Introduction
Lung cancer is one of the most common types of
cancer and is the leading cause of cancer-associated mortality
worldwide, accounting for >25% of all deaths due to cancer
(1). Approximately 85% of lung
cancers are non-small cell lung cancer (NSCLC) (2). In turn, NSCLCs are comprised of lung
adenocarcinoma (LUAD, 40-50% of lung cancers), lung squamous cell
carcinoma (LUSC, 20-30% of lung cancers) and large cell carcinoma
(9% of lung cancers) (2). Despite
advances in diagnostics, surgery and medication in recent decades,
the average 5-year survival rate of patients with NSCLC remains as
low as 15% (1). This poor prognosis
is a consequence of high rates of tumor metastasis and recurrence,
and numerous signaling pathways having been identified to be
involved in these processes (1).
Thus, an enhanced understanding of the molecular mechanisms
controlling NSCLC progression is required to improve the low
survival rate. The development of high-throughput sequencing has
allowed for comprehensive comparisons of gene expression profiles,
thereby identifying differentially expressed genes (DEGs) between
tumor and normal tissues. Changes in expression levels usually
indicate pathological states, as proteins encoded by DEGs may be
involved in tumorigenesis and tumor progression (3).
microRNAs (miRNAs/miRs) are short non-coding RNA
molecules that mediate the post-transcriptional regulation of mRNAs
via binding to complementary sequences in the 3'-untranslated
region (UTR) of mRNAs and suppressing their translation or
mediating their degradation (4). An
individual miRNA may regulate hundreds of different mRNA molecules,
highlighting the existence of miRNA-mRNA regulatory networks.
Depending on how it is expressed, a specific miRNA may therefore
act to suppress or promote oncogenesis via specific effects on
relevant target mRNAs (5). Indeed,
miRNA profiling efforts have been used to identify specific miRNA
signatures associated with particular tumor subtypes, thereby
allowing for cancer diagnosis, treatment planning and prediction of
patient prognosis (6).
Bioinformatics analysis of gene expression microarray data provides
a useful tool for revealing numerous previously unrecognized mRNAs
and miRNAs that may be implicated in the pathogenesis of cancer or
other diseases (7).
In the present study, 3 gene expression datasets
were analyzed using an integrated bioinformatics approach in order
to identify DEGs and differentially expressed miRNAs (DEMs) between
NSCLC tumors and healthy control tissues. Functional enrichment and
protein-protein interaction (PPI) network analyses were performed
to better establish the functions of these mRNAs and miRNA, and
these approaches were combined with an analysis of mRNA-miRNA
interactions to screen hub genes and miRNAs in this regulatory
network. Through this approach, the present study aimed to further
elucidate the molecular mechanisms of NSCLC to identify potentially
novel therapeutic strategies for its treatment.
Materials and methods
Microarray data information
The datasets used in the present study were obtained
from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) (8). The original gene expression profiles
from the datasets GSE18842(9),
GSE32863(10) and GSE29250(11) were used and the clinical information
of the patients was obtained from the original research articles.
The GSE18842 dataset included 91 samples (46 tumors and 45
controls) and all samples were paired except 2 tumors and 1
control. The platform used for the GSE18842 dataset was GPL570
(HG-U133_Plus_2) Affymetrix Human Genome U133 Plus 2.0 Array. The
GSE32863 dataset included 116 samples (58 tumors and 58 controls)
and all samples were paired. The platform used for the GSE32863
dataset was the GPL6884 Illumina Human WG-6 v.3.0 expression
beadchip. The GSE29250 dataset included 12 samples (6 tumors and 6
controls) and all samples were paired. The platform used for the
GSE29250 dataset was the GPL8179 Illumina Human v.2 MicroRNA
expression beadchip.
Identification of DEGs
To compare gene expression profiles, the GEO2R tool
(http://www.ncbi.nlm.nih.gov/geo/geo2r/; accessed March
2019) (12), which is based on the
limma package in R, was used to individually identify DEMs and DEGs
in each dataset. To control for type I error as a result of
multiple comparisons within each dataset, the false discovery rate
(FDR) determination feature automatically included in the GEO2R
tool was employed. Significant DEGs were those that remained
significant after FDR correction when tested via
multiple-comparisons t-tests, and fold change (FC)>2 and
P<0.05 were set as the cut-off criteria. Any probes that did not
correspond to a specific gene symbol were then filtered from the
resultant data.
Gene function analysis
For gene ontology (GO) and Kyoto Encyclopedia of
Genes and Genomes (KEGG) pathway analyses of DEMs and DEGs, the
database for annotation, visualization and integrated discovery
(DAVID) database (v6.8; http://david.abcc.ncifcrf.gov/) was used, focusing
specifically on humans and using all genes as an enrichment
background. Significant enrichment in these analyses was determined
based on an adjusted P-value of 0.05 as established via the
Benjamini-Hochberg method (13).
These P-values were determined on the basis of a cumulative
hypergeometric distribution, calculating q-values based on the
Benjamini-Hochberg procedure as a means of controlling for multiple
testing (13). For comparisons of
hierarchical clustering of enriched terms, clusters were designated
as groupings that had a similarity score >0.3, with the most
significant term within a given cluster being selected to represent
the cluster as a whole.
PPI network construction and
analysis
The online Search Tool for the Retrieval of
Interacting Genes and proteins (STRING) database (v.11.0;
https://string-db.org/) was used for PPI network
construction. PPI pairs with a combined score ≥0.7 were used to
generate the network. Cytoscape (v.3.4.0; https://cytoscape.org/) was used to visualize the
regulatory interactions between these genes and CentiScaPe (v.2.2;
Center for Biomedical Computing, University of Verona, Italy)
(14) was used to analyze network
distributions based on topological properties. The Molecular
Complex Detection application (MCODE; v.1.6) was used to identify
and extract subnetworks from the global PPI network based on the
k-core algorithm (15). The genes
with a degree ≥30 in this regulatory network were identified as hub
genes, as described previously (16).
Prediction of the mRNA-miRNA
interactions
An online tool called miRWalk 3.0 (http://mirwalk.umm.uni-heidelberg.de/),
which integrates predictive outputs of TargetScan (17) and miRDB (18), was used to predict DEG and DEM
interactions. A score ≥0.95 was considered as the critical
criterion for the miRWalk predictive analysis. Only the target
mRNAs predicted by all 3 tools (miRWalk, TargetScan and miRDB) were
used for further analysis. By overlaying identified DEGs and these
predicted mRNA targets, a miRNA-mRNA regulatory network was
constructed and then visualized using Cytoscape.
