Introduction
Marfan syndrome (MFS) is an autosomal dominant multisystemic disorder of connective tissue that is caused by mutations in the fibrillin-1 (FBN1) gene. The FBN1 gene encodes FBN1, which is a glycoprotein of the extracellular matrix (ECM) that is involved in microfibril construction. MFS has a prevalence of ~1:5,000, and its cardinal clinical manifestations are primarily associated with the cardiovascular, ocular and skeletal systems, whereas lung, skin and dura are less frequently affected (1,2). Currently, the clinical diagnosis of MFS is performed using the diagnostic criteria provided by the Ghent nosology, which was revised in 2010 (3,4). Previous studies have demonstrated that matrix metalloproteinases (MMPs) are closely associated with the development of MFS, by causing a loss of elastic fibers and in ECM structures (5,6). In addition, increased microRNA-29b expression, which is a key molecule in the pathogenesis of early aneurysms in MFS, has been identified to regulate aortic wall apoptosis and ECM abnormalities (7). A series of studies have indicated that MFS is altered and may be associated with a variety of pathological processes (1,8). It is therefore important to improve the understanding behind the molecular alterations in MFS, especially the key molecules and fundamental interaction networks.
Long noncoding RNAs (lncRNAs) are non-protein coding transcripts, which are >200 nucleotides in length. Accumulating evidence has demonstrated that lncRNA transcripts can participate in almost every aspect of the cell biological functions, including cell differentiation, metabolism and apoptosis, by coordinating gene expression (9,10). An aortic aneurysm is a primary clinical manifestation that is identified in patients with MFS (11,12). Previous studies have shown that numerous lncRNAs are differentially expressed in aortic aneurysms. Yang et al (13) revealed that novel lncRNA candidates are associated with the pathogenesis of abdominal aortic aneurysm and that lnc-arginase 1 (ARG) can decrease the mRNA expression levels of arachidonate 5-lipoxygenase (ALOX5) and decrease the production of reactive oxygen species in HeLa cells. Similarly, He et al (14) demonstrated that the lncRNA HIF 1 α-antisense RNA 1 (HIF1α-AS1) exhibited a raised expression level in thoracoabdominal aortic aneurysm samples and that the interaction between HIF1a-AS1 and apoptotic proteins serves a key role in the proliferation and apoptosis of vascular smooth muscle cells (VSMCs) (14). The potential role of lncRNAs in the pathogenesis of MFS; however, has not yet been determined.
In the present study, the expression profiles of lncRNA and mRNA in aortic tissues with MFS and normal aorta (NA) tissues were analyzed using microarray analysis. Bioinformatics analysis was performed to predict the potential functions of the differentially expressed mRNAs. A lncRNA-mRNA co-expression network was constructed to determine the potential target genes of the aberrantly expressed lncRNAs and mRNAs. Subsequently, a total of five pivotal lncRNAs were selected for further validation using reverse transcription-quantitative (RT-q) PCR.
Materials and methods
Patients and sample collection and ethics statement
The present study protocol was approved by the Institutional Ethics Review Board of the First Affiliated Hospital of Wenzhou Medical University and written informed consent was obtained from family members for all donors and patients, prior to their inclusion in the current study. NA (n=10) and MFS tissue (n=9) were obtained from donors from the First Affiliated Hospital of Wenzhou Medical University between January 2016 and June 2018, of which 4 NA tissues and 3 MFS tissues were used for microarray analysis and the rest of the samples, 6 NA tissues and 6 MFS tissues, were used for RT-qPCR analysis. Inclusion and exclusion criteria of MFS: Diagnosis of MFS was confirmed following the 2010 Ghent criteria (4), and the reason for surgery for MFS was severe ascending aortic dilatation without aortic dissection. Hereditary aortic diseases of Ehlers-Danlos syndrome (EDS), Loeys-Dietz syndrome (LDS) were excluded. Inclusion and exclusion criteria of NA: Multi-organ donors who were terminally brain dead without hereditary aortic diseases, including MFS, EDS and LDS. The aortic tissue samples were prepared into smaller sample sizes, placed in freezing tubes and frozen immediately in liquid nitrogen until RNA extraction. The demographic information of MFS and NAs are listed in Table I.
 |
Table I
Demographic and clinical characteristics of patients with MFS or NAs.
|
Table I
Demographic and clinical characteristics of patients with MFS or NAs.
| Parameter |
MFS (n=9) |
NA (n=10) |
P-value |
| Age (year) |
|
|
<0.05 |
| Mean |
36.3 |
54.8 |
|
| Sex |
|
|
0.76 |
| Male |
6 |
6 |
|
| Female |
3 |
4 |
|
| Hypertension |
|
|
0.70 |
| Yes |
2 |
3 |
|
| No |
7 |
7 |
|
| Aortic sinus diameter (mm) |
|
|
<0.05 |
| Mean |
45 |
32 |
|
| Aortic insufficiency |
|
|
0.09 |
| Yes |
4 |
1 |
|
| No |
5 |
9 |
|
Hematoxylin and eosin (H&E) and Verhoeff-Van Gieson (VG) connective tissue staining
Samples were fixed with 4% paraformaldehyde for 4 h at room temperature and then embedded in paraffin. Subsequently, paraffin-embedded samples were cut into 4 µm-thick sections and stained with H&E and VG.
H&E
The dried tissue slices, were immediately dewaxed using xylene for 5-10 min followed by a descending ethanol gradient. The slices were then stained with moved into hematoxylin for 5 min at room temperature and differentiated in 1% hydrochloric acid alcohol, for up to 30 sec. before being stained with eosin stain for 1 min at room temperature.
VG
Samples were dehydrated using an ascending ethanol gradient followed by xylene. The slides were then stained with hematoxylin for 20 min at room temperature and into VG (Beijing Leagene Biotech Co., Ltd.) for 1 min at room temperature before being differentiated rapidly in 95% alcohol solution for 5 sec. Aortic tissue slices from 3 patients with MFS and 3 control patients were observed under a light microscope (Leica EZ4 W; Leica Microsystems GmbH) at x200 magnification.
Immunohistochemical staining
Samples were fixed with 4% paraformaldehyde for 4 h at room temperature and then embedded in paraffin. Paraffin-embedded samples were cut into 4 µm-thick sections. The characterization of the inflammatory infiltrate was carried out by immunohistochemistry. Slides from 3 control patients and 3 patients with MFS were put into heated citrate buffer at 98˚C for 15 min and cooled to room temperature. Endogenous peroxidase was blocked with 3% H2O2 solution (diluted in distilled water) for 10 min at room temperature. Sections were immunostained overnight at room temperature with anti-CD68 (cat. no. ZM-0060; 1:100; OriGene Technologies, Inc.; macrophage marker) (15), anti-CD3 (cat. no. kit-0003; 1:100; MXB Biotechnologies; T cell marker); anti-smooth muscle actin (SMA; cat. no. kit-0006; 1:1,000; MXB Biotechnologies) and anti-desmin (cat. no. ZA-0610; 1:100; OriGene Technologies, Inc.). The sections were then incubated using the Histostain™ SAP kit (cat. no. SAP-9100; 1:1,000; OriGene Technologies, Inc.) at 4˚C overnight. Sections were treated with streptavidin-biotin-peroxidase for 10 min at room temperature. Diamino-benzidine was used as a chromogenic substrate. Finally, sections were washed with distilled water. The slices were observed under a light microscope (Leica EZ4 W; Leica Microsystems GmbH) at x100 magnification.
