wy14643) on AC16 cells under euglycemia condition.
AC16 cells were divided into 3 groups, and received the following
treatments: 5.5 mM glucose (control group); 5.5 mM glucose + 30 mM
3-TYP (G (5.5 mM) + 3-TYP group); and 5.5 mM glucose + 30 mM 3-TYP
+ 100 µM Wy14643 [G (5.5 mM) + 3-TYP + Wy14643 group]. AC16 cells
in each group were treated with glucose (added into glucose-free
DMEM media) for 24 h. Subsequently, the indicated chemicals were
added and cells were cultured for 72 h. Proliferation of AC16 cells
was assessed at 0, 24, 48 and 72 h after treatment. Cell apoptosis,
reactive oxygen species levels, and JNK1/2 and p-JNK1/2 protein
levels were examined at the end of the experiment.
Interaction between PPAR-α protein and SIRT3
promoter. Cells were treated with Wy14643 (100 µM), GW6471 (10
µM) or DMSO for 48 h. Chromatin immunoprecipitation (ChIP) and
Dual-Lucy Assay kit (Promega, Beijing, China) were employed to test
the interaction between PPAR-α and SIRT3.
Experimental methods
Cell proliferation and apoptosis assay.
Cultured AC16 cells were harvested at relative time-points.
Proliferation of AC16 cells was examined using CCK8 assay kit
following the manufacturer's instructions. Cell absorbance was
measured at 450 nm using a microplate reader (Thermo Fisher
Scientific, Inc.).
Cultured AC16 cells were harvested and incubated
with FITC-labelled Annexin V and PI at 25˚C for 20 min following
the manufacturer's instructions. Subsequently, the intensity of
Annexin V or PI fluorescence was analyzed by FACScan
(Becton-Dickinson). For each sample, 10,000 cells were tested.
Dichlorodihydrofluorescein diacetate (DCFH-DA)
flow cytometry. ROS production was assessed using DCFH-DA assay
according to a previous study (19).
Briefly, AC16 cells (106/ml) were cultured with 5 µM of
2'-7'-dichlorodihydrofluorescein diacetate (DCFH-DA) for 20
min at 37˚C (forward-reverse mixing once per 3 min). DCFH-DA
(non-fluorescent) entered cells and hydrolyzed into
cell-impermeable and non-fluorescent DCFH. DCFH was oxidized into
highly fluorescent dichloroflurescein (DCF) by intracellular ROS.
The green fluorescence intensity was proportional to ROS level. DCF
fluorescence was assayed at 525 nm after excitation of cells at 480
nm using flow cytometry analysis (Becton-Dickinson). Results were
expressed as the ratio of fluorescence intensity of AC16 cells in
the other groups to that in control group.
ChIP and luciferase assay. ChIP is a powerful
tool to investigate interactions between intracellular DNA and
specific proteins, and detect their genomic localization (20). In the present study, interaction
between PPAR-α protein and SIRT3 promoter was tested by ChIP assay
according to a previous study (21).
Briefly, cell samples were subjected to standard cross-linking,
chromatin shearing, immunoprecipitation, reverse cross-linking, DNA
precipitation and PCR analysis. The PCR products were determined
using semi-quantitative RT-PCR (primers are shown in Table I).
Cells were co-transfected with a dual-luciferase
reporter plasmid containing SIRT3 promoter-reporter plasmid (JRDun
Biotech), in combination with Wy14643 or GW6471 treatment for 48 h
in 24-well plates. Luciferase activity was measured using the
Dual-Lucy Assay Kit (Promega) following the manufacturer's protocol
(primers are shown in Table I).
RT-qPCR. Relative mRNA expression was
determined by RT-qPCR. Total mRNA was isolated using TRIzol, and 2
µg of total RNA from each sample was reverse transcribed into cDNA
using First Strand cDNA Synthesis kit (Thermo Fisher Scientific,
Inc.) on RT-qPCR machine (ABI-7300; Applied Biosystems).
Primers used for the RT-qPCR are shown in Table II. Relative mRNA expression was
evaluated by 2-ΔΔCt relative quantitative analysis
against GAPDH.
| Table IIPrimers used in real-time fluorogenic
PCR assays. |
Table II
Primers used in real-time fluorogenic
PCR assays.
| Gene name | Primer sequence
(5'-3') |
|---|
| SIRT3 | F:
CCTTGGCTTGGCATCCTC |
| SIRT3 | R:
GCACAAGGTCCCGCATCTC |
| PPAR-α | F:
TCACGGACACGCTTTCACC |
| PPAR-α | R:
CCCCGCAGATTCTACATTCG |
| GAPDH | F:
AATCCCATCACCATCTTC |
| GAPDH | R:
AGGCTGTTGTCATACTTC |
Western blot analysis. Total protein from
each sample was extracted, and protein concentration was measured
using BCA protein assay kit. Protein (30 µg) was separated by 10%
SDS-PAGE and transferred onto PVDF membranes, which were then
blocked in 5% skimmed milk at 25˚C for 1 h, followed by incubation
with primary antibodies overnight at 4˚C. After washing, membranes
were incubated with secondary antibodies for 1 h. Finally, protein
bands were visualized using ECL-detection kit (Beyotime
Biotechnology). The following primary antibodies were used: PPAR-α
(1:1,000), SIRT3 (1:1,000), cleaved caspase-3 (1:1,000), Bax
(1:1,000), Bcl2 (1:1,000), JNK1/2 (1:1,000), p-JNK1/2 (1:1,000) and
GAPDH (1:1,000; CST). GAPDH served as a loading control.
