Introduction
Proteus syndrome (PS) is a sporadic hamartomatous
syndrome that manifests as asymmetric overgrowth of body parts,
hyperplasia of connective tissues, hyperostosis, hemangiomas,
lipomas, tumors, disordered adipose tissue, nervous system
complications, and vessel malformations (1,2). This
syndrome was first described by Cohen and Hayden in 1979(3), and named ‘Proteus syndrome’ by Wiedeman
in 1983(4). It causes overgrowth of
multiple tissues in a patchy or mosaic pattern, and commonly
affects tissues that include but are not limited to the skin,
connective tissue, bones, nervous system, and eyes, which determine
disease severity in different patients (2,5). The
onset of PS typically occurs in childhood, with more complex
manifestations developing over time (6). Its approximate incidence is one per 1
million (7). Clinical manifestations
perceived to be diacritic include macrodactyly deformities,
unilateral hypertrophy, palmar or plantar cerebriform hyperplasia,
subcutaneous tumors, verrucous nevus, exostosis, and vessel
hamartomas (8,9). At present, somatic mosaicism is the
widely accepted hypothesis for the etiology of PS (7); however, this disorder has not been
fully understood. In the present report, a rare case of PS that
presented with massive excrescence and cerebriform plantar
hyperplasia of the left foot was documented. Furthermore, given
that PS overgrowth typically manifests between the ages of 6 and 18
months, this case is unusual owing to the solitary cutaneous
finding and delayed presentation in the fourth decade of life.
Current available literature on the syndrome was also
discussed.
Materials and methods
Sections of 4 µm thickness were made from
formalin-fixed (10% neutral-buffered formalin at 4˚C for 24 h)
paraffin-embedded block. The sections were then rehydrated by
xylene and a descending ethanol gradient, following which they were