Analysis of datasets from the cancer
genome atlas (TCGA)
TCGA is an online database that may be used to
research and explore publicly available datasets (https://cancergenome.nih.gov/; accessed March 2019)
(19), including RNA sequencing
(RNA-seq) data from TCGA samples of 31 different types of cancer.
In the present study, the tumor types were limited to LUAD and
LUSC. RNA-seq data and clinical data from 478 LUAD and 482 LUSC
samples from TCGA datasets were used. Based on the approach
previously outlined by Li and Dewey (20), a PERL program was used to multiply
the ‘scaled estimate’ by 106, yielding transcripts per
million (TPM) values for all gene expression, as TPM values were
thought to be a more reliable means of comparing gene expression
than fragments per kilobase of TPM-mapped reads or reads per
kilobase of TPM-mapped reads values (21). In the present study, to improve the
reliability of the analysis, the expression of hub genes was
validated in TCGA datasets using Gene Expression Profiling
Interactive Analysis (GEPIA; v1.0; http://gepia.cancer-pku.cn/). For each of the hub
genes, patients were stratified into 2 groups based on expression
levels of each gene and differences in patient survival were
analyzed, generating hazard ratio (HR) and 95% CI values, as well
as log-rank P-values for each comparison.
Results
Identification of DEGs and DEMs
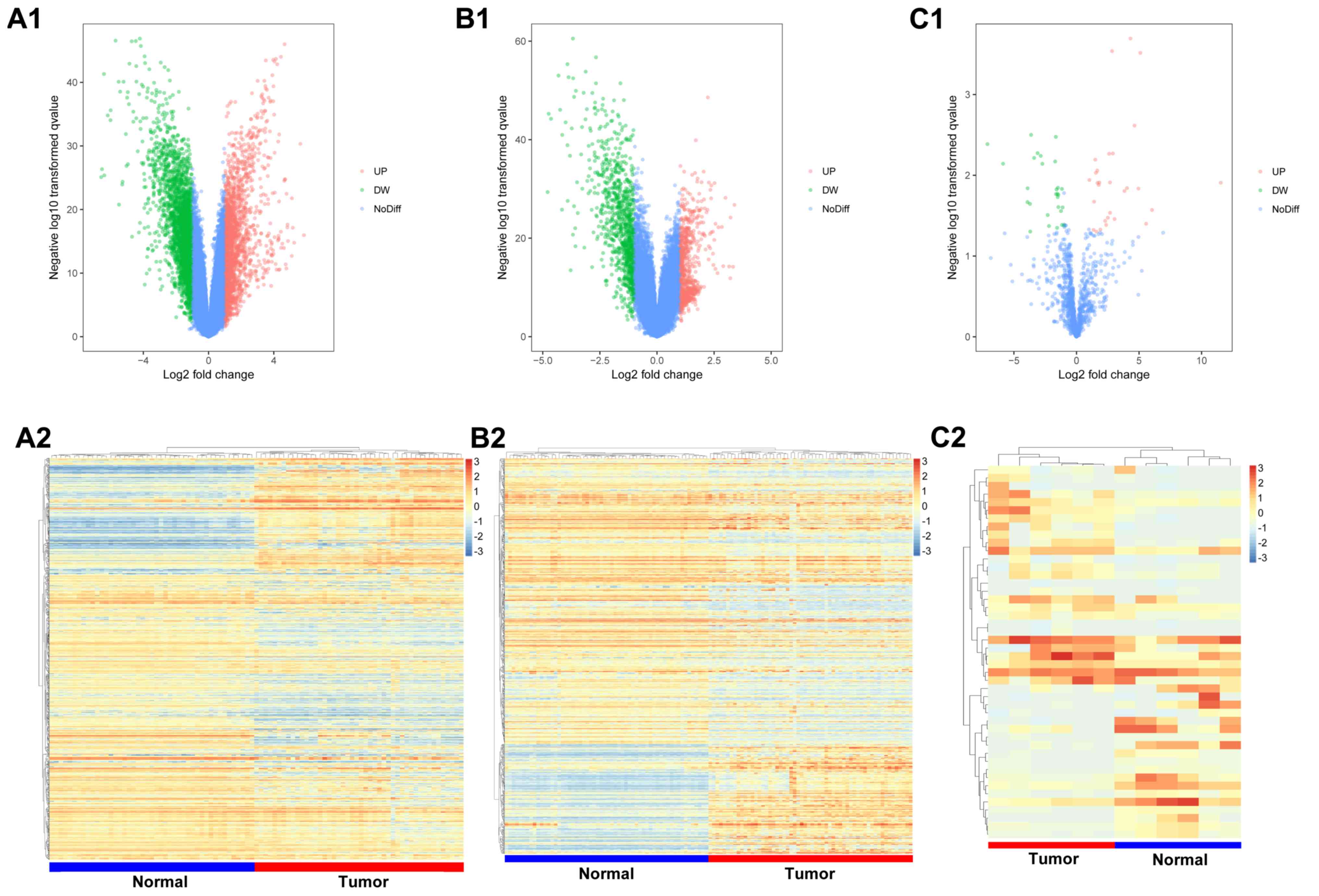
In the GSE18842 and GSE32863 datasets, 3,167 and
1,270 DEGs were identified, respectively, of which 1,395 and 514
were upregulated, and 1,772 and 756 were downregulated (Fig. 1A and B). A total of 782 DEGs were shared between
these 2 datasets (232 upregulated and 550 downregulated). The
GSE32863 dataset yielded a list of 46 DEMs, of which 26 were
upregulated and 20 were downregulated (Fig. 1C).