Statistical analysis
Data were expressed as mean ± SD (n=3). Difference
was evaluated using ANOVA. Ducan multiple range test was determined
using SPSS 20.0 software (IBM Corp). P<0.05 was considered as
statistically significant.
Results
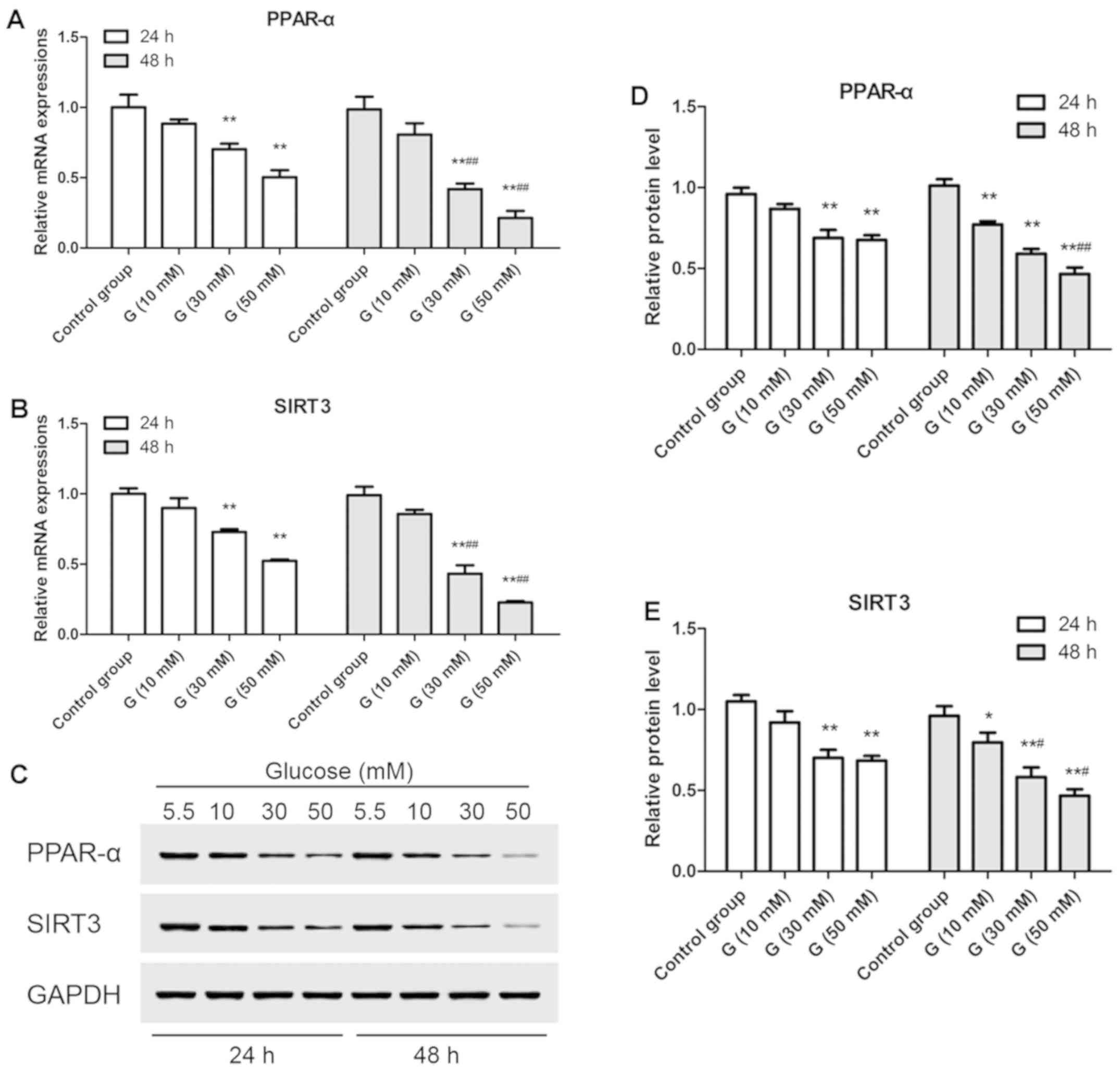
Glucose treatment downregulates
expression of SIRT3 and PPAR-α in AC16 cells
In the current study, both mRNA expression and
protein levels of SIRT3 and PPAR-α in AC16 cells showed no change
with time under euglycemia condition (5.5 mM of glucose). However,
with increasing dose (≥30 mM), SIRT3 and PPAR-α mRNA expression
(Fig. 1A and B) and protein levels (Fig. 1C-E) were reduced, and further
diminished over time (Fig. 1).