stained with Verhoeff-van Gieson method under the standard protocol
for confirmation of diagnosis (10).
The stained slides were observed under a light microscope. Two
independent observers evaluated all the slides to reduce subjective
bias. The elastic fibers were analyzed at magnifications, x10 and
x40 for density, morphology, pattern of grouping. Statistical
analysis was conducted using χ2 test.
Case report
A 35-year-old woman visited First Affiliated
Hospital of Kunming Medical University (Kunming, China) for
treatment of progressive postnatal overgrowth involving her left
foot. The patient observed swellings and excrescences on the fourth
and fifth toes of the left foot for the past 10 years, but did not
exhibit any other subjective symptoms. Despite undergoing surgical
excision at a local hospital at Kaiyuan, Yunnan, the patient
experienced recurrence with enlarged hyperplasia and an expanded
rash. She terminated further treatments. However, the affected
tissue continued to grow over the years, seriously affecting the
daily life of the patient. Thus, the patient was admitted to our
hospital in July 2018. Cutaneous examination revealed the presence
of a massive soft tissue hyperplasia in the fourth and fifth toes
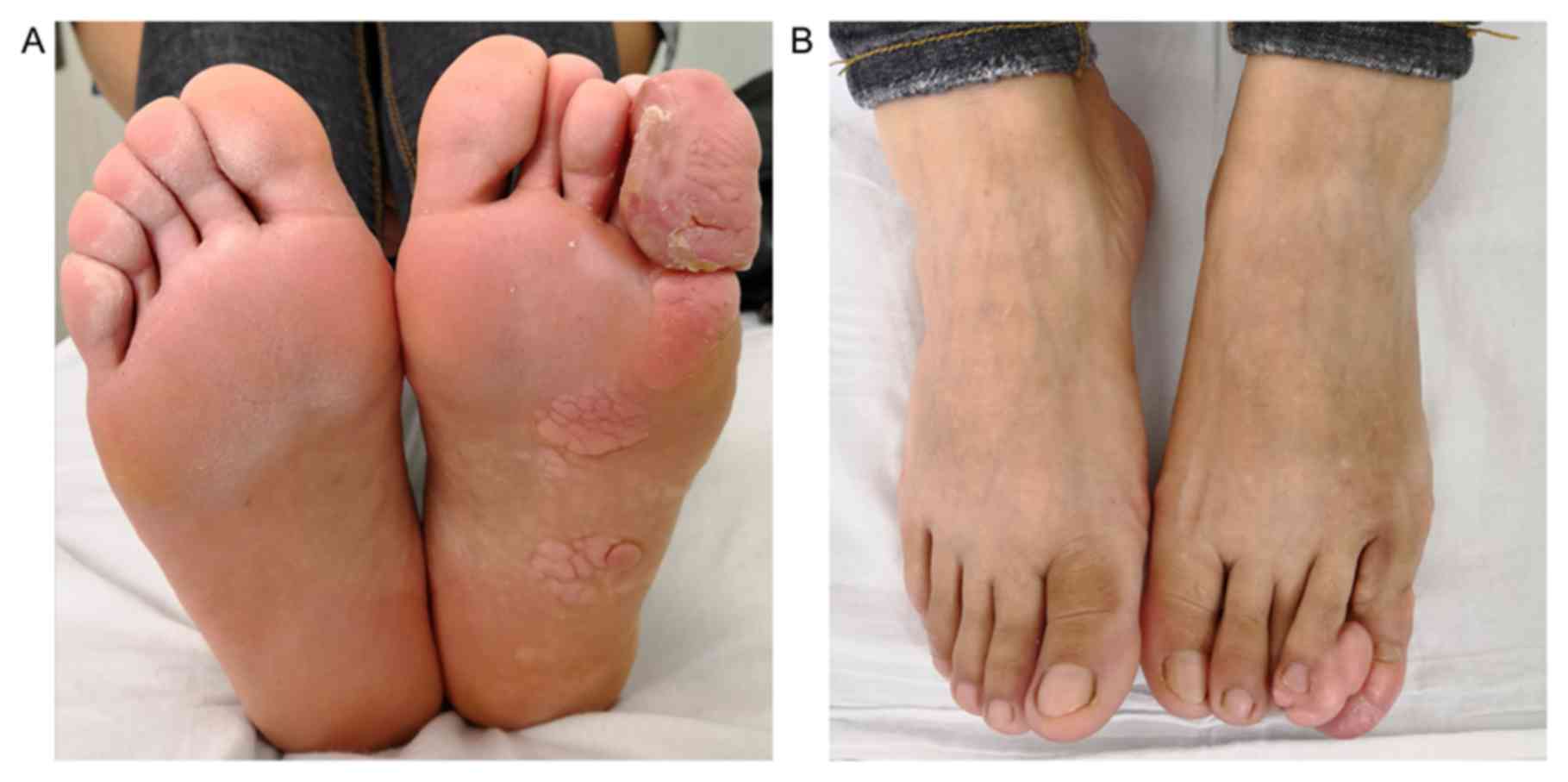
and classic cerebriform plantar hyperplasia of the foot (Fig. 1A). There was no similar overgrowth
observed in any other part of the body, and no other member of the
patient's family exhibited similar features.
Physical examination showed apparent discrepancy in
the thickness of both lower limbs, varicosity, joint deformity, or
scoliosis. Cerebriform hyperplastic tissues of different size were
observed in the planta. The epidermis of the hyperplastic area was
significantly thickened owing to keratinization; however, there was
no evidence of ulceration (Fig. 1B).
There were no port-wine stains, hemangiomas, or purpura.