Functional enrichment analysis of
overlapped DEGs and target genes of DEMs
To assess the biological roles of these DEGs and
target genes of DEMs, KEGG and GO enrichment analyses were
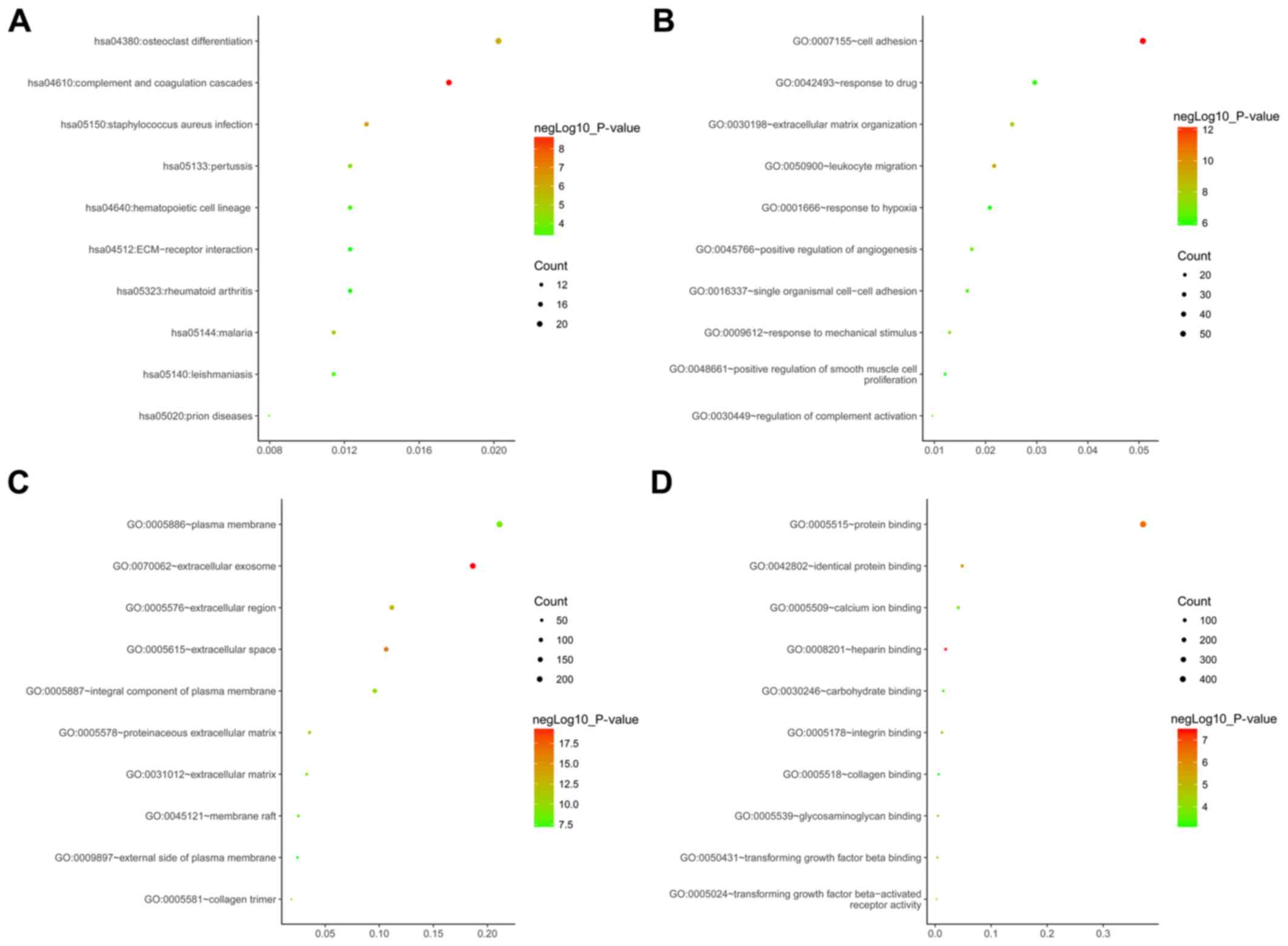
performed. The top 10 enriched terms for each analysis were
compiled in Fig. 2. KEGG pathway
enrichment analysis indicated that the DEGs and target genes of
DEMs were mainly enriched in osteoclast differentiation, complement
and coagulation cascades, Staphylococcus aureus infection
and pertussis (Fig. 2A). GO analysis
in the category biological process suggested these DEGs and target
genes of DEMs were primarily enriched in ‘cell adhesion,’ ‘response
to drugs’ and ‘extracellular matrix organization’ (Fig. 2B). GO analysis in the category
cellular component suggested that the DEGs and target genes of DEMs
were mainly enriched in ‘plasma membrane,’ ‘extracellular exosome’
and ‘extracellular localization’ (Fig.
2C). In the category molecular function, the DEGs and target
genes of DEMs were mostly enriched in ‘protein binding,’ ‘identical
protein binding’ and ‘calcium ion binding’ (Fig. 2D).
PPI network construction and analysis
of modules
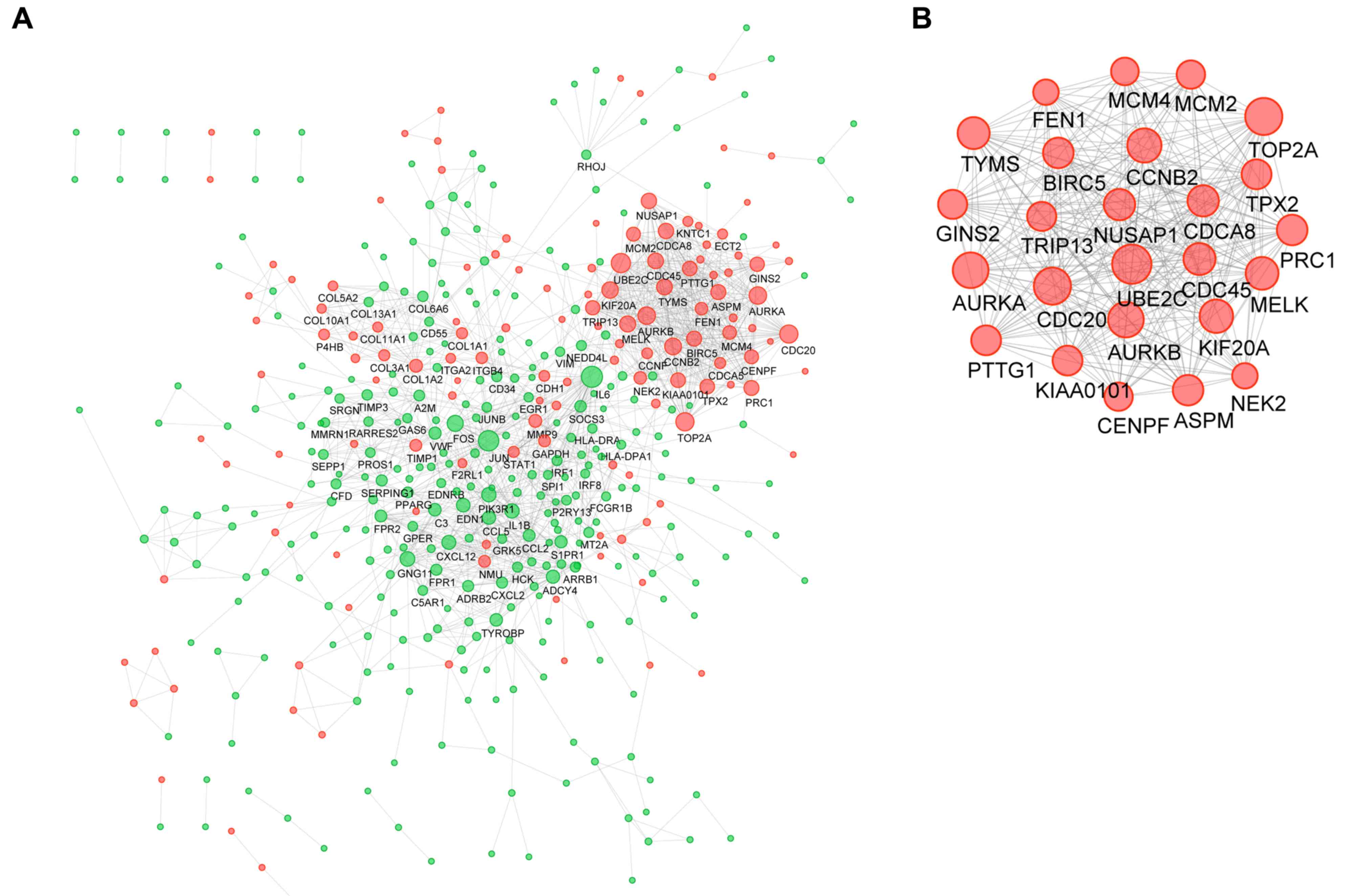
The 782 overlapping DEGs which were shared between
the 2 datasets (GSE18842 and GSE32863) indicated a distinct set of
interactions and networks. PPI pairs with a combined score ≥0.7
were used to generate the network. A PPI network was constructed
using 445 of the 782 DEGs and the resultant network had 445 nodes
and 1,490 edges (Fig. 3A). There
were 137 upregulated and 308 downregulated genes among the 445
DEGs. A total of 11 nodes had a degree of >30 and were
designated as hub genes, including interleukin 6, Jun
proto-oncogene (JUN), ubiquitin E2 ligase (UBE2C), cell division