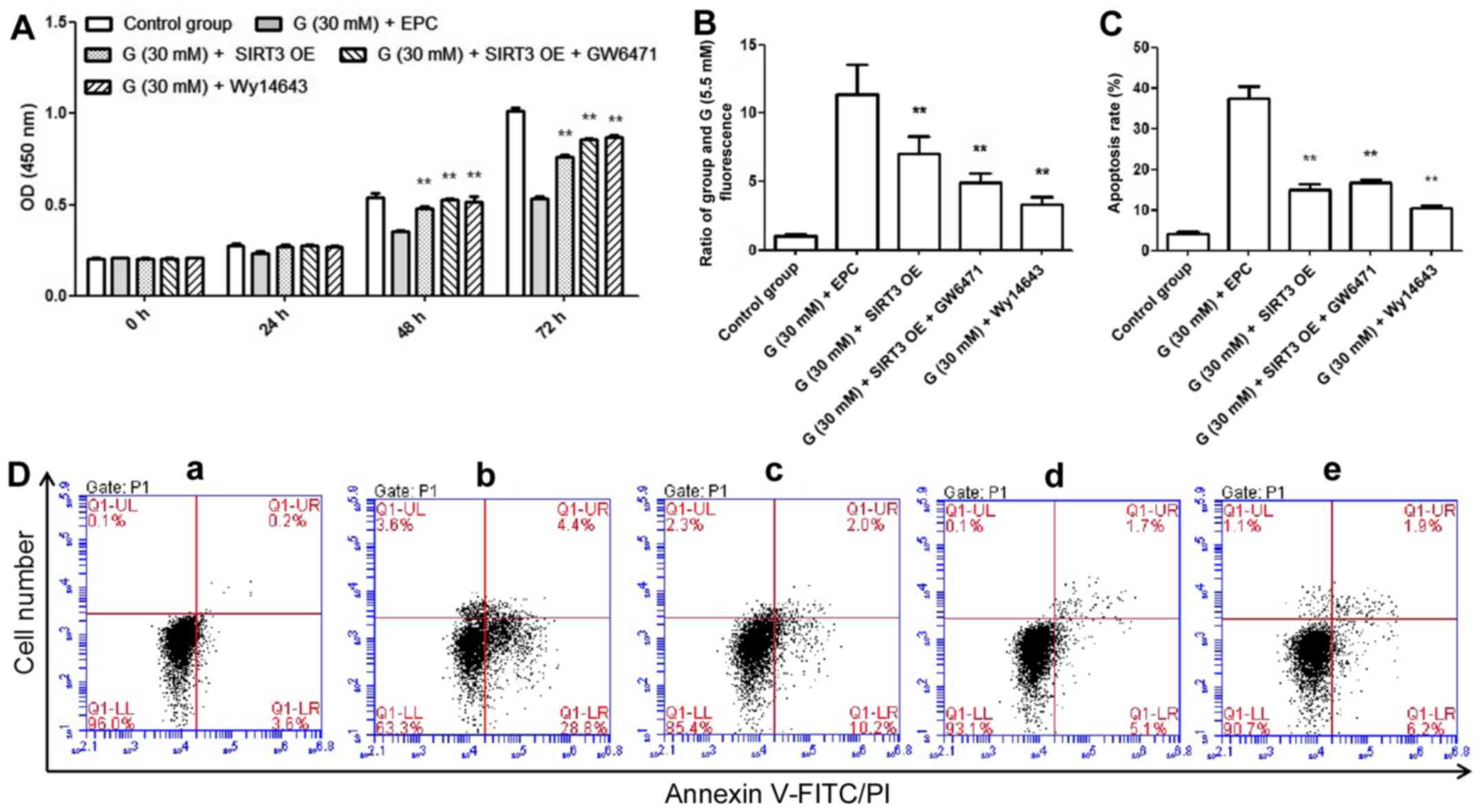
Effects of SIRT3 OE and PPAR-α
activation/inhibition on AC16 cells under high glucose (30 mM)
condition
The regulatory function of SIRT and whether it was
regulated by PPAR-α under high glucose (30 mM) condition were
investigated. The present results indicated that the proliferation
of AC16 cells was inhibited by high glucose (30 mM) treatment at 48
h (34.67±3.21%) and 72 h (47.13±5.42%), but was antagonized by
SIRT3 OE, SIRT3 OE + GW6471 and Wy14643 treatment (Fig. 2A). Moreover, intracellular hydrogen
peroxide production and apoptosis were induced by glucose (30 mM)