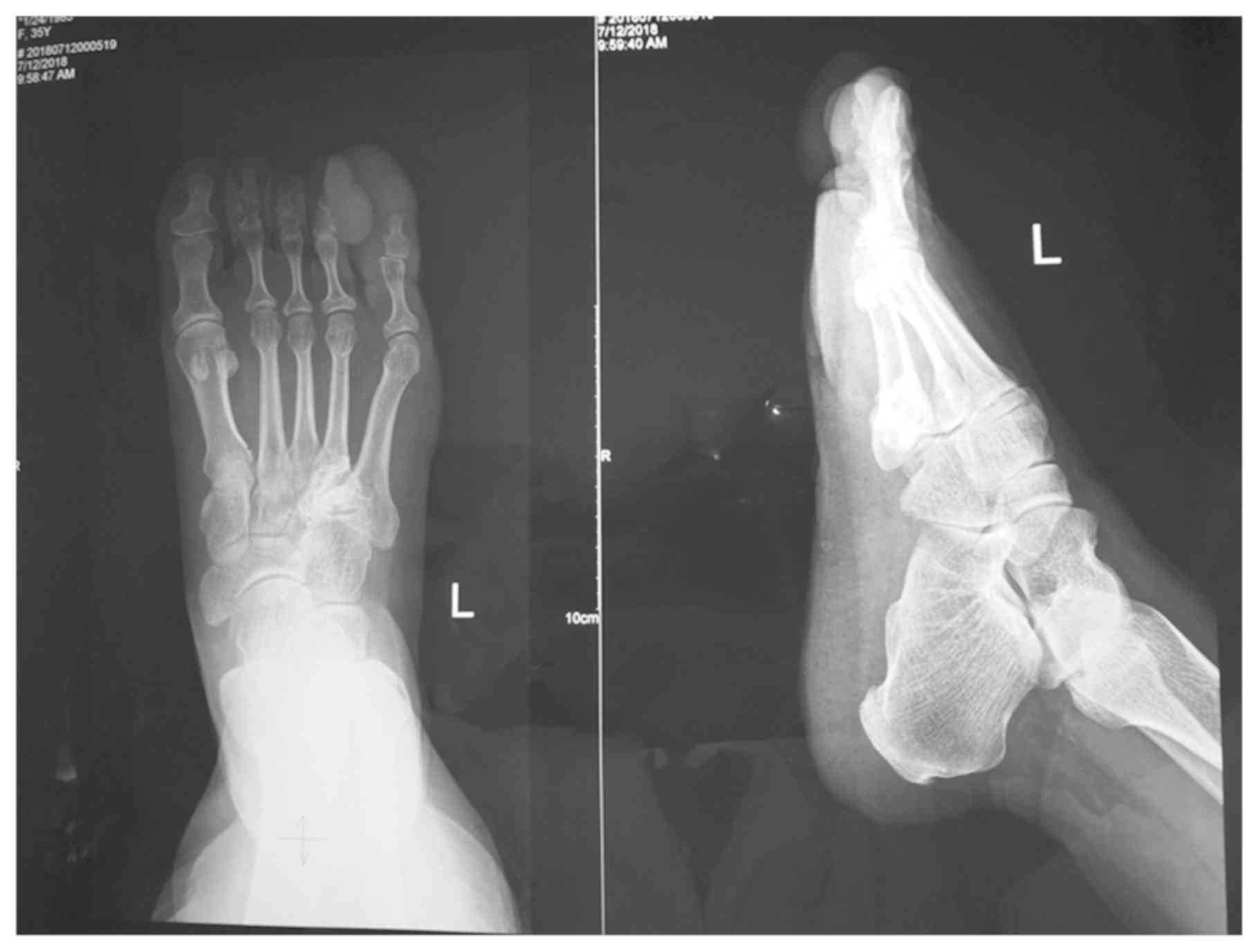
Radiography of the affected lower limb showed soft
tissue masses around the phalanxes of the fourth and fifth toes,
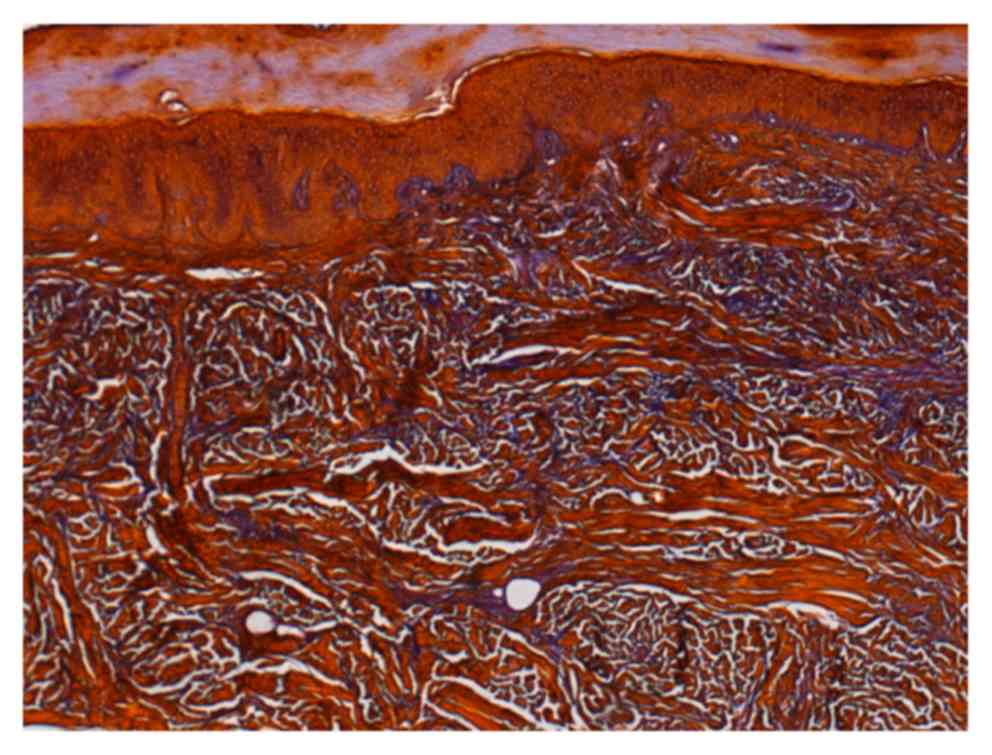
and irregular bone structure of the phalanxes (Fig. 2). Histopathologic examination of the
hyperplastic tissues revealed normal structure of the epidermis.