RNA extraction and lncRNA microarray analysis
RNA was extracted from the frozen aortic tissue using Trizol® reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocols. RNA samples were analyzed using the microarray assays. An Arraystar Human Microarray lncRNA v3.0 (array format: 8 x 60K; Arraystar, Inc.), which can probe more than 30,000 lncRNAs, covering all lncRNAs from authoritative databases RefSeq (http://www.ncbi.nlm.nih.gov/projects/RefSeq) (16), UCSC Known Genes (http://genome.ucsc.edu) (17), LNCipedia (http://www.lncipedia.org) (18), NONCODEv4 (http://www.noncode.org) (19) and Ensembl (https://asia.ensembl.org) (20) and their coding proteins, were used for microarray analysis. Microarray hybridization was performed based on the manufacturer's instructions. Quantile normalization and subsequent data analysis were performed using the GeneSpring GX v11.5.1 software package (Agilent Technologies, Inc.).
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses
GO and pathway enrichment analyses were applied to predict the functions of the differentially expressed genes. GO analysis was performed based on GO (www.geneontology.org), which comprises three integrated domains (molecular function, biological process and cellular component) to describe gene product functions. KEGG (www.genome.jp/kegg) enrichment analysis was employed to analyze the biological pathways associated with the differentially expressed lncRNAs and mRNAs.
lncRNA-mRNA coexpression network analysis
A lncRNA-mRNA coexpression network was built according to the normalized signal intensity of the aberrantly expressed lncRNAs and mRNAs. To further identify the interactions between the lncRNAs and mRNAs and to locate core regulatory factors (genes) among genes the Pearson correlation coefficient was calculated and significant correlation pairs (P<0.05) were selected to construct the network. In this network, the number of links from each lncRNA to mRNA or to another lncRNA was computed and defined as the central degree. A higher degree for a lncRNA indicated that the lncRNA serves a more vital role in the network.
RT-qPCR validation
RT-qPCR was used to verify the differentially expressed genes that were detected on the microarray. Total RNA was extracted by TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. RNA concentration and quality were analyzed by detecting the absorbance at 260 nm using Beckman DU6400 spectrophotometer (Beckman Coulter, Inc.). RNA was treated at 42˚C for 2 min to remove gDNA, following which cDNA was synthesized at 42˚C for 15 min before inactivation at 95˚C for 3 min using the QuantiTect® reverse transcription kit (Qiagen, Inc). qPCR was performed in triplicate using SsoAdvance™ Universal SYBR® Green Supermix (Bio-Rad Laboratories, Inc.) following manufacturer instructions on a Bio-Rad CFX96 Real-Time PCR system. The primers used for RT-qPCR are listed in Table II. The thermocycling conditions for the qPCR: Initial denaturation at 95˚C for 3 min; followed by 40 cycles of 95.0˚C for 10 sec, 59.1˚C for 30 sec and 72.0˚C for 30 sec. The relative expression levels of the genes were calculated using the 2-ΔΔCq method (21), and P<0.05 was considered to indicate a statistically significant difference.
|
Table II
Primers used for reverse transcription-quantitative PCR.
|
Table II
Primers used for reverse transcription-quantitative PCR.
| Sequence name |
Forward primer (5'-3') |
Reverse primer (5'-3') |
| GADPH |
CGGATTTGGTCGTATTG |
GAAGATGGTGATGGGATT |
| uc003jka_1 |
TGCCATTGATACAGTGATGACTTC |
GGAGAAGGTCAGTTGACAGCATAC |
| uc003jox_1 |
GTCTGGTTTGGAAGTGATTGCT |
CACAGCCACTAACTCTTGATGTTG |
| XIST |
GTGGGTTGTTGCACTCTCTG |
CATTCTCTGCCAAAGCGGTAG |
| linc-LPA-1 |
GCAGAAATGGCAAGACGATTAAC |
GCTTCTCTCCTCTCTAGCAGATCTC |
| linc-PPWD1 |
GCATGTGAACAGCATCAGACAG |
GGTGATTTCCACCAGAGAAGG |
Statistical analysis
All statistical analyses were performed using SPSS version 16.0 (SPSS, Inc.). Demographic and clinical characteristic analysis of populations were performed using descriptive statistics. The differential expression levels of the tested lncRNAs and mRNAs between MFS and NA tissues were assessed using Student's t-tests and a fold change (FC) ≥1.5 and P<0.05 was considered to indicate a statistically significant difference. Fisher's exact tests were used for GO and KEGG pathway analyses. Similarly, a P<0.05 was considered to indicate a statistically significant difference.
Results
HE and VG staining of aortic tissues
HE and VG staining indicated that NAs had a complete and continuous aortic structure (Fig. 1A). MFS specimens exhibited separation and loosening of each of the aortic structures (the intima, tunica media and adventitia; Fig. 1B). In addition, collagen and muscle fibers were stained red and yellow, respectively, in VG staining. NA exhibited abundant collagen fibers and dense tissue structure in the aortic intima, tunica media and adventitia, which were decreased and destroyed in the aortic tissues of MFS specimens (Fig. 1C and D).
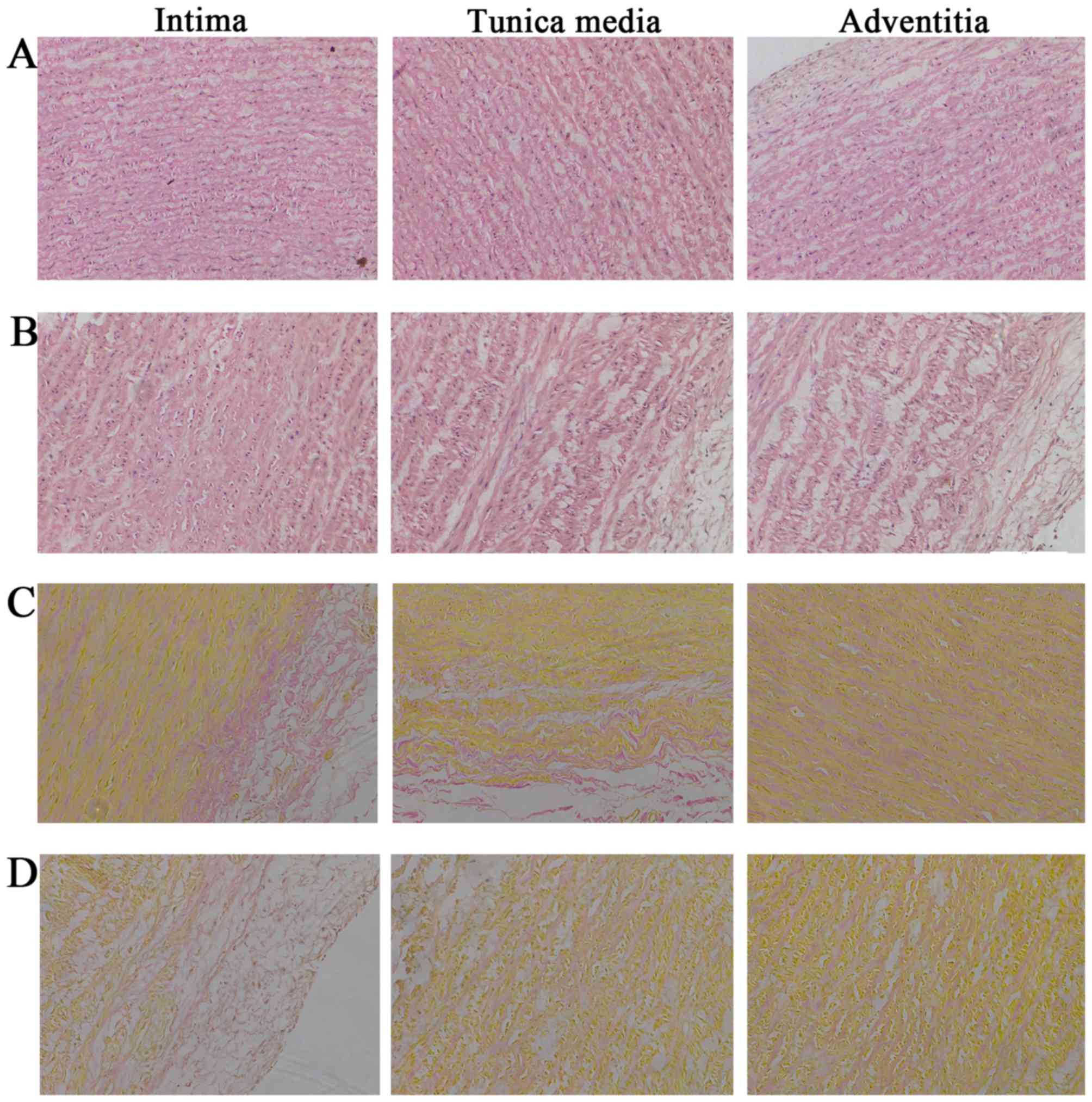 |
Figure 1
H&E and VG staining of aortic tissue. (A) H&E staining of normal aortic tissue. (B) H&E staining of MFS aortic tissue. (C) VG staining of normal aortic tissue. (D) VG staining of MFS aortic tissue. Collagen and muscle fibers were stained red and yellow, respectively. Magnification, x200. H&E, hematoxylin and eosin; MFS, Marfan syndrome, VG, Verhoeff-Van Gieson.