treatment for 72 h, but reduced by SIRT3 OE, SIRT3 OE + GW6471 and
Wy14643 treatment (Fig. 2B-D). In
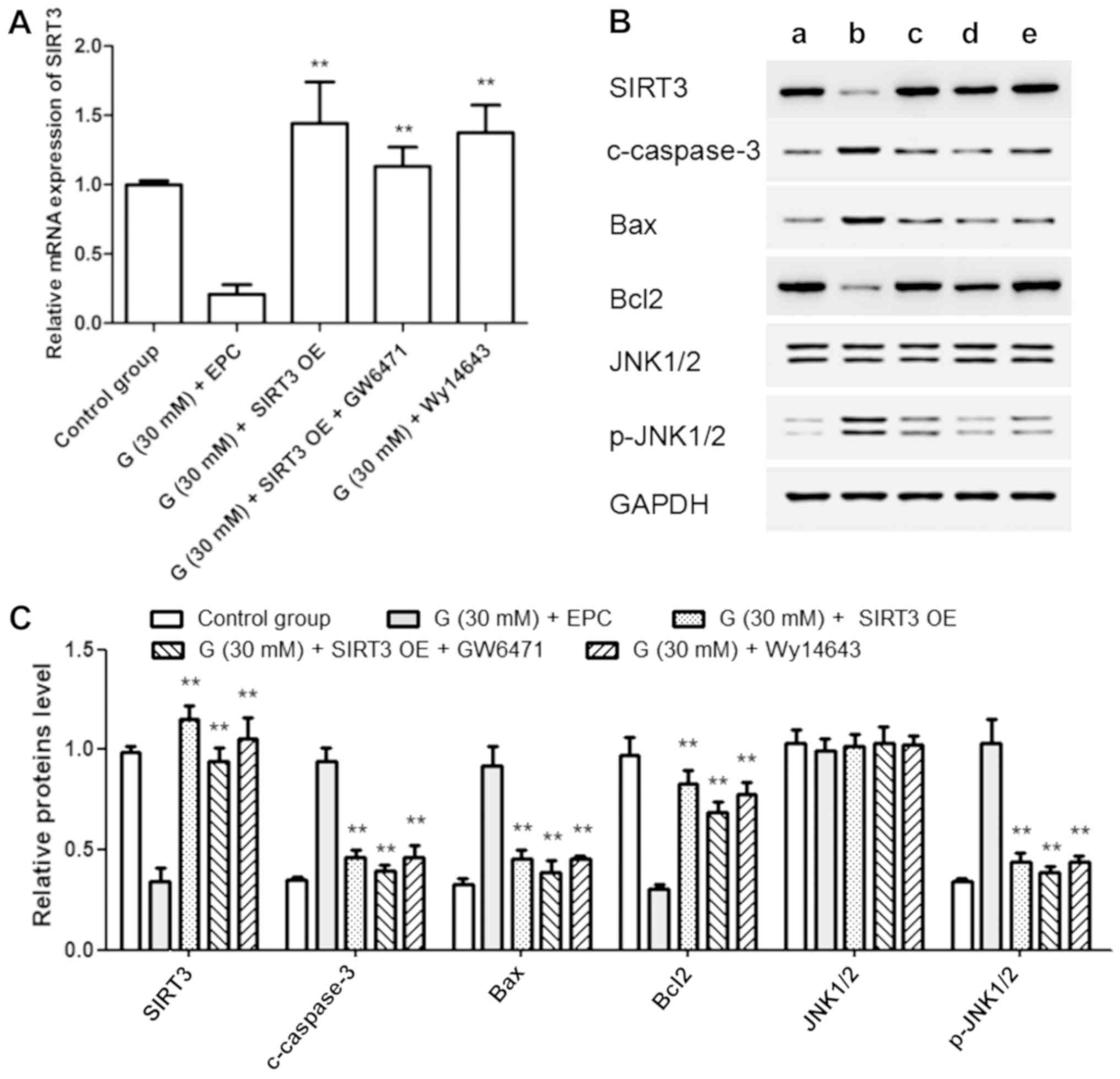
addition, SIRT3 mRNA expression and protein level were diminished
by high glucose (30 mM) but increased by SIRT3 OE, SIRT3 OE +
GW6471 and Wy14643 treatment (Fig.
3A-C). Moreover, the protein levels of cleaved caspase-3, Bax
and p-JNK1/2 displayed trends consistent with apoptosis, whereas
Bcl2 protein level showed a trend opposite to Bax. These results
suggested that SIRT3 might be positively regulated by PPAR-α in
AC16 cells (Fig. 3B and C).