cycle protein (CDC)20, DNA topoisomerase IIα (TOP2A), aurora kinase
A (AURKA), AURKB, cyclin B2 (CCNB2), kinesin family member 20A, FBJ
osteosarcoma oncogene and maternal embryonic leucine zipper kinase
(MELK). The topology parameters of these hub genes in the PPI
network are presented in Table I.
Furthermore, a subnetwork containing 25 nodes and 284 edges was
extracted from the global PPI network (Fig. 3B). The results of the KEGG and GO
analyses for the genes in the subnetwork are presented in Table II. The most significantly enriched
terms in this network were ‘cell cycle, division’ and ‘DNA
replication’ associated with cancer, confirming the relevance of
the present analysis to NSCLC progression and prognosis.
 | Table ITopology parameters of the genes with
a degree ≥30 in the protein-protein interaction network. |
Table I
Topology parameters of the genes with
a degree ≥30 in the protein-protein interaction network.
| Gene | Closeness | Betweenness | Degree | Stress | MCODE_Score | Regulation |
|---|
| IL6 |
9.74x10-4 | 29543.72 | 44 | 157806 | 4.545455 | Down |
| JUN |
9.19x10-4 | 20492.43 | 42 | 105026 | 5.066667 | Down |
| UBE2C |
8.01x10-4 | 10608.03 | 39 | 65066 | 20 | Up |
| CDC20 |
7.15x10-4 | 2025.242 | 36 | 13016 | 20 | Up |
| TOP2A |
7.67x10-4 | 8145.198 | 36 | 53776 | 20 | Up |
| AURKA |
6.94x10-4 | 1793.101 | 34 | 12868 | 20 | Up |
| AURKB |
7.17x10-4 | 2846.847 | 34 | 23916 | 20 | Up |
| CCNB2 |
7.04x10-4 | 1426.159 | 32 | 12102 | 20 | Up |
| KIF20A |
7.20x10-4 | 5841.105 | 31 | 43260 | 20 | Up |
| FOS |
8.72x10-4 | 7497.451 | 30 | 41706 | 5.785714 | Down |
| MELK |
6.97x10-4 | 549.8218 | 30 | 6356 | 20 | Up |
| Table IIKEGG pathway and GO enrichment
analysis for the genes in the subnetwork. |
Table II
KEGG pathway and GO enrichment
analysis for the genes in the subnetwork.