|
Immunohistochemical staining
Immunohistochemistry was performed using monoclonal antibodies directed against markers for T lymphocytes and macrophages to characterize the cells that were present in the aortic tissue of NA and MFS samples. CD3 T cells were clearly observed throughout the sections of the aortic media and adventitia of MFS but not in the control aortas (Fig. 2A), as was also confirmed by immunostaining with the macrophage-specific marker, CD68 (Fig. 2B). SMA and desmin immunostaining was used to mark the expression of smooth muscle cells (SMCs) and was strongly positive in all aortic tissues (Fig. 2C and D). However, structural degeneration and disintegration of SMCs was observed in the MFS tissue.
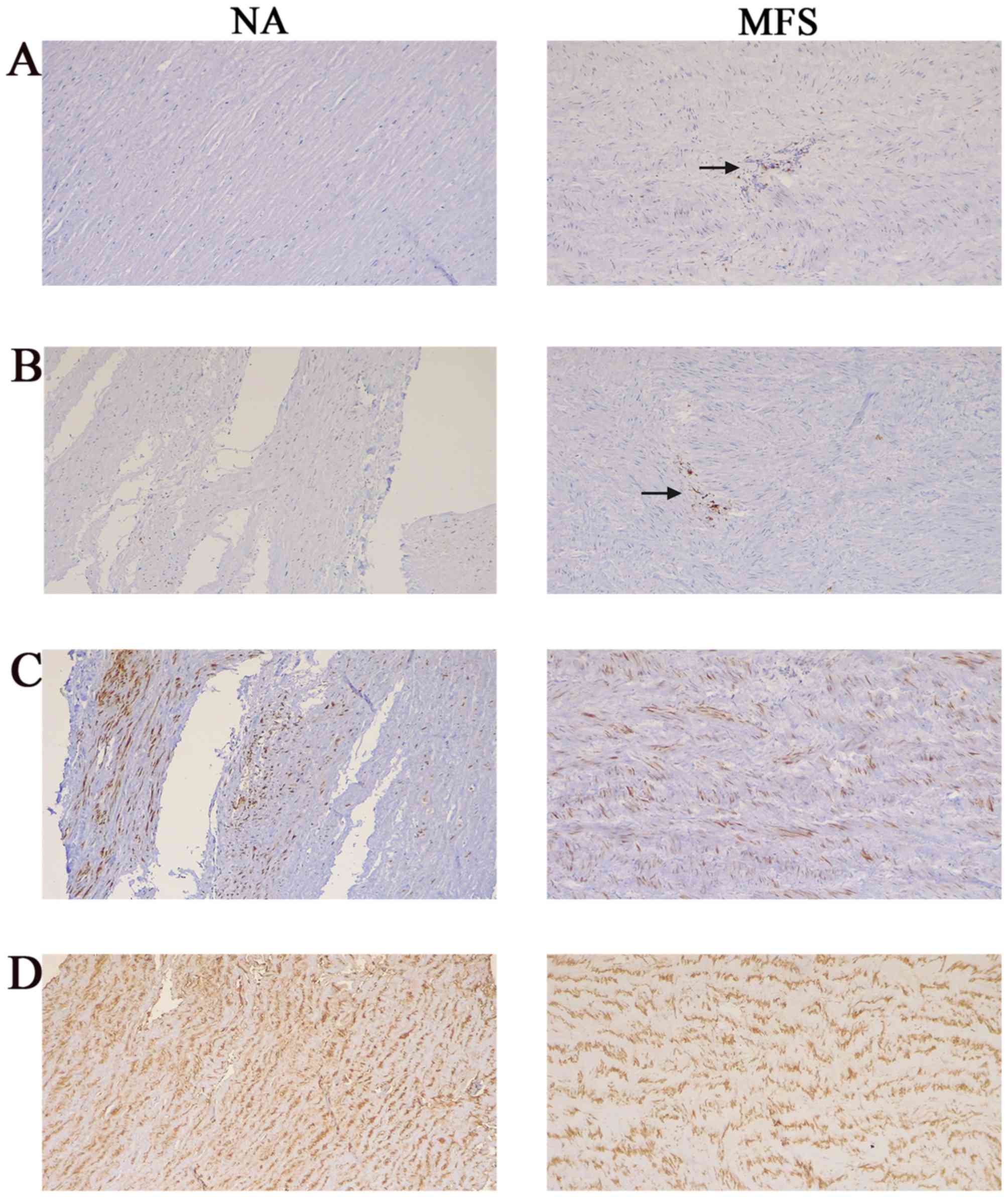 |
Figure 2
Immunohistochemistry staining of aortic tissue. (A) CD3 T cells in aortic tissue of NA and MFS samples. (B) CD68 macrophages in the aortic tissue of NA and MFS samples. Immunohistochemical staining indicated that inflammatory cells (CD3 and CD68) were present diffusely in the media and adventitia of the aortas from patients with MFS, but were rarely found in control aortas. (C) SMA immunostaining in the aortic tissue of NA and MFS samples. (D) Desmin immunostaining in aortic tissue of NA and MFS samples. SMCs were both strongly positive in all aortic tissues, but the structure was degenerative and disintegrative in MFS samples compared with NAs. Magnification x100. MFS, Marfan syndrome; NA, normal aorta; SMA, smooth muscle actin; SMCs, smooth muscle cells.
|
The profiling of differentially expressed genes
Arraystar microarray chips were used to identify the differentially expressed lncRNAs and mRNAs in pathological aortic tissue of MFS and NA specimens (Table SI). In total, 294 lncRNAs were identified; 245 lncRNAs were upregulated and 49 were downregulated. Additionally, 644 differentially expressed mRNAs were found to be dysregulated, of which 455 were upregulated and 189 were downregulated (FC ≥1.5; P<0.05). Finally, to detect the relationships between specimens, hierarchical clustering and scatter plots were performed to identify the distinguishable expression patterns among samples (Fig. 3B and C). Subsequently, log2 (FC) was indicated as the X-axis and log10 (P-value) as the Y-axis to indicate the distribution of the lncRNAs and mRNAs in a volcano plot (Fig. 3A).