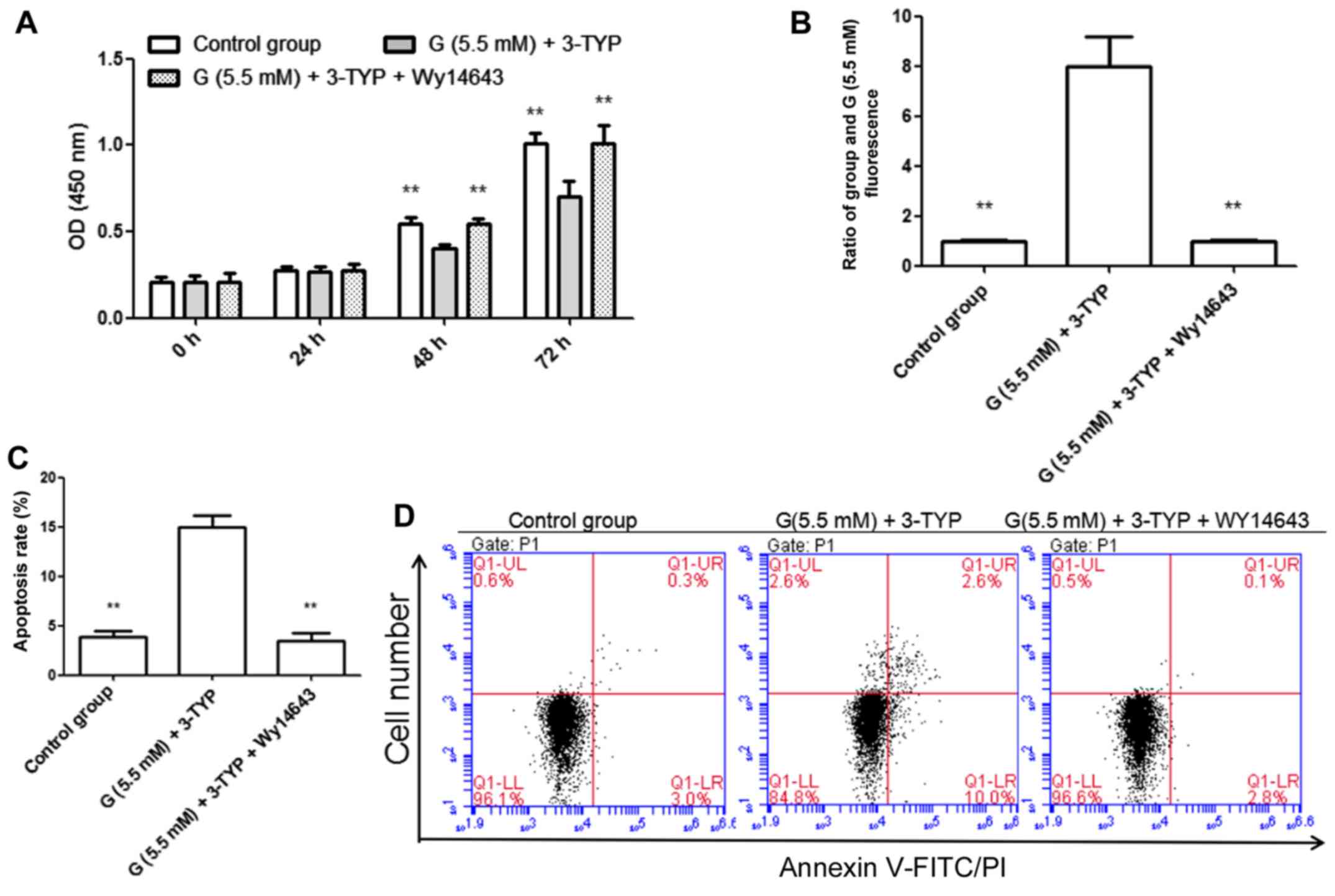
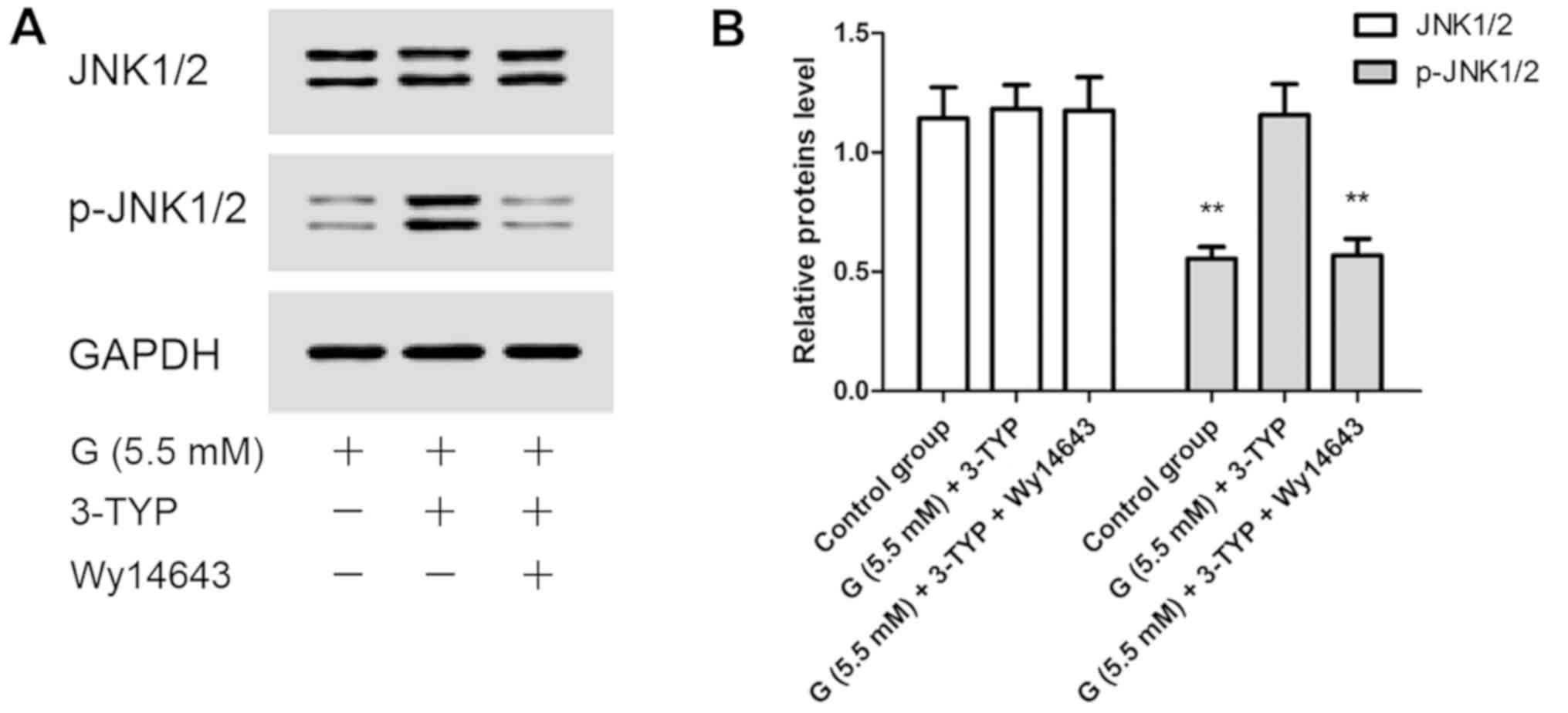
Effects of 3-TYP and Wy14643
treatments on AC16 cells under euglycemia condition
Based on the above findings, it was hypothesized
that the downregulation of SIRT3 might play an important role
during the process of high glucose-induced myocardial injury.
Therefore, the effects of 3-TYP and Wy14643 treatments on AC16
cells under euglycemia condition were analyzed. In the present
study, the inhibition of SIRT3 (by treatment with 3-TYP) in AC16
cells led to similar results under euglycemia condition as high
glucose treatment, including inhibition of cell proliferation
(Fig. 4A), stimulation of