| A, KEGG
pathways |
|---|
| Term | Gene ratio | P-value | Genes |
|---|
| hsa04110: Cell
cycle | 0.13132 |
4.03x10-7 | CDC45, CCNB2,
CDC20, MCM2, PTTG1, MCM4 |
| hsa03030: DNA
replication | 0.06566 |
1.16x10-3 | MCM2, MCM4,
FEN1 |
| hsa04114: Oocyte
meiosis | 0.06566 |
1.02x10-2 | AURKA, CDC20,
PTTG1 |
| B, GO analysis in
category BP |
| Term | Gene ratio | P-value | Genes |
| GO:0007067 -
mitotic nuclear division | 0.21887 |
3.12x10-11 | CCNB2, NEK2, TPX2,
CENPF, BIRC5, AURKA, CDC20, PTTG1, AURKB, ASPM |
| GO:0051301 - cell
division | 0.21887 |
6.65x10-10 | CDCA8, CCNB2, NEK2,
TPX2, CENPF, BIRC5, AURKA, CDC20, PTTG1, UBE2C |
| GO:0000086 - G2/M
transition of mitotic cell cycle | 0.13132 |
1.26x10-6 | CCNB2, NEK2, TPX2,
BIRC5, AURKA, MELK |
| GO:0006260 - DNA
replication | 0.13132 |
2.32x10-6 | GINS2, CDC45,
KIAA0101, MCM2, MCM4, FEN1 |
| GO:0031145 -
anaphase-promoting complex-dependent catabolic process | 0.109433 |
4.49x10-6 | AURKA, CDC20,
PTTG1, AURKB, UBE2C |
| GO:0007062 - sister
chromatid cohesion | 0.109433 |
1.29x10-5 | CDCA8, CENPF,
BIRC5, CDC20, AURKB |
| GO:0042787 -
protein ubiquitination involved in ubiquitin-dependent protein
catabolic process | 0.10943 |
6.11x10-5 | AURKA, CDC20,
PTTG1, AURKB, UBE2C |
| GO:0006268 - DNA
unwinding involved in DNA replication | 0.06566 |
8.75x10-6 | MCM2, MCM4,
TOP2A |
| GO:0007051 -
spindle organization | 0.06566 |
2.32x10-4 | AURKA, AURKB,
ASPM |
| GO:0000082 - G1/S
transition of mitotic cell cycle | 0.08755 |
4.01x10-4 | TYMS, CDC45, MCM2,
MCM4 |
| C, GO analysis in
category CC |
| Term | Gene ratio | P-value | Genes |
| GO:0005819 -
spindle | 0.19698 |
2.01x10-12 | PRC1, TPX2, NUSAP1,
CENPF, BIRC5, AURKA, CDC20, AURKB, KIF20A |
| GO:0030496 -
midbody | 0.19698 |
3.38x10-12 | CDCA8, PRC1, NEK2,
CENPF, BIRC5, AURKA, AURKB, ASPM, KIF20A |
| GO:0005654 -
nucleoplasm | 0.41585 |
1.04x10-10 | GINS2, PRC1, TPX2,
KIAA0101, CENPF, BIRC5, AURKA, CDC20, AURKB, MCM2, UBE2C, MCM4,
TYMS, CDC45, CDCA8, CCNB2, TOP2A, FEN1, KIF20A |
| GO:0005634 -
nucleus | 0.50339 |
3.50x10-10 | GINS2, PRC1, NEK2,
TPX2, KIAA0101, NUSAP1, CENPF, BIRC5, AURKA, CDC20, PTTG1, AURKB,
MCM2, MCM4, TYMS, CDC45, CDCA8, CCNB2, TOP2A, ASPM, MELK, FEN1,
TRIP13 |
| GO:0032133 -
chromosome passenger complex | 0.08755 |
2.00x10-8 | CDCA8, BIRC5,
AURKA, AURKB |
| GO:0005876 -
spindle microtubule | 0.10943 |
3.03x10-7 | PRC1, NUSAP1,
BIRC5, AURKA, AURKB |
| GO:0000922 -
spindle pole | 0.10943 |
1.17x10-5 | PRC1, NEK2, TPX2,
CENPF, CDC20 |
| GO:0015630 -
microtubule cytoskeleton | 0.10943 |
2.89x10-5 | CCNB2, PRC1, TPX2,
AURKA, MCM2 |
| GO:0005874 -
microtubule | 0.13132 |
4.56x10-5 | NEK2, TPX2, NUSAP1,
BIRC5, AURKA, KIF20A |
| GO:0005813 -
centrosome | 0.13132 |
2.01x10-4 | CDC45, CCNB2, NEK2,
CENPF, AURKA, CDC20 |
| D, GO analysis in
category MF |
| Term | Gene ratio | P-value | Genes |
| GO:0005524 - ATP
binding | 0.24075 |
1.77x10-5 | NEK2, TPX2, AURKA,
MCM2, AURKB, UBE2C, MCM4, TOP2A, MELK, TRIP13, KIF20A |
| GO:0005515 -
protein binding | 0.50339 |
3.97x10-5 | GINS2, PRC1, NEK2,
TPX2, KIAA0101, NUSAP1, CENPF, BIRC5, AURKA, CDC20, PTTG1, AURKB,
MCM2, UBE2C, MCM4, CDC45, CDCA8, CCNB2, TOP2A, MELK, FEN1, KIF20A,
TRIP13 |
| GO:0008017 -
microtubule binding | 0.08755 |
3.08x10-3 | PRC1, NUSAP1,
BIRC5, KIF20A |
| GO:0035174 -
histone serine kinase activity | 0.04377 |
7.09x10-3 | AURKA, AURKB |
| GO:0043138-3’-5’
DNA helicase activity | 0.04377 |
9.91x10-3 | GINS2, CDC45 |
| GO:0019899 - enzyme
binding | 0.087547 |
1.13x10-2 | BIRC5, CDC20, MCM2,
TOP2A |
| GO:0004672 -
protein kinase activity | 0.08755 |
1.39x10-2 | NEK2, AURKA, AURKB,
MELK |
| GO:0003688 - DNA
replication origin binding | 0.04377 |
1.55x10-2 | CDC45, MCM2 |
| GO:0004674 -
protein serine/threonine kinase activity | 0.08755 |
1.57x10-2 | NEK2, AURKA, AURKB,
MELK |
| GO:0019901 -
protein kinase binding | 0.08755 |
1.57x10-2 | PRC1, TPX2, AURKA,
KIF20A |
Survival analysis of hub genes
Survival analysis was performed for the 11 hub genes
based on TCGA data. Increased expression of 7 of the hub genes
(UBE2C, CDC20, TOP2A, AURKA, AURKB, CCNB2 and MELK) was
significantly associated with poorer overall survival (OS) in
patients with LUAD (Fig. 4).