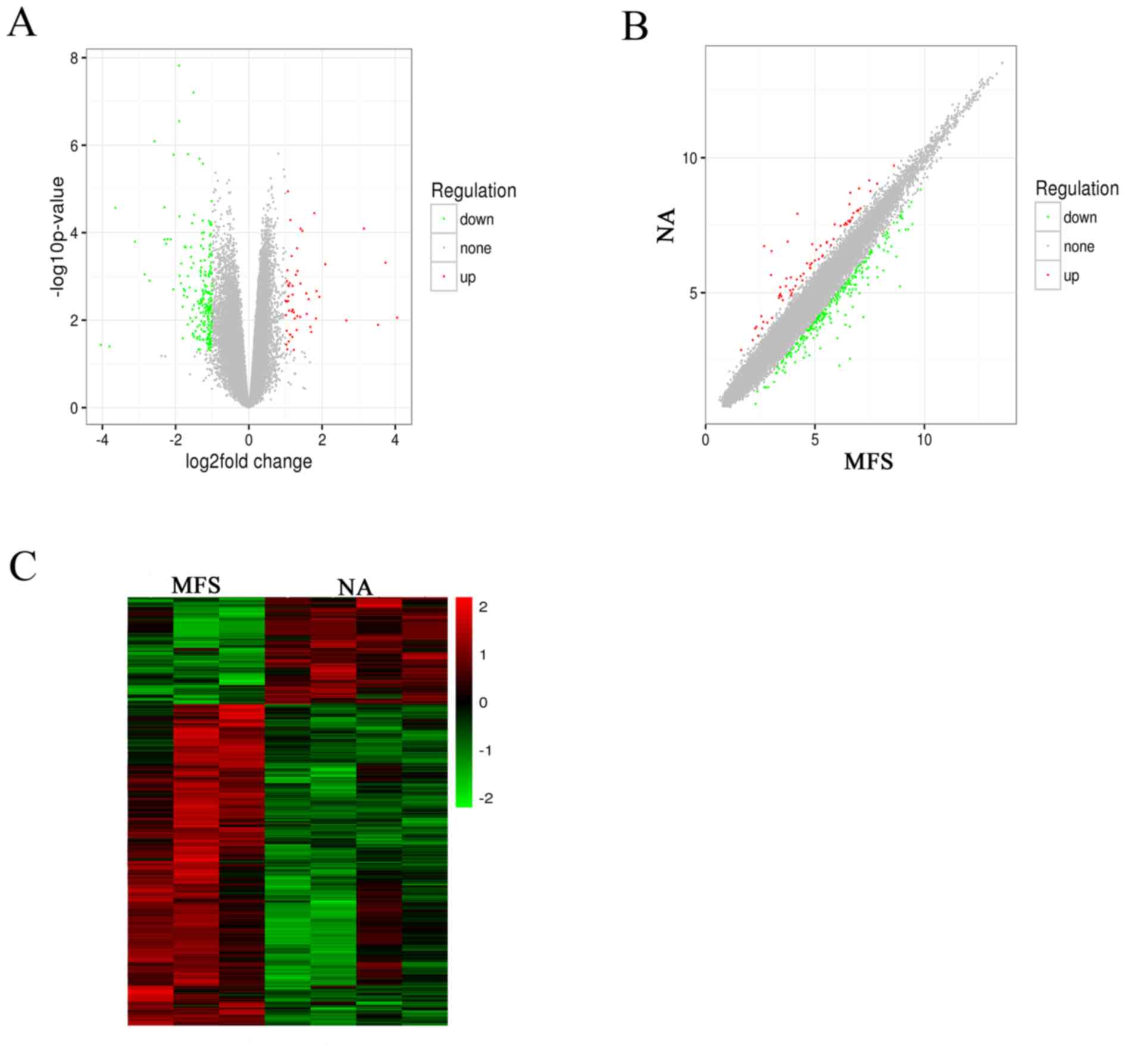 |
Figure 3
Microarray profiling of lncRNAs in the MFS and NA tissue specimens. (A) Volcano plots, (B) scatter plots and (C) hierarchical clustering showing the lncRNA expression profiling (P<0.05 and fold change ≥1.5). Upregulated lncRNAs are denoted in red and downregulated in green. lncRNA, long noncoding RNA; MFS, Marfan syndrome; NA, normal aorta.
|
GO and KEGG pathway analyses
GO and KEGG pathway analyses were used to analyze the potential roles that the differentially expressed mRNAs play in biological processes. Firstly, GO analysis was performed to enrich the significant functions of the differentially expressed genes (Table SII). A total of 122 GO functions were obtained according to the P-value (P<0.01), including 75 upregulated GO functions and 47 downregulated GO functions. Moreover, the-lg P-value was used to describe the significance level of the GO enrichment, with a higher-lg P-value indicating a higher significance level. Some important upregulated GO functions may be associated with MFS and are mainly involved in ‘cell adhesion’, ‘elastic fiber assembly’ and ‘extracellular matrix organization’. In contrast, ‘response to virus’ and ‘inflammatory response’ were downregulated (Fig. 4A and B). These results indicated that the components of ECM organization were significantly altered in patients with MFS.
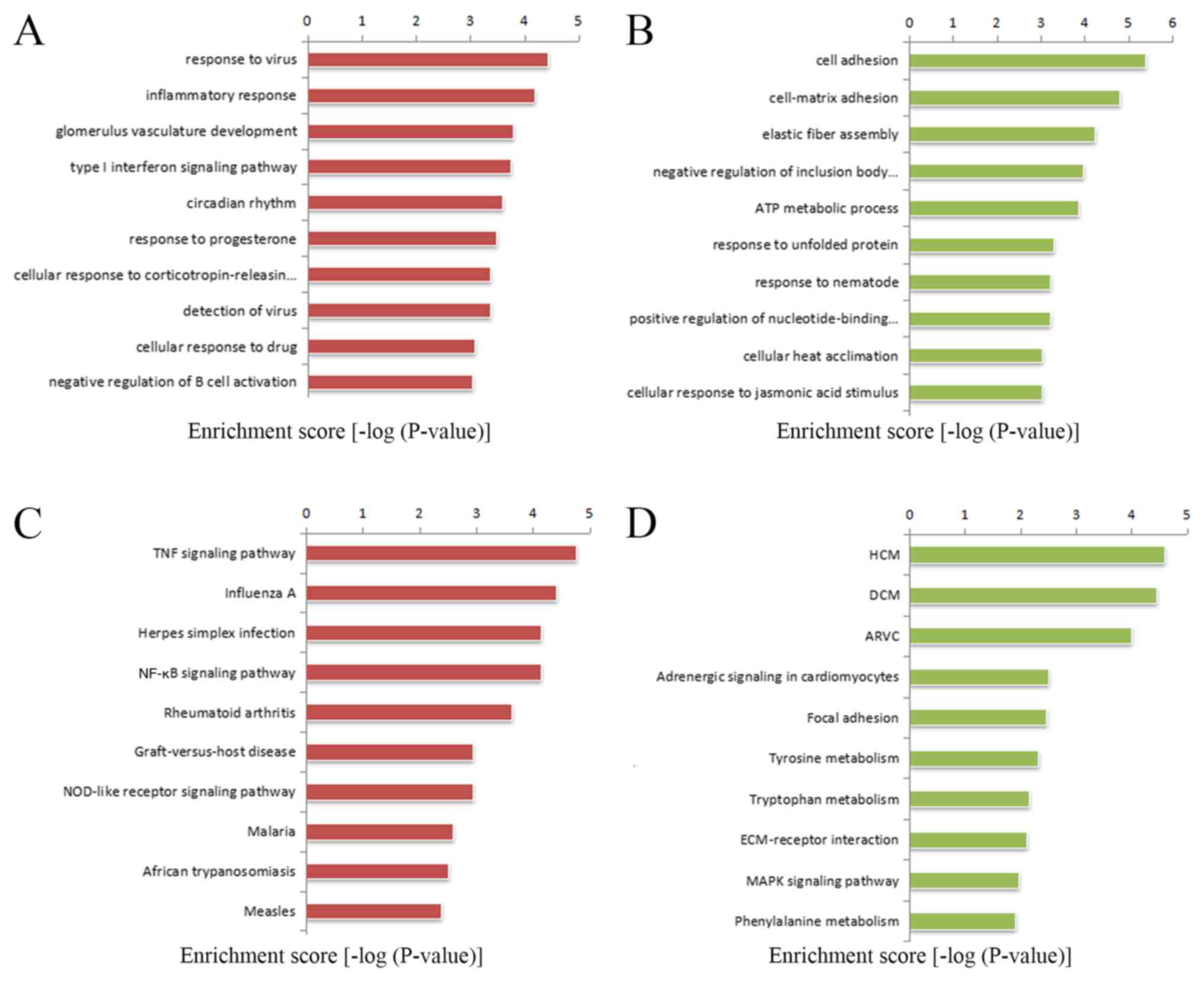 |
Figure 4
GO and KEGG pathway analysis of differentially expressed mRNAs in Marfan syndrome compared with normal aortas. The top 10 GO terms that were associated with the gene function of (A) upregulated mRNAs and (B) downregulated mRNAs are listed. The top 10 pathways that were associated with the (C) upregulated mRNAs and (D) downregulated mRNAs are listed. ARVC, Arrhythmogenic right ventricular cardiomyopathy; ECM, extracellular matrix; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; MAPK, mitogen-activated protein kinase; TNF, tumor necrosis factor; NOD, nucleotide binding oligomerization domain containing.