intracellular hydrogen peroxide production and induction of
apoptosis (Fig. 4B-D), and increased
phosphorylation of JNK1/2. Furthermore, these phenomena could be
relieved by activation of PPAR-α (Fig.
5A and B).
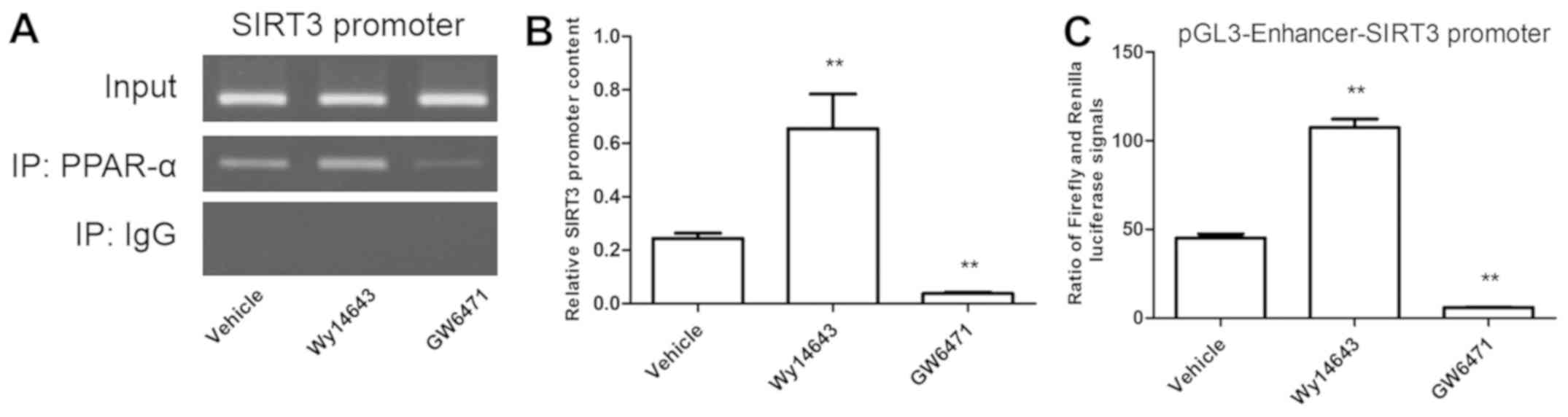
SIRT3 is a direct, positively
regulated target of PPAR-α
The intracellular interaction between SIRT3 promoter
and PPAR-α was tested using ChIP and luciferase assay. The present
results suggested that intracellular PCR production of SIRT3
promoter was increased by PPAR-α agonist and decreased by PPAR-α
antagonist (Fig. 6A and B). Furthermore, the ratio of Firefly and
Renilla luciferase signals showed similar trends (Fig. 6C).