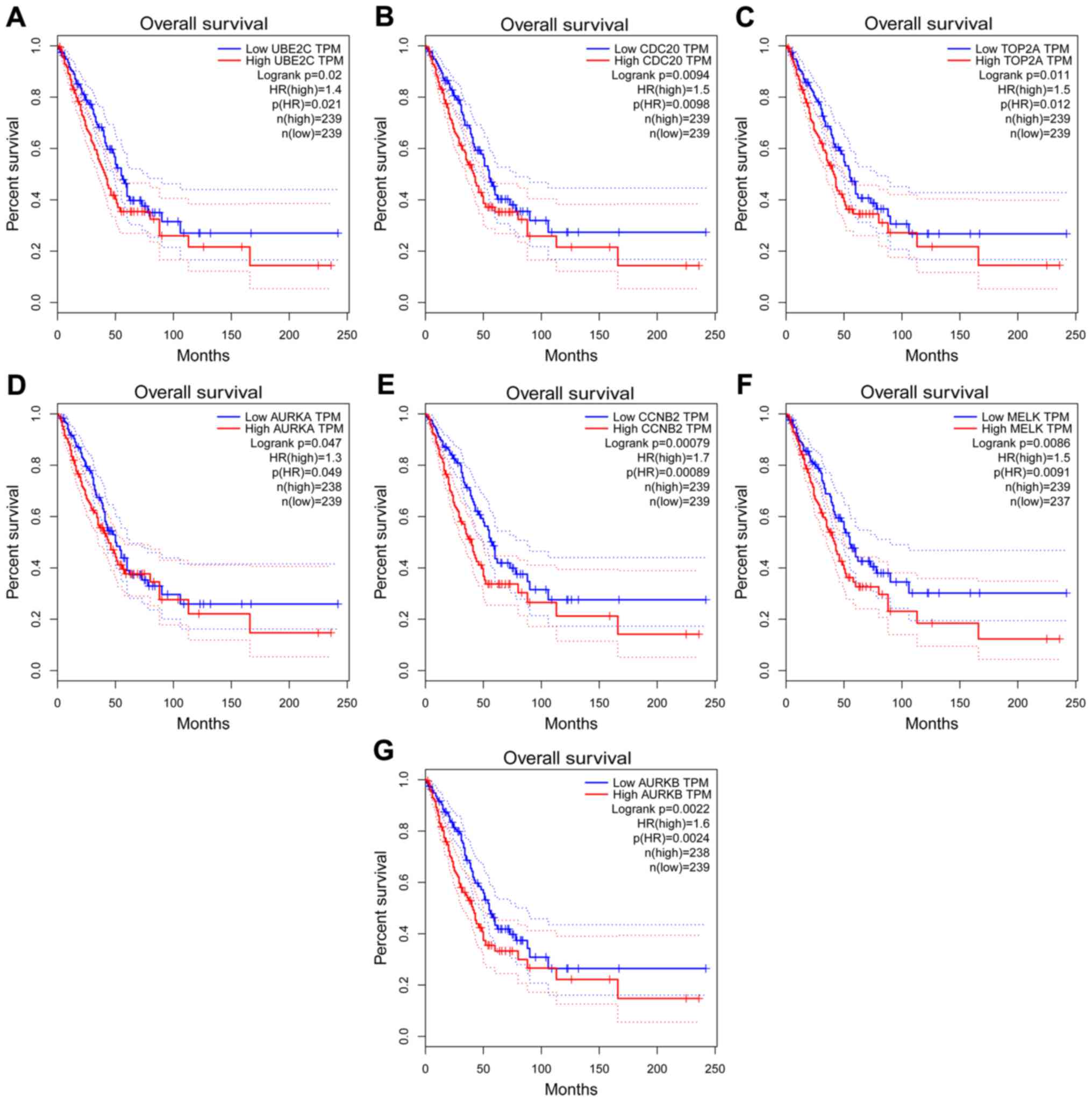 | Figure 4Kaplan-Meier survival analysis of the
hub genes in the protein-protein interaction network. The
Kaplan-Meier survival analysis based on the expression of (A)
UBE2C, (B) CDC20, (C) TOP2A, (D) AURKA, (E) CCNB2, (F) MELK and (G)
AURKB. The horizontal axis represents overall survival (months) and
the vertical axis represents the percentage of survival. The dotted
line indicates the upper and lower boundaries of the 95% confidence
interval. UBE2C, ubiquitin E2 ligase; CDC20, cell division cycle
20; TOP2A, DNA topoisomerase IIα; AURKA, aurora kinase A; CCNB2,
cyclin B2; MELK, maternal embryonic leucine zipper kinase; HR,
hazard ratio; TPM, expression. |
miRNA-gene network
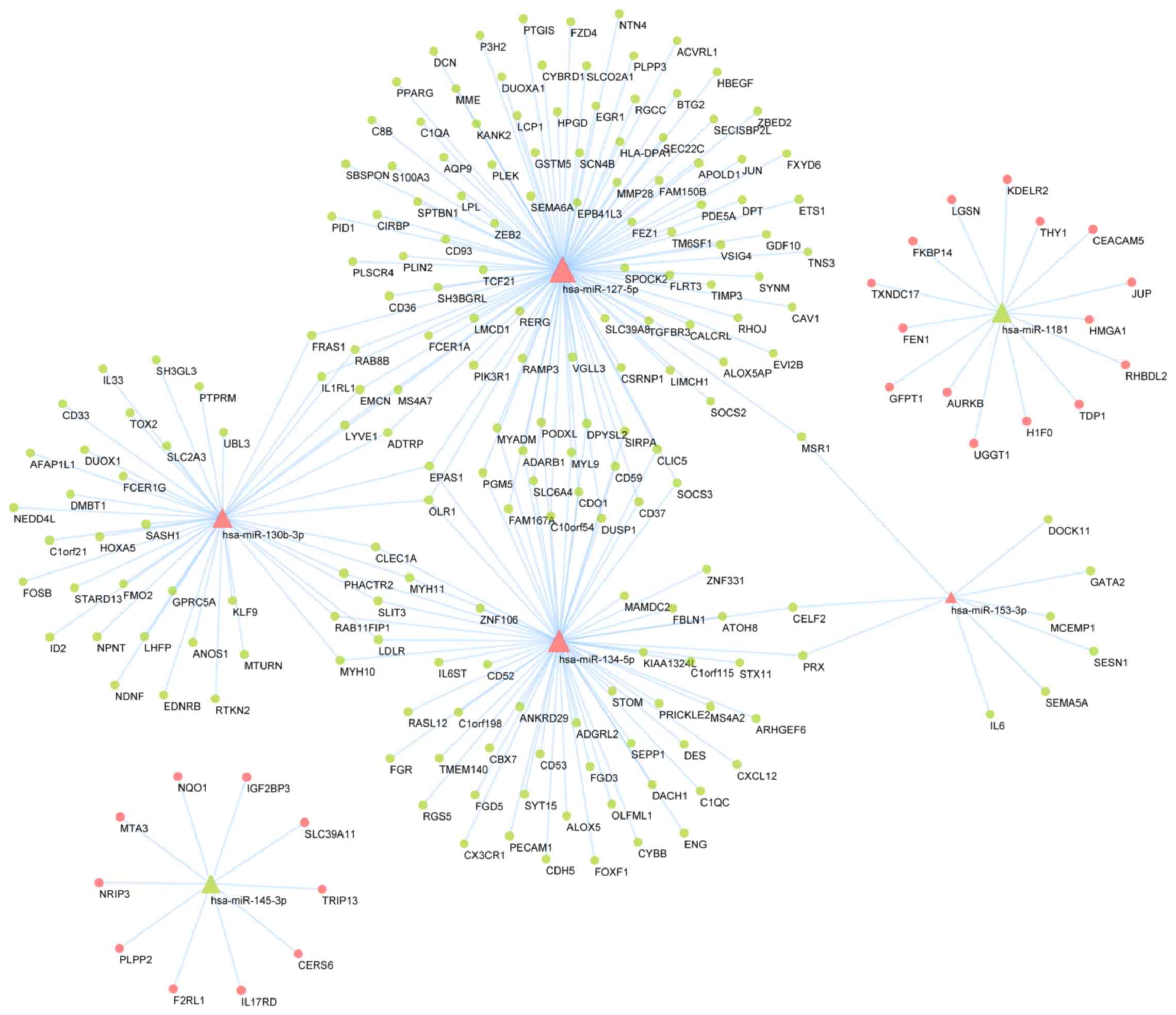
Using the miRWalk application, putative DEM targets
as established by the TargetScan and miRDB databases were
identified. A total of 210 putative target mRNAs overlapped with
the DEG dataset, yielding 247 miRNA-gene pairs based on 6 DEMs and
the 210 DEGs that were putative targets. These were used to
construct an overlapping regulatory network (Fig. 5). A total of 4 upregulated DEMs were