|
The significantly altered pathways that mediated the functions of the differentially expressed genes based on the KEGG database were the enriched (Table SIII). The analysis showed that 26 pathways corresponded to increased mRNAs and 19 pathways corresponded to decreased mRNAs, with P-values <0.05. Some important pathways that may be connected to MFS were enriched in upregulated genes that participate in the tumor necrosis factor (TNF) signaling pathway and cell adhesion molecules. The downregulated genes were involved in focal adhesion, the ECM-receptor interaction and the mitogen-activated protein kinase (MAPK) signaling pathway (Fig. 4C and D). The -lg P-value was used to describe the significance level of pathway enrichment.
lncRNA and mRNA coexpression network analysis and prediction of target lncRNAs
A lncRNA-mRNA coexpression network was constructed to identify the interactions between mRNAs and lncRNAs (Table SIV). The degree of centrality was calculated to assess the significance of these lncRNAs and mRNAs in the coexpression network (Fig. S1). In Fig. 5, the size of a circle indicates the ability of a gene to interact according to its degree of quantification. Among the network, a total of 5 lncRNAs [uc003jka.1, uc003jox.1, X-inactive specific transcript (XIST), linc-lysophosphatidic acid receptor 1 (linc-LPA-1) and linc-peptidylprolyl isomerase domain and wd repeat containing 1 (linc-PPWD1)] with the highest degree of differentiation were selected as the target lncRNAs, implying that these lncRNAs may serve a significant role in the pathogenesis and development of MFS.
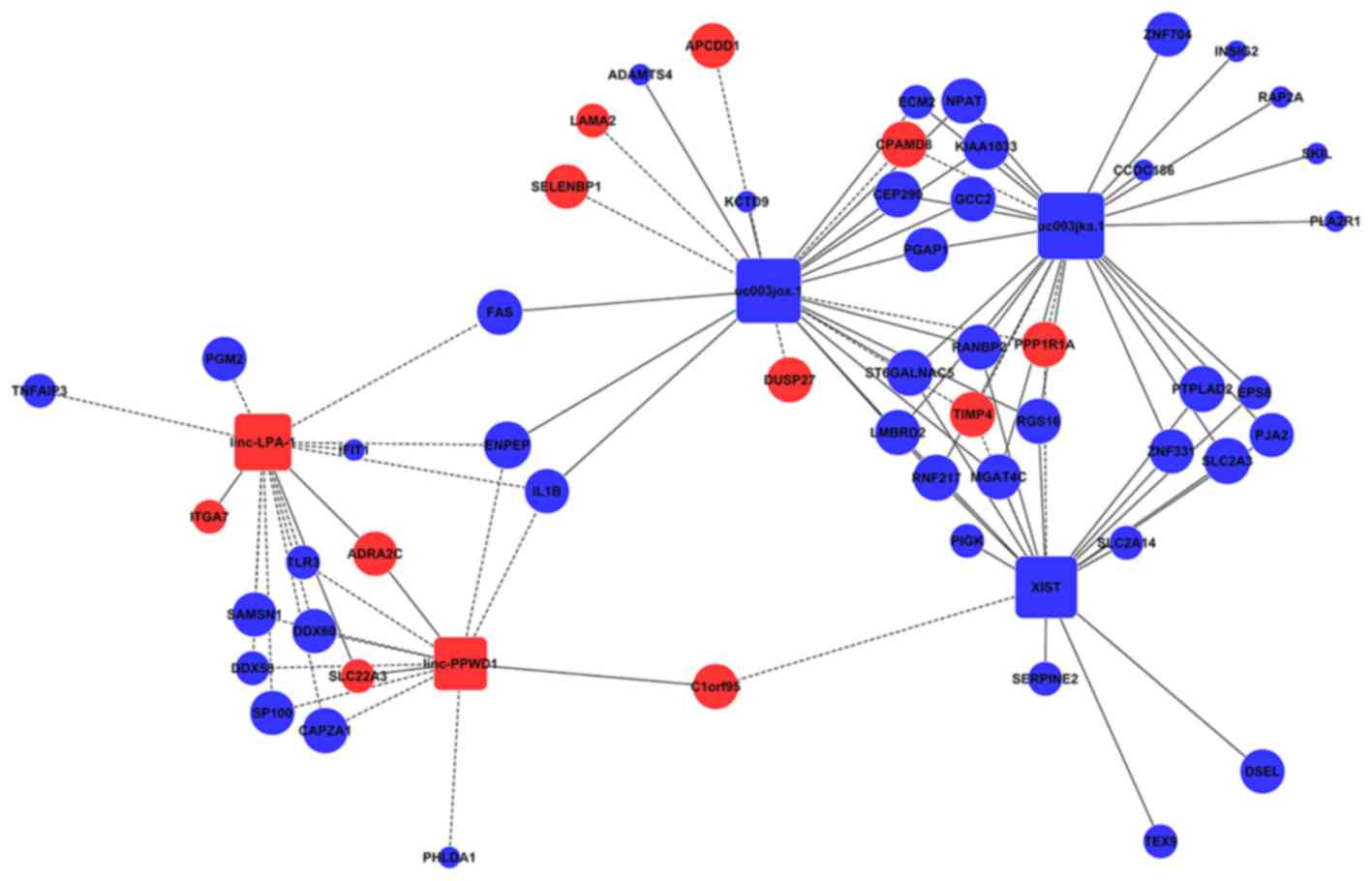 |
Figure 5
The lncRNAs and mRNA co-expression network. The interaction network of differentially expressed genes (lncRNA: uc003jka.1, uc003jox.1, XIST, linc-LPA-1, linc-PPWD1). Round nodes represent protein-coding genes and square nodes represent lncRNAs. Blue nodes represent upregulated genes or lncRNAs, red nodes represent downregulated genes or lncRNAs. The node size represents the connectivity, with larger node showing that more genes or lncRNAs are co-expressed with this gene or lncRNA. Solid lines represent positive correlation and dotted lines negative correlation. lncRNA, long noncoding RNA; XIST, X-inactive specific transcript; linc-LPA-1, linc-lysophosphatidic acid receptor 1; linc-PPWD1, linc-peptidylprolyl isomerase domain and wd repeat containing 1.
|
RT-qPCR validation
RT-qPCR was performed to verify the reliability of the microarray data. A total of five differentially expressed lncRNAs (uc003jka.1, uc003jox.1, XIST, linc-LPA-1 and linc-PPWD1) that exhibited the highest expression in the coexpression network were selected for validation (Fig. 6). The MFS and NA tissue samples (all, n=6) were used for validation. Altered expression levels of the candidates revealed that the results of RT-qPCR analysis were similar to those observed in the microarray data and demonstrated that the result of the microarray data were reliable and could be used for bioinformatics analysis in subsequent experiments.