Discussion
DCM has become a worldwide public health concern,
yet the mechanisms and occurrence of DCM remain unknown (2,5). Herein,
it was found that abnormally low expression of SIRT3 may play an
important role in DCM. SIRT3 expression was weakened under high
glucose condition in AC16 cells. Whereas, high glucose inhibited
proliferation of AC16 cells and enhanced apoptosis and
intracellular hydrogen peroxide production. These phenomena could
be improved by SIRT3 overexpression. SIRT3 inhibition led to
similar phenomena in AC16 cells under euglycemia condition as under
high glucose treatment.
The production of ROS is considered to be a
contributing factor in the occurrence and progression of diabetic
cardiomyopathy (22). The strategy
of enhancing mitochondrial ROS scavenge system has been clinically
proven to be effective in reducing cardiac dysfunction caused by
diabetes (22). SIRT3 plays a key
role in improving mitochondrial dysfunction and oxidative stress
via the MAPK pathway (23,24). SIRT3 is able to maintain ROS
homeostasis by regulating diverse mitochondrial enzymes, including
superoxide dismutase 2 (SOD2), which can transform harmful
superoxide free radicals into non-toxic hydrogen peroxide or oxygen
(25). In the present study, ROS
production was negatively correlated with SIRT3 expression, which
may be related to the downregulation of JNK1/2 phosphorylation.
PPAR-α is a member of PPAR nuclear transcription
factor family that is enriched in myocardium (26). PPAR-α maintains antioxidant defense
and oxidant equilibrium, and exhibits anti-inflammatory action
(26). PPAR-α also plays a crucial
role in the regulation of lipoprotein transport and assembly, as
well as mitochondrial fatty acid oxidation (27). Decrease in PPAR-α expression has been
suggested to be a self-adaptation process that transforms the
metabolic substrate of cardiac energy from fatty acid to glucose
(28). It was found in this study
that PPAR-α expression was downregulated in AC16 cells under high
glucose condition. Furthermore, PPAR-α agonists improved myocardial
cell injury induced by high glucose. Results from ChIP and
luciferase assay attested that SIRT3 was a direct, positively
regulated target of PPAR-α.
In conclusion, the present results indicated that
diminished expression of SIRT3 played an important role during high
glucose-induced injury in AC16 cells. The function of SIRT3 and its
interaction with PPAR-α were further evaluated in AC16 cells, and
the results demonstrated that SIRT3 was a downstream target of
PPAR-α.
Acknowledgements
Not applicable.
Funding
This study was supported by Science and Technology
Committee of Minhang District, Shanghai (grant no. 2017MHZ72).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZ contributed to the conception and design of the
study, supervised the progress of individual study experiments and
wrote the manuscript. KC and GY performed cell proliferation and
apoptosis assay and flow cytometry. ZW, QS and DY were responsible
for RT-qPCR and western blot analysis. PL, WH and YC contributed to
observation indexes analysis. The final version was read and
adopted by all the authors. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
Central Hospital of Minhang District (Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Li D, Han N and Cui Z: Application of
latent class model in the classification of the patients with
diabetes vulnerability. Chin J Health Stat. 1:11–13. 2018.
|
|
2
|
Pappachan JM, Varughese GI, Sriraman R and
Arunagirinathan G: Diabetic cardiomyopathy: Pathophysiology,
diagnostic evaluation and management. World J Diabetes. 4:177–189.
2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Rubler S, Dlugash J, Yuceoglu YZ, Kumral
T, Branwood AW and Grishman A: New type of cardiomyopathy
associated with diabetic glomerulosclerosis. Am J Cardiol.
30:595–602. 1972.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Rutter MK, Parise H, Benjamin EJ, Levy D,
Larson MG, Meigs JB, Nesto RW, Wilson PW and Vasan RS: Impact of
glucose intolerance and insulin resistance on cardiac structure and
function: Sex-related differences in the Framingham Heart Study.
Circulation. 107:448–454. 2003.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Aneja A, Tang WH, Bansilal S, Garcia MJ
and Farkouh ME: Diabetic cardiomyopathy: Insights into
pathogenesis, diagnostic challenges, and therapeutic options. Am J
Med. 121:748–757. 2008.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Rossen JD: Abnormal microvascular function
in diabetes: Relationship to diabetic cardiomyopathy. Coron Artery
Dis. 7:133–138. 1996.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Hattori Y, Kawasaki H, Abe K and Kanno M:
Superoxide dismutase recovers altered endothelium-dependent
relaxation in diabetic rat aorta. Am J Physiol. 261:H1086–H1094.