predicted to downregulate 185 DEGs, whereas decreased expression of
2 DEMs was predicted to be associated with increased expression of
25 DEGs. TCGA-based survival analysis suggested that none of the
hub genes were associated with OS in patients with NSCLC. However,
specifically among patients with LUAD, those with elevated
expression of slit guidance ligand 3 (SLIT3), phosphoglucomutase 5
(PGM5), endomucin (EMCN), cysteine dioxygenase type 1 (CDO1),
dihydropyrimidinase-like 2 (DPYSL2), miR-130b and miR-1181 had a
significantly higher OS compared with patients who had low
expression of these genes. By contrast, elevated miR-130b and
miR-127 expression was associated with poorer OS in patients with
LUAD (Fig. 6). Furthermore, the
differences of the DEGs and DEMs in miRNA-gene networks of NSCLCs
with or without KRAS mutation were examined. The results suggested
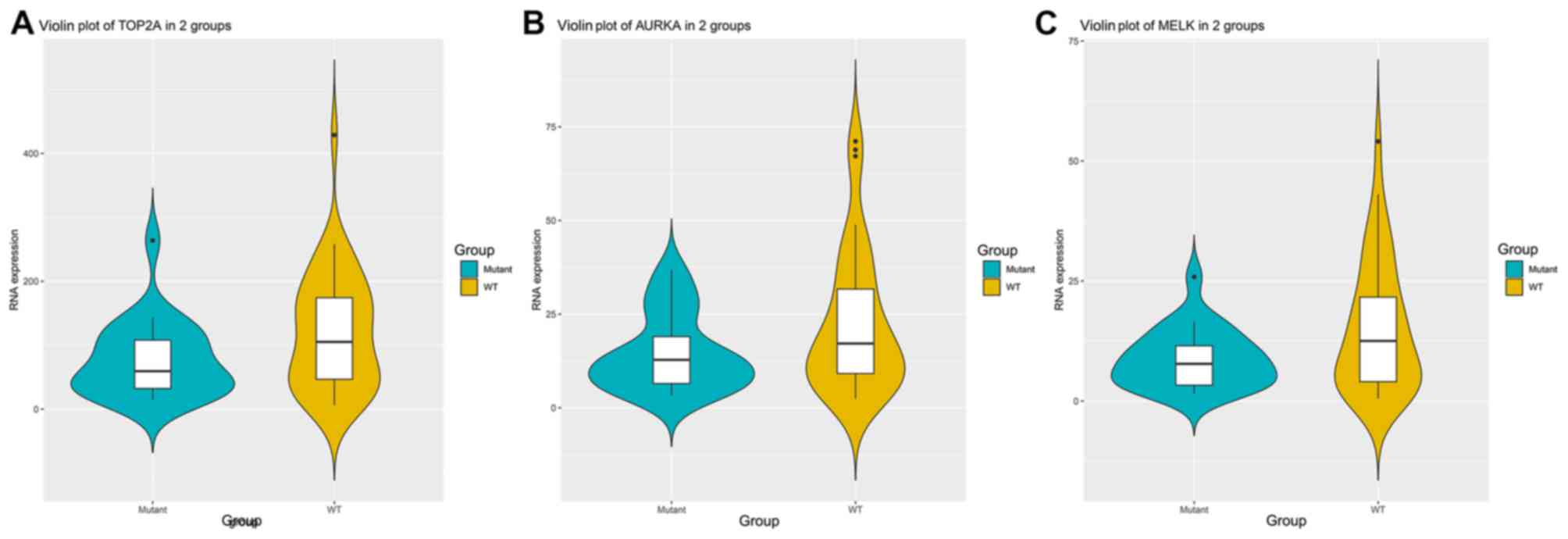
that the expression levels of TOP2A (P=0.0357; Fig. 7A), AURKA (P=0.0409; Fig. 7B) and MELK (P=0.0190; Fig. 7C) were significantly lower in KRAS
mutation groups compared to KRAS wild-type groups (Fig. 7).