However, collagen in the dermis was thickened and disorderly
arranged. In addition, elastic fibers were also decreased in the
affected dermis (Fig. 3). Based on
the clinical features and results from the histopathological and
radiological evaluation, the patient was diagnosed with PS.
Discussion
PS affects more male than female patients with a
ratio of approximately 2:1(11).
However, its prevalence is evenly distributed among ethnic groups
(12). Recently reported PS cases
associated with the foot is summarized in Table I. This disease is hardly noticeable
at birth; however, its diagnosis is challenging in the early stage
owing to the involvement of multiple tissues (13). Moreover, the vast clinical
variability in PS probably results in misdiagnosis (14).
 | Table IAll recent published research reports
regarding Proteus syndrome (PS) cases with foot involvement. |
Table I
All recent published research reports
regarding Proteus syndrome (PS) cases with foot involvement.
| Title of article
(refs.) | Year |
|---|
| Reassessment of the
Proteus syndrome literature: Application of diagnostic criteria to
published cases (11) | 2004 |
| A mosaic activating
mutation in AKT1 associated with the Proteus syndrome (19) | 2011 |
| Proteus syndrome:
Three case reports with a review of the literature (28) | 2012 |
| Proteus syndrome:
Clinical profile of six patients and review of literature (29) | 2013 |
| Thoracolumbar
scoliosis in a patient with Proteus syndrome: A case report and
literature review (30) | 2015 |
| Island nail flap in
the treatment of foot macrodactyly of the first ray in children:
report of two cases (31) | 2015 |
| Recurrent cerebriform
connective tissue nevus on the foot of a patient with Proteus
syndrome (32) | 2016 |
| Proteus syndrome: A
case report and review of the literature (14) | 2017 |
| Proteus syndrome
(33) | 2017 |
| Case report:
‘Incognito’ Proteus syndrome (34) | 2018 |
Macrodactyly is characterized by hypertrophy of the
bone with hamartomatous overgrowth of several fibro-adipose
structures without any other associated skin lesions and systemic
involvement. Bannayan-Zonana syndrome associates macrocephaly,
polyposis of the colon and subcutaneous lipomas (15). Maffucci syndrome combines
enchondromatosis and haemangioma (16). Ollier disease shows enchondromatosis
with possible malignant transformation and cerebral neoplasms
(17). PS exhibits more features of
reticular connective tissue naevi, which are not manifested in
Klippel-Trenaunay-Weber syndrome (18).