1991.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bucala R, Tracey KJ and Cerami A: Advanced
glycosylation products quench nitric oxide and mediate defective
endothelium-dependent vasodilatation in experimental diabetes. J
Clin Invest. 87:432–438. 1991.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Tarquini R, Lazzeri C, Pala L, Rotella CM
and Gensini GF: The diabetic cardiomyopathy. Acta Diabetol.
48:173–181. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Huynh K, Bernardo BC, McMullen JR and
Ritchie RH: Diabetic cardiomyopathy: Mechanisms and new treatment
strategies targeting antioxidant signaling pathways. Pharmacol
Ther. 142:375–415. 2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Cai L, Li W, Wang G, Guo L, Jiang Y and
Kang YJ: Hyperglycemia-induced apoptosis in mouse myocardium:
Mitochondrial cytochrome C-mediated caspase-3 activation pathway.
Diabetes. 51:1938–1948. 2002.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Dreyer C, Krey G, Keller H, Givel F,
Helftenbein G and Wahli W: Control of the peroxisomal
beta-oxidation pathway by a novel family of nuclear hormone
receptors. Cell. 68:879–887. 1992.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Finck BN, Lehman JJ, Leone TC, Welch MJ,
Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, et
al: The cardiac phenotype induced by PPARalpha overexpression
mimics that caused by diabetes mellitus. J Clin Invest.
109:121–130. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Finley LWS, Haas W, Desquiret-Dumas V,
Wallace DC, Procaccio V, Gygi SP and Haigis MC: Succinate
dehydrogenase is a direct target of sirtuin 3 deacetylase activity.
PLoS One. 6(e23295)2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Qiu X, Brown K, Hirschey MD, Verdin E and
Chen D: Calorie restriction reduces oxidative stress by
SIRT3-mediated SOD2 activation. Cell Metab. 12:662–667.
2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Tao R, Coleman MC, Pennington JD, Ozden O,
Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, et
al: Sirt3-mediated deacetylation of evolutionarily conserved lysine
122 regulates MnSOD activity in response to stress. Mol Cell.
40:893–904. 2010.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Lombard DB, Alt FW, Cheng HL, Bunkenborg
J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D,
Murphy A, et al: Mammalian Sir2 homolog SIRT3 regulates global
mitochondrial lysine acetylation. Mol Cell Biol. 27:8807–8814.
2007.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kendrick AA, Choudhury M, Rahman SM,
McCurdy CE, Friederich M, Van Hove JL, Watson PA, Birdsey N, Bao J,
Gius D, et al: Fatty liver is associated with reduced SIRT3
activity and mitochondrial protein hyperacetylation. Biochem J.
433:505–514. 2011.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Caldefie-Chézet F, Walrand S, Moinard C,
Tridon A, Chassagne J and Vasson MP: Is the neutrophil reactive
oxygen species production measured by luminol and lucigenin
chemiluminescence intra or extracellular? Comparison with DCFH-DA
flow cytometry and cytochrome c reduction. Clin Chim Acta.
319:9–17. 2002.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Gilmour DS and Lis JT: RNA polymerase II
interacts with the promoter region of the noninduced hsp70 gene in
Drosophila melanogaster cells. Mol Cell Biol. 6:3984–3989.
1986.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Us-Camas R and De-la-Peña C: Chromatin
immunoprecipitation (ChiP) protocol for the analysis of gene
regulation by histone modifications in Agave angustifolia
haw. Methods Mol Biol. 1815:371–383. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Cai L, Wang Y, Zhou G, Chen T, Song Y, Li
X and Kang YJ: Attenuation by metallothionein of early cardiac cell
death via suppression of mitochondrial oxidative stress results in
a prevention of diabetic cardiomyopathy. J Am Coll Cardiol.
48:1688–1697. 2006.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zheng J, Shi L, Liang F, Xu W, Li T, Gao
L, Sun Z, Yu J and Zhang J: Sirt3 ameliorates oxidative stress and
mitochondrial dysfunction after intracerebral hemorrhage in
diabetic rats. Front Neurosci. 12(414)2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wang G, Fu XL, Wang JJ, Guan R, Sun Y and
Tony To SS: Inhibition of glycolytic metabolism in glioblastoma
cells by Pt3glc combinated with PI3K inhibitor via SIRT3-mediated
mitochondrial and PI3K/Akt-MAPK pathway. J Cell Physiol.
234:5888–5903. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Bause AS and Haigis MC: SIRT3 regulation
of mitochondrial oxidative stress. Exp Gerontol. 48:634–639.
2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Lee TI, Kao YH, Chen YC, Huang JH, Hsiao
FC and Chen YJ: Peroxisome proliferator-activated receptors
modulate cardiac dysfunction in diabetic cardiomyopathy. Diabetes
Res Clin Pract. 100:330–339. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Desvergne B and Wahli W: Peroxisome
proliferator-activated receptors: Nuclear control of metabolism.
Endocr Rev. 20:649–688. 1999.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Barger PM and Kelly DP: PPAR signaling in
the control of cardiac energy metabolism. Trends Cardiovasc Med.
10:238–245. 2000.PubMed/NCBI View Article : Google Scholar
|