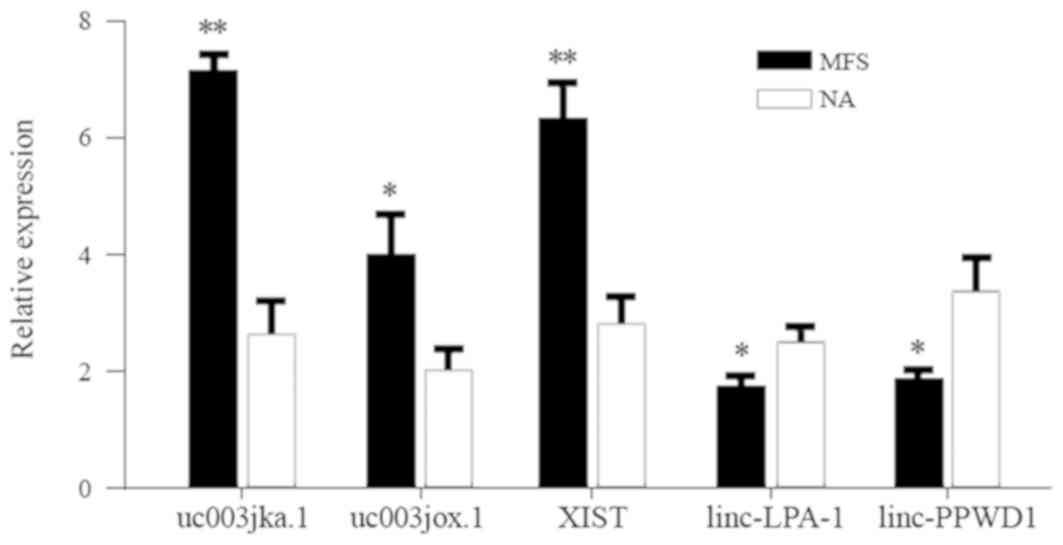 |
Figure 6
Reverse transcription-quantitative PCR validation. Compared to healthy controls, 5 long noncoding RNAs (uc003jka.1, uc003jox.1, XIST, linc-LPA-1, linc-PPWD1) with highest degree were selected. Results were consistent with the findings obtained from the microarray chip analysis (n=6). Data are presented as the mean ± standard deviation. *P<0.05, **P<0.01 vs. NA samples. MFS, Marfan syndrome; NA, normal aorta; XIST, X-inactive specific transcript; linc-LPA-1, linc-lysophosphatidic acid receptor 1; linc-PPWD1, linc-peptidylprolyl isomerase domain and WD repeat containing 1.
|
Discussion
MFS is an autosomal dominant multisystem disorder with manifestations typically involving the cardiovascular, skeletal and ocular systems (1,22). Acute aortic dissection is the leading cause of premature death in untreated individuals with MFS worldwide (8). Currently, pathogenesis studies on MFS are comprehensive, but research into the molecular mechanisms of action are still scarce. lncRNAs have long been considered to be transcriptional noise; however, they may serve a novel role in the pathogenesis of MFS (23). lncRNAs may provide basic information that can be used to recognize the disease course of MFS and to design more effective targeted therapies.
In the present study, the expression profiles of lncRNAs and mRNAs in MFS and NA tissues were investigated using microarray analysis, and the present study revealed potential roles for lncRNA in the pathogenesis of MFS. In total, 294 lncRNAs and 644 mRNAs were identified (FC ≥1.5; P<0.05). GO and KEGG pathway analyses were used to explore the possible biological functions and potential mechanisms of the mRNAs in MFS.
The results of the present study demonstrated that different biological processes, including cell adhesion, elastic fiber assembly and ECM organization, are among the significantly enriched mRNAs in MFS. Most of these functions are involved in ECM organization, and this is consistent with previous studies (22,24,25). The components of the ECM that contain microfibrils and proteoglycans are fundamental units of the cytoskeleton and regulate the structure of the vascular wall (26). Severe elastolysis, adjacent breaks and breakthroughs caused by a mutation in FBN1 were present in the aortic tissues of MFS specimens (26,27). In the present study, it was indicated that cell adhesion, elastic fiber assembly and ECM organization were remarkably downregulated by GO analyses in the aortic tissue samples from individuals with MFS compared with those with NA. A number of core mRNAs are associated with the function of the ECM, including Microfibril Associated Protein 4(28), Fibulin 5(29), Laminin Subunit α2(30), Tenascin XB (31) and Integrin Subunit α7(32).
A number of previous studies have documented an inflammatory component in medial degeneration composed primarily of T cells and macrophages, and it was identified that inflammation aggravates disease severity in patients with MFS (33-35). Ju et al (36), indicated that the activation of interleukin (IL)6-STAT3 signaling contributed to aneurysmal dilation in mgR/mgR mice through increased MMP-9 activity, which aggravated ECM degradation. In the current study, the inflammatory response was revealed to be the most important downregulated GO, with 21 enriched genes, including interleukin (IL)-6, IL-1B, toll-like receptor (TLR)3, Fas Cell Surface Death Receptor (FAS), and E-Selectin (SELE).
The MAPK signaling pathway, which includes ERK, JNK and p38-MAPK, serves a pivotal role in cell proliferation, differentiation, survival and death (37). A previous study indicated that the improper activation of p38 MAPK is a precursor of constitutive Smad2/3 signaling in the aortic wall of a mouse model of neonatal lethal MFS (38). In the present study, the MAPK signaling pathway was prominently changed, with 7 upregulated and 14 downregulated genes enriched. In addition, the important downregulated pathways, including the TNF, the NF-κB and the NOD-like receptor signaling pathways, were not reported to be associated with MFS in previous studies. However, the present study indicated that these pathways may be potential mechanisms of action behind the process and progression of MFS. However, further experiments are required to validate this observation.
A previous study indicated that the transcription of lncRNAs may affect the expression of their nearby mRNAs at the level of chromatin modification, transcription and posttranscriptional processing (39). The functions of most associated aberrantly expressed lncRNAs are unknown. Hence, in the present study, these lncRNAs were revealed based on the functions of their associated mRNAs. Therefore, a novel lncRNA-mRNA coexpression network was constructed to predict the significant core lncRNAs with the highest degree of centrality, including uc003jka.1, uc003jox.1, XIST, linc-LPA-1 and linc-PPWD1. The five lncRNAs were validated using RT-qPCR, which confirmed the reliability of the microarray results. The three lncRNAs (XIST, uc003jka.1 and linc-LPA-1) and their coexpressed mRNAs were analyzed in detail.