Lindhurst et al (19), found that a somatic activating
mutation (c. G49A, p. E17K) in the oncogene AKT1 was
associated with the severity of PS. Subsequently, Valera et
al (6) proposed that a genetic
test for the AKT1 mutation in affected and adjacent
non-affected tissues could be a more accurate approach to confirm
the diagnosis. However, a few cases did not show any association
between the AKT1 mutation and PS, including three of the 29
patients with PS reported by Ou et al (14) and Lindhurst et al (19). It was speculated that somatic
mutations in another gene-phosphatase and tensin homolog
(PTEN)-causing dysfunction of the PI3K-AKT pathway may be
potential causes of PS (20,21). However, this hypothesis was rejected
based on recently published evidence (22,23),
wherein numerous affected individuals with PTEN mutations
and clinical features of ‘Proteus-like syndrome’ were eventually
diagnosed with SOLAMEN (segmental overgrowth, lipomatosis,
arteriovenous malformation, and epidermal nevus) or Cowden
syndromes. In 1987, Happle suggested a pathogenetic theory stating
that PS is caused by somatic alteration in a gene resulting in
mosaic effects that could be lethal if the mutation presented in a
non-mosaic pattern (24); however,
no PS-associated gene has been identified thus far. Comparison of
the present case with another one published by Ou et al
(14) revealed a considerable
difference in severity. According to Happle's theory, an earlier
postzygotic alteration in the patient with PS would result in more
severe manifestations, as the earlier mutation would have affected
a larger number of cell lineages. Thus, further investigation is
required to identify the specific genes associated with the
development of PS.
At present, the diagnosis of PS is mainly based on
clinical manifestations and imaging techniques (25). In 2004, Turner et al revised
the diagnostic criteria for the syndrome (11), which comprise three general and three
specific criteria categories (Table
II). For a confirmed diagnosis, the patient should fulfill all
the general criteria, namely mosaic distribution of the phenotype,
sporadic emergence, and progressive course. Subsequently, an
additional single sign from category A, two signs from category B,
or three signs from category C are sufficient to establish the
diagnosis of PS (26). In the
present case, the diagnosis was based on the patient fulfilling all
three general criteria and three of the specific criteria, namely
cerebriform connective tissue nevus (category A), asymmetric and
disproportionate overgrowth of the digits (category B), and
dysregulated adipose tissue (category C).
| Table IIDiagnostic criteria of Proteus
syndrome (11). |
Table II
Diagnostic criteria of Proteus
syndrome (11).
| General criteria
(diagnosis includes all of the following): |
|---|
| • Mosaic distribution
of lesions |
| • Sporadic
occurrence |
| • Progressive
course |
| Specific criteria
categories (Either Category A or 2 from Category B or 3 from
category C) |
| A. 1. Cerebriform
connective tissue nevus (skin lesions characterized by deep grooves
and gyrations as seen on the surface of the brain). |
| B. 1. Linear
epidermal nevus |
| Asymmetric,
disproportionate overgrowth (asymmetric, disproportionate
overgrowth should be carefully distinguished from asymmetric,
proportionate overgrowth) ≥1 of: |
|
a. Limbs:
Arms/legs/hands/feet/digits |
|
b. Skull
(hyperostosis) |
|
c. External
auditory meatus (hyperostosis) |
|
d. Vertebra
(megaspondylodysplasia) |
|
e. Viscera:
Spleen and/or thymus |
| Specific tumors
before 2nd decade |
| One of the
following: |
|
a. Ovarian
cystadenoma |
|
b. Paratid
monomorphic adenoma |
| C. 1. Dysregulated
adipose tissue |
| Either one: |
|
a.
Lipomas |
|
b. Regional
absence of fat |
| Vascular
malformations |
| One or more: |
|
a. Capillary
malformation |
|
b. Venous
malformation |
|
c. Lymphatic
malformation |
| Lung cysts |
| Facial phenotype (the
criteria have been found, to date, only in PS patients who have
mental deficiency, and in some cases, seizures and/or brain
malformations). |
| All: |
|
a.
Dolichocephaly |
|
b. Long
face |
|
c. Down
slanting palpebral fissures and/or minor ptosis |
|
d. Low nasal
bridge |
|
e. Wide or
anteverted nares |
|
f. Open
mouth at rest |
The treatment of PS is challenging owing to its
varied clinical features. The patients ought to be periodically
followed up for the development of complications (27). Surgical treatment is generally
applied for the removal of symptomatic lesions. Given the
increasing operative difficulty and rate of complications over
time, early surgical interventions are vital to reduce extra
malformations, physical defects, or loss of movement (6). In addition, it is necessary to monitor
the patients for potential tumor development. Moreover, the
disfiguring impairments characteristic of PS can place enormous
psychological burden on patients and their families; thus,
psychological counseling is of great importance (28).
Prognosis is closely related to the severity of
complications. Approximately 20% of patients with PS expire
prematurely, commonly due to venous thromboembolism or pulmonary
embolism, pneumonia, or surgical complications (1,28).