The lncRNA XIST is the master regulator of X inactivation in mammals and is exclusively transcribed from the inactive X chromosome (40). According to recent studies, XIST is highly expressed in some malignant tumors, including breast (41), ovarian (42) and non-small-cell lung cancer (43). The results of the present study revealed that differentially expressed XIST, with a FC=16.47, is the most upregulated lncRNA in MFS. The coexpression network identified tissue inhibitors of metalloproteinase 4 (TIMP-4) as a mRNA adjacent to XIST. TIMP-4, which is a tissue inhibitor of MMPs, is believed to serve a central role in the pathogenesis of MFS (7). Therefore, it was predicted that XIST may participate in the mechanism of action behind MFS by regulating the function of TIMP. To date, the detailed roles and underlying mechanisms of XIST remain unclear. The lncRNA uc003jka.1, with the highest degree of differentiation in the coexpression network, to the best of our knowledge, has not been reported in the literature; therefore, its biological function and regulatory mechanism in MFS remain unclear. SKI Like Proto-Oncogene (SKIL) is one of the mRNAs that was coexpressed. Another alias for SKIL is SnoN. SnoN is a component of the Smad pathway, which regulates cell growth and differentiation through transforming growth factor-β (44). This finding is consistent with the pathogenesis of MFS as described by a previous study (45). This result implies that uc003jka.1 may participate in MFS by influencing the expression of SnoN. The linc-LPA-1 is a lncRNA that to the best of our knowledge, has never been reported. The present study identified a total of 11 coexpressed mRNAs. IL1B (46), SP100 Nuclear Antigen (47), FAS (48) and TLR3(49) are mRNAs that are regulators that modulate the inflammatory and immune responses. The possibility that linc-LPA-1 serves a role in the inflammatory and immune responses in MFS by regulating mRNA expression levels requires further investigation.
There are two limitations in the present study. First, due to the difficulty in obtaining tissue samples, a relatively small sample size was used. Further studies with a larger sample size should be performed to verify the relevant results. Furthermore, the present study primarily focused on the value of bioinformatics-based analyses for discovering novel or important lncRNAs, but direct experimental evidence is still lacking to support these hypotheses. Therefore, further investigations should be performed to establish and clarify the detailed molecular mechanism of action behind the lncRNA-mediated regulation of potential coding genes in MFS.
In conclusion, the present data implied that lncRNAs may be associated with a variety of biological and pathological processes in MFS. The potential roles of aberrantly expressed lncRNAs were also predicted with their underlying molecular network. The findings may provide novel targets for further investigation for both the molecular mechanisms and effective targeted therapies for MFS.
Supplementary Material
Figure S1. Co.expression network of the differentially expressed long noncoding RNAs and mRNAs.
Table SI. Differentially expressed genes between normal aortas and Marfan syndrome tissues.
Table SII. Gene Ontology enrichment analysis of differentially expressed genes between normal aortas and Marfan syndrome tissues.
Table SIII. Pathway enrichment analysis of differentially expressed genes between normal aortas and Marfan syndrome tissues.
Table SIV. lncRNA-mRNA coexpression network analysis of differentially expressed genes between normal aortas and Marfan syndrome tissues.
Acknowledgements
Not applicable.
Funding
This study was supported by National Natural Science Foundation of China (grant no. 81670255) and the Zhejiang Provincial Natural Science Foundation of China (grant no. LY14H020008).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
JW and LG conceived the study and participated in the design of the study. JN and SS performed the experiments. KZ and CS analyzed the microarray data and drew the figures and tables. JW and LG performed statistical analysis and wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study protocol was approved by the Institutional Ethics Review Board of the First Affiliated Hospital of Wenzhou Medical University (Wenzhou, China) and all patients gave informed consent prior to their inclusion in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
1
|
Cañadas V, Vilacosta I, Bruna I and Fuster V: Marfan syndrome. Part 1: Pathophysiology and diagnosis. Nat Rev Cardiol. 7:256–265. 2010.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Romaniello F, Mazzaglia D, Pellegrino A, Grego S, Fiorito R, Ferlosio A, Chiariello L and Orlandi A: Aortopathy in Marfan syndrome: An update. Cardiovasc Pathol. 23:261–266. 2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
De Paepe A, Devereux RB, Dietz HC, Hennekam RC and Pyeritz RE: Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet. 62:417–426. 1996.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, et al: The revised ghent nosology for the Marfan syndrome. J Med Genet. 47:476–485. 2010.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Xiong W, Meisinger T, Knispel R, Worth JM and Baxter BT: MMP-2 regulates Erk1/2 phosphorylation and aortic dilatation in Marfan syndrome. Circul Res. 110:e92–e101. 2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Chung AW, Au Yeung K, Sandor GG, Judge DP, Dietz HC and van Breemen C: Loss of elastic fiber integrity and reduction of vascular smooth muscle contraction resulting from the upregulated activities of matrix metalloproteinase-2 and -9 in the thoracic aortic aneurysm in Marfan syndrome. Circ Res. 101:512–522. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Merk DR, Chin JT, Dake BA, Maegdefessel L, Miller MO, Kimura N, Tsao PS, Iosef C, Berry GJ, Mohr FW, et al: MiR-29b participates in early aneurysm development in Marfan syndrome. Circ Res. 110:312–324. 2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Nienaber CA, Clough RE, Sakalihasan N, Suzuki T, Gibbs R, Mussa F, Jenkins MP, Thompson MM, Evangelista A, Yeh JS, et al: Aortic dissection. Nat Rev Dis Primers. 2(16053)2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Quinn JJ and Chang HY: Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 17:47–62. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ponting CP, Oliver PL and Reik W: Evolution and functions of long noncoding RNAs. Cell. 136:629–641. 2009.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Milewicz DM, Prakash SK and Ramirez F: Therapeutics targeting drivers of thoracic aortic aneurysms and acute aortic dissections: Insights from predisposing genes and mouse models. Annu Rev Med. 68:51–67. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
David TE, David CM, Manlhiot C, Colman J, Crean AM and Bradley T: Outcomes of aortic valve-sparing operations in Marfan syndrome. J Am Coll Cardiol. 66:1445–1453. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yang YG, Li MX, Kou L, Zhou Y, Qin YW, Liu XJ and Chen Z: Long noncoding RNA expression signatures of abdominal aortic aneurysm revealed by microarray. Biomed Environ Sci. 29:713–723. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
He Q, Tan J, Yu B, Shi W and Liang K: Long noncoding RNA HIF1A-AS1A reduces apoptosis of vascular smooth muscle cells: Implications for the pathogenesis of thoracoabdominal aorta aneurysm. Pharmazie. 70:310–315. 2015.PubMed/NCBI
|
|
15
|