In summary, PS is a very rare, highly variable, and
progressive tissue overgrowth disorder. The exact pathogenesis and
etiology of this disease remain incompletely understood.
Considering the complications and early mortality observed in
patients, we emphasize the significance of early diagnosis of PS
and the need for interventions through a multi-disciplinary
approach must be emphasized.
Acknowledgements
Not applicable.
Funding
No Funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this article.
Authors' contributions
MH made significant contributions to data analysis
and literature search, and was a major contributor in writing the
manuscript. WZ made substantial contributions to conception and
design, and revised the manuscript critically for important
intellectual content. Each author agreed to be accountable for all
aspects of the work related to the accuracy or integrity of any
part of the work, and approved the final version of the
manuscript.
Ethics approval and consent to
participate
This study was approved by the Institutional Review
Board of The First Affiliated Hospital of Kunming Medical
University (Kunming, China). Informed consent was obtained from the
patient in the present study.
Patient consent for publication
Patient consent was obtained from the patient for
publication of this report and any accompanying images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Biesecker LG, Happle R, Mulliken JB,
Weksberg R, Graham JM Jr, Viljoen DL and Cohen MM Jr: Proteus
syndrome: Diagnostic criteria, differential diagnosis, and patient
evaluation. Am J Med Genet. 84:389–395. 1999.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Cohen MM Jr: Proteus syndrome: An update.
Am J Med Genet C Semin Med Genet. 137C:38–52. 2005.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Cohen MM Jr and Hayden PW: A newly
recognized hamartomatous syndrome. Birth Defects Orig Artic Ser.
15:291–296. 1979.PubMed/NCBI
|
|
4
|
Wiedemann HR, Burgio GR, Aldenhoff P,
Kunze J, Kaufmann HJ and Schirg E: The proteus syndrome. Partial
gigantism of the hands and/or feet, nevi, hemihypertrophy,
subcutaneous tumors, macrocephaly or other skull anomalies and
possible accelerated growth and visceral affections. Eur J Pediatr.
140:5–12. 1983.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Furquim I, Honjo R, Bae R, Andrade W,
Santos M, Tannuri U and Kim C: Proteus syndrome: Report of a case
with recurrent abdominal lipomatosis. J Pediatr Surg. 44:E1–E3.
2009.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Valéra MC, Vaysse F, Bieth E, Longy M,
Cances C and Bailleul-Forestier I: Proteus syndrome: Report of a
case with AKT1 mutation in a dental cyst. Eur J Med Genet.
58:300–304. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Biesecker L: The challenges of proteus
syndrome: Diagnosis and management. Eur J Hum Genet. 14:1151–1157.
2006.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Cavero JA, Castro EG and Junco L: Proteus
syndrome. Int J Dermatol. 39:707–709. 2000.PubMed/NCBI
|
|
9
|
Yasuda H, Yamamoto O, Hirokawa H, Asahi M,
Kashimura M and Sakai A: Proteus syndrome. Dermatology.
203:180–184. 2001.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Dahal S, Swaminathan G, Carney S,
Broekelmann T, Mecham R and Ramamurthi A: Pro-elastogenic effects
of mesenchymal stem cell derived smooth muscle cells in a 3D
collagenous milieu. Acta Biomater. 105:180–190. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Turner JT, Cohen MM Jr and Biesecker LG:
Reassessment of the proteus syndrome literature: Application of
diagnostic criteria to published cases. Am J Med Genet A.
130A:111–122. 2004.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Satter E: Proteus syndrome: 2 case reports
and a review of the literature. Cutis. 80:297–302. 2007.PubMed/NCBI
|
|
13
|
Sethi SK, Yadav D, Garg P, Chawla J and
Goyal D: A child with mental retardation and asymmetrical
hypertrophy of limbs. Eur J Pediatr. 170:813–814. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ou M, Sun Z, Zhu P, Sun G and Dai Y:
Proteus syndrome: A case report and review of the literature. Mol
Clin Oncol. 6:381–383. 2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Bialer MG, Riedy MJ and Wilson WG: Proteus
syndrome versus bannayan-zonana syndrome: A problem in differential
diagnosis. Eur J Pediatr. 148:122–125. 1988.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Haga N, Nakamura K, Taniguchi K and
Nakamura S: Enchondromatosis with features of
dysspondyloenchondromatosis and maffucci syndrome. Clin Dysmorphol.
7:65–68. 1998.PubMed/NCBI
|
|
17
|
Klein C, Delcourt T, Salon A, Finidori G,
Glorion C and Pannier S: Surgical treatment of enchondromas of the
hand during childhood in ollier disease. J Hand Surg Am.
43:946.e1–946.e5. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Krengel S, Fustes-Morales A, Carrasco D,
Vázquez M, Durán-McKinster C and Ruiz-Maldonado R: Macrodactyly:
Report of eight cases and review of the literature. Pediatr
Dermatol. 17:270–276. 2000.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Lindhurst MJ, Sapp JC, Teer JK, Johnston
JJ, Finn EM, Peters K, Turner J, Cannons JL, Bick D, Blakemore L,
et al: A mosaic activating mutation in AKT1 associated with the
proteus syndrome. N Engl J Med. 365:611–619. 2011.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhou X, Hampel H, Thiele H, Gorlin RJ,
Hennekam RC, Parisi M, Winter RM and Eng C: Association of germline
mutation in the PTEN tumour suppressor gene and proteus and
proteus-like syndromes. Lancet. 358:210–211. 2001.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Smith JM, Kirk EP, Theodosopoulos G,
Marshall GM, Walker J, Rogers M, Field M, Brereton JJ and Marsh DJ:
Germline mutation of the tumour suppressor PTEN in proteus
syndrome. J Med Genet. 39:937–940. 2002.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Caux F, Plauchu H, Chibon F, Faivre L,
Fain O, Vabres P, Bonnet F, Selma ZB, Laroche L, Gérard M and Longy
M: Segmental overgrowth, lipomatosis, arteriovenous malformation
and epidermal nevus (SOLAMEN) syndrome is related to mosaic PTEN
nullizygosity. Eur J Hum Genet. 15:767–773. 2007.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Loffeld A, McLellan NJ, Cole T and Moss C:
Type 2 segmental cowden disease vs. proteus syndrome: Reply from
authors. Br J Dermatol. 158:410–411. 2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Happle R: Lethal genes surviving by
mosaicism: A possible explanation for sporadic birth defects
involving the skin. J Am Acad Dermatol. 16:899–906. 1987.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Alves C, Acosta AX and Toralles MB:
Proteus syndrome: Clinical diagnosis of a series of cases. Indian J
Endocrinol Metab. 17:1053–1056. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Nguyen D, Turner JT, Olsen C, Biesecker LG
and Darling TN: Cutaneous manifestations of proteus syndrome:
Correlations with general clinical severity. Arch Dermatol.
140:947–953. 2004.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Talari K, Subbanna PK, Amalnath D and Suri
SD: Proteus syndrome: A rare case report. Indian J Hum Genet.
18:356–358. 2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Thomason JL, Abramowsky CR, Rickets RR,
Culbertson JH, Clifton MS and Shehata BM: Proteus syndrome: Three
case reports with a review of the literature. Fetal Pediatr Pathol.
31:145–153. 2012.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Angurana SK, Angurana RS, Panigrahi I and
Marwaha RK: Proteus syndrome: Clinical profile of six patients and
review of literature. Indian J Hum Genet. 19:202–206.
2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Li Z, Shen J and Liang J: Thoracolumbar
scoliosis in a patient with proteus syndrome: A case report and
literature review. Medicine (Baltimore). 94(e360)2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Downey Carmona FJ, Lagares A,
Farrington-Rueda D and Lirola-Criado J: Island nail flap in the
treatment of foot macrodactyly of the first ray in children: Report
of two cases. J Child Orthop. 9:281–285. 2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Wu J, Wang Q, Cui P, Wu X and Yan Z:
Recurrent cerebriform connective tissue nevus on the foot of a
patient with Proteus syndrome. Cutis. 98:E16–E19. 2016.PubMed/NCBI
|
|
33
|
Rocha RCC, Estrella MPS, Amaral DMD,
Barbosa AM and Abreu MAM: Proteus syndrome. An Bras Dermatol.
92:717–720. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Vestita M, Filoni A, Arpaia N, Ettorre G
and Bonamonte D: Case report: ‘Incognito’ proteus syndrome.
F1000Res. 7:228–236. 2018.PubMed/NCBI View Article : Google Scholar
|