Gill AJ, Garza R, Ambegaokar SS, Gelman B and Kolson DL: Heme oxygenase-1 promoter region (GT)n polymorphism associates with increased neuroimmune activation and risk for encephalitis in HIV infection. J Neuroinflammation. 15(70)2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, Lomsadze A, Pruitt KD, Borodovsky M and Ostell J: NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44:6614–6624. 2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM and Haussler D: The human genome browser at UCSC. Genome Res. 12:996–1006. 2002.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Volders PJ, Anckaert J, Verheggen K, Nuytens J, Martens L, Mestdagh P and Vandesompele J: LNCipedia 5: Towards a reference set of human long non-codingRNAs. Nucleic Acids Res. 47:D135–D139. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Fang S, Zhang L, Guo J, Niu Y, Wu Y, Li H, Zhao L, Li X, Teng X, Sun X, et al: NONCODEV5: A comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 46:D308–D314. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Chen Y, Cunningham F, Rios D, McLaren WM, Smith J, Pritchard B, Spudich GM, Brent S, Kulesha E, Marin-Garcia P, et al: Ensembl variation resources. BMC Genomics. 11(293)2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Cook JR, Carta L, Bénard L, Chemaly ER, Chiu E, Rao SK, Hampton TG, Yurchenco P, GenTAC Registry Consortium Costa KD, et al: Abnormal muscle mechanosignaling triggers cardiomyopathy in mice with Marfan syndrome. J Clin Invest. 124:1329–1339. 2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Evans JR, Feng FY and Chinnaiyan AM: The bright side of dark matter: lncRNAs in cancer. J Clin Invest. 126:2775–2782. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Perrucci GL, Rurali E, Gowran A, Pini A, Antona C, Chiesa R, Pompilio G and Nigro P: Vascular smooth muscle cells in Marfan syndrome aneurysm: Thebroken bricks in the aortic wall. Cell Mol Life Sci. 74:267–277. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Jensen SA and Handford PA: New insights into the structure, assembly and biological roles of 10-12 nm connective tissue microfibrils from fibrillin-1 studies. Biochem J. 473:827–838. 2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
El-Hamamsy I and Yacoub MH: Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat Rev Cardiol. 6:771–786. 2009.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Marshall LM, Carlson EJ, O'Malley J, Snyder CK, Charbonneau NL, Hayflick SJ, Coselli JS, Lemaire SA and Sakai LY: Thoracic aortic aneurysm frequency and dissection are associated with fibrillin-1 fragment concentrations in circulation. Circ Res. 113:1159–1168. 2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Pilecki B, Holm AT, Schlosser A, Moeller JB, Wohl AP, Zuk AV, Heumüller SE, Wallis R, Moestrup SK, Sengle G, et al: Characterization of microfibrillar-associated protein 4 (MFAP4) as a tropoelastin- and fibrillin-binding protein involved in elastic fiber formation. J Biol Chem. 291:1103–1114. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Padhy B, Kapuganti RS, Hayat B, Mohanty PP and Alone DP: De novo variants in an extracellular matrix protein coding gene, fibulin-5 (FBLN5) are associated with pseudoexfoliation. Eur J Hum Genet. 27:1858–1866. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Van Wijk XM, Döhrmann S, Hallström BM, Li S, Voldborg BG, Meng BX, McKee KK, van Kuppevelt TH, Yurchenco PD, Palsson BO, et al: Whole-Genome sequencing of invasion-resistant cells identifies laminin α2 as a host factor for bacterial invasion. mBio. 8:e02128–e02116. 2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Chen WY, Perritt AF, Morissette R, Dreiling JL, Bohn MF, Mallappa A, Xu Z, Quezado M and Merke DP: Ehlers-Danlos syndrome caused by biallelic TNXB variants in patients with congenital adrenal hyperplasia. Hum Mutat. 37:893–897. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Klaffky E, Williams R, Yao CC, Ziober B, Kramer R and Sutherland A: Trophoblast-Specific expression and function of the integrin alpha 7 subunit in the peri-implantation mouse embryo. Dev Biol. 239:161–175. 2001.PubMed/NCBI View Article : Google Scholar
|
|
33
|
He R, Guo DC, Sun W, Papke CL, Duraisamy S, Estrera AL, Safi HJ, Ahn C, Buja LM, Arnett FC, et al: Characterization of the inflammatory cells in ascending thoracic aortic aneurysms in patients with Marfan syndrome, familial thoracic aortic aneurysms, and sporadic aneurysms. J Thorac Cardiovasc Surg. 136:922–929. 2008.PubMed/NCBI View Article : Google Scholar
|
|
34
|
He R, Guo DC, Estrera AL, Safi HJ, Huynh TT, Yin Z, Cao SN, Lin J, Kurian T, Buja LM, et al: Characterization of the inflammatory and apoptotic cells in the aortas of patients with ascending thoracic aortic aneurysms and dissections. J Thorac Cardiovasc Surg. 131:671–678. 2006.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Radonic T, de Witte P, Groenink M, de Waard V, Lutter R, van Eijk M, Jansen M, Timmermans J, Kempers M, Scholte AJ, et al: Inflammation aggravates disease severity in Marfan syndrome patients. PLoS One. 7(e32963)2012.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Ju X, Ijaz T, Sun H, Lejeune W, Vargas G, Shilagard T, Recinos A III, Milewicz DM, Brasier AR and Tilton RG: IL-6 regulates extracellular matrix remodeling associated with aortic dilation in a fibrillin-1 hypomorphic mgR/mgR mouse model of severe Marfan syndrome. J Am Heart Assoc. 3(e000476)2014.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Kim EK and Choi EJ: Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 1802:396–405. 2010.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Carta L, Smaldone S, Zilberberg L, Loch D, Dietz HC, Rifkin DB and Ramirez F: P38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J Biol Chem. 284:5630–5636. 2009.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Mercer TR, Dinger ME and Mattick JS: Long non-coding RNAs: Insights into functions. Nat Rev Genet. 10:155–159. 2009.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Lee HJ, Gopalappa R, Sunwoo H, Choi SW, Ramakrishna S, Lee JT, Kim HH and Nam JW: En bloc and segmental deletions of human XIST reveal X chromosome inactivation-involving RNA elements. Nucleic Acids Res. 7:3875–3887. 2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Huang YS, Chang CC, Lee SS, Jou YS and Shih HM: Xist reduction in breast cancer upregulates AKT phosphorylation via HDAC3-mediated repression of PHLPP1 expression. Oncotarget. 7:43256–43266. 2016.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Huang KC, Rao PH, Lau CC, Heard E, Ng SK, Brown C, Mok SC, Berkowitz RS and Ng SW: Relationship of XIST expression and responses of ovarian cancer to chemotherapy. Mol Cancer Ther. 1:769–776. 2002.PubMed/NCBI
|
|
43
|
Wang H, Shen Q, Zhang X, Yang C, Cui S, Sun Y, Wang L, Fan X and Xu S: The long non-coding RNA XIST controls non-small cell lung cancer proliferation and invasion by modulating miR-186-5p. Cell Physiol Biochem. 41:2221–2229. 2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Bonni S, Wang HR, Causing CG, Kavsak P, Stroschein SL, Luo K and Wrana JL: TGF-Beta induces assembly of a smad2-smurf2 ubiquitin ligase complex that targets SnoN for degradation. Nat Cell Biol. 3:587–595. 2001.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, van Erp C, Lindsay ME, Kim D, Schoenhoff F, Cohn RD, et al: Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 332:358–361. 2011.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Pulugulla SH, Packard TA, Galloway NL, Grimmett ZW, Doitsh G, Adamik J, Galson DL, Greene WC and Auron PE: Distinct mechanisms regulate IL1B gene transcription in lymphoid CD4 T cells and monocytes. Cytokine. 111:373–381. 2018.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Lorenz DR, Misra V and Gabuzda D: Transcriptomic analysis of monocytes from HIV-positive men on antiretroviral therapy reveals effects of tobacco smoking on interferon and stress response systems associated with depressive symptoms. Hum Genomics. 13(59)2019.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Jangalwe S, Kapoor VN, Xu J, Girnius N, Kennedy NJ, Edwards YJK, Welsh RM, Davis RJ and Brehm MA: Cutting edge: Early attrition of memory T cells during inflammation and costimulation blockade is regulated concurrently by proapoptotic proteins fas and bim. Immunol. 202:647–651. 2019.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Shaalan A, Carpenter G and Proctor G: Caspases are key regulators of inflammatory and innate immune responses mediated by TLR3 in vivo. Mol Immunol. 94:190–199. 2018.PubMed/NCBI View Article : Google Scholar
|