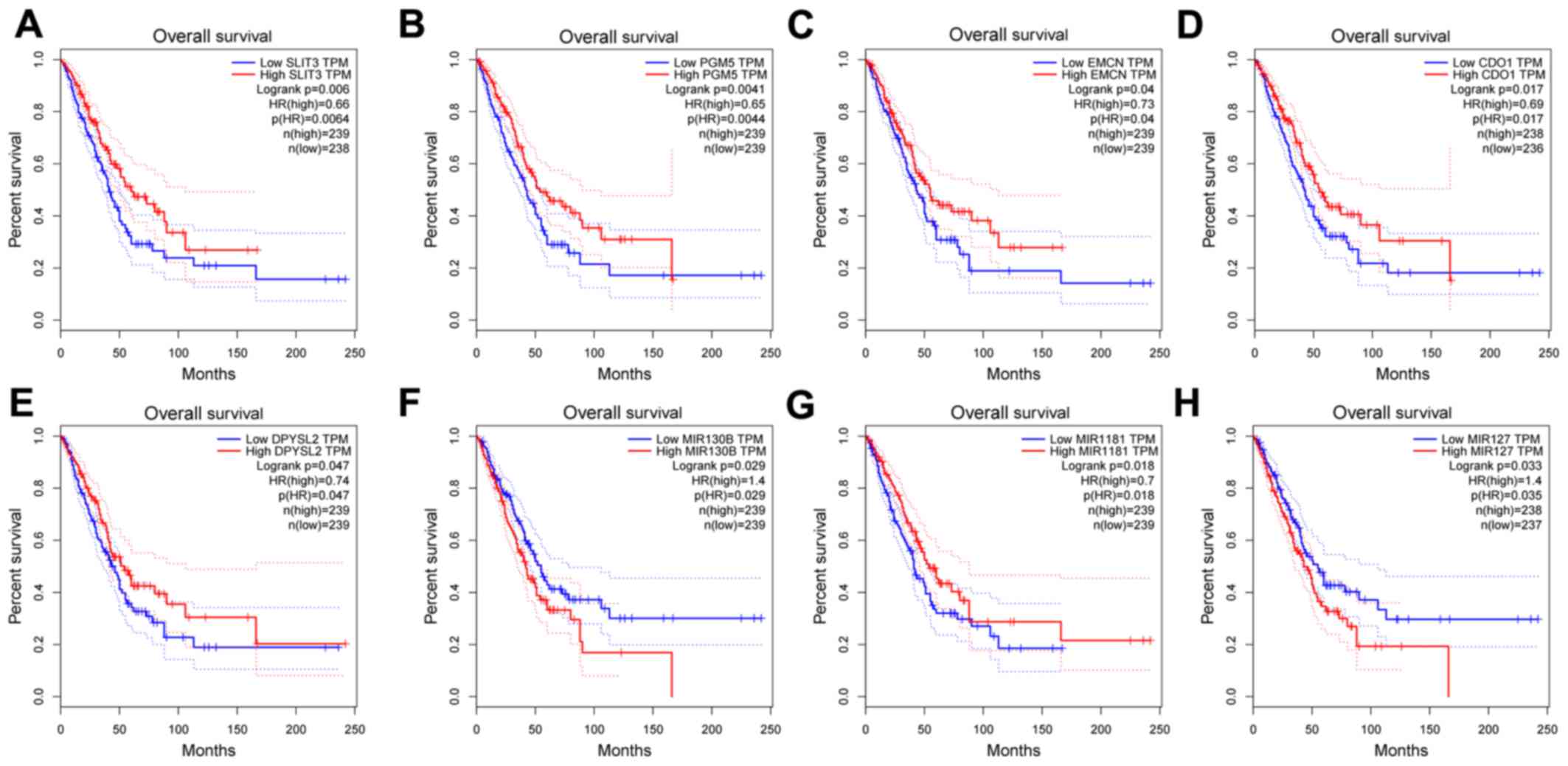 | Figure 6Kaplan-Meier survival analysis of the
hub genes in the miRNA-mRNA regulatory network. The Kaplan-Meier
survival analysis based on the expression of (A) SLIT3, (B) PGM5,
(C) EMCN, (D) CDO1, (E) DPYSL2, (F) MIR130B, (G) MIR1181 and (H)
MIR127. The horizontal axis represents overall survival (months)
and the vertical axis represents the percentage of survival. The
dotted line indicates the upper and lower boundaries of the 95%
confidence interval. miRNA/miR, microRNA; SLIT3, slit guidance
ligand 3; PGM5, phosphoglucomutase 5; EMCN, endomucin; CDO1,
cysteine dioxygenase type 1; DPYSL2, dihydropyrimidinase-like 2;
TPM, transcripts per million; HR, hazard ratio. |
Discussion
Cancer is a genetic disease wherein cumulative
mutations drive the multi-step progression towards oncogenesis,
eventually culminating in unrestrained cancer growth. NSCLC remains
one of the most common and deadliest forms of cancer, making the
elucidation of the molecular mechanisms governing this disease
paramount (22). In the present
study, DEMs and DEGs associated with NSCLC were identified via a
bioinformatics analysis, yielding 782 DEGs and 46 DEMs based on
overlapping hits in the GSE18842, GSE32863 and GSE29250 datasets.
These hits included 232 upregulated and 550 downregulated genes, as
well as 26 upregulated and 20 downregulated miRNAs. Through
functional enrichment analyses, it was determined that these DEGs
were primarily associated with processes including ‘osteoclast
differentiation’, ‘complement and coagulation cascades’, ‘cell
adhesion, drug responses’, ‘plasma membrane’, ‘extracellular
exosome’ and ‘protein binding’. In addition, a DEG PPI network was
generated and a significant subnetwork module was identified that
contained genes associated with the cell cycle, DNA replication and
oocyte meiosis, with the GO terms enrichment for ‘mitotic nuclear
division’, ‘cell division’, ‘G2/M transition of mitotic cell
cycle’, ‘spindle’, ‘midbody’, ‘nucleoplasm’, ‘ATP binding’,
‘protein binding’ and ‘microtubule binding’. Cell cycle
dysregulation is known to be a key factor linked to tumor
development and progression (23,24).
Recent studies indicated that microtubule binding is linked to
tumor metastasis and drug resistance (25,26).
Complement activation and coagulation cascade activation are
similarly able to promote tumor development as a consequence of
their ability to mediate the recruitment of myeloid cells that
support tumor growth (27). To
summarize, the identified DEGs may regulate the proliferation,
invasion, migration and drug-resistance of cancer cells through
these pathways, thus affecting the occurrence and development of
NSCLC. The investigation of these DEGs may pave a way towards novel
targeted therapies for NSCLC.
Based on the PPI network, 11 hub genes with high
degrees of interaction (degree ≥30) were extracted. A prognostic
analysis of these 11 hub genes was performed using the online tool
GEPIA. The results revealed that patients with LUAD who had
upregulation of UBE2C, CDC20, TOP2A, AURKA, AURKB, CCNB2 and MELK
had a worse prognosis.
The expression of UBE2C has been previously
indicated to be upregulated in lung cancer (28), and the results of the present study
suggested that it was associated with poor survival. Similar
observations have previously been made in ovarian cancer (29), breast cancer (30) and gastric cancer (31). UBE2C is involved in the progression
of the cell cycle and transcription, and upregulation of UBE2C may
induce an enhanced growth and colony formation of tumors (32), as well as decreased autophagy in
cancer cells (33). Furthermore,
UBE2C reduces the sensitivity of cells to common chemotherapy drugs
for lung cancer, including cisplatin (34) and docetaxel (35). UBE2C may be used as a therapeutic
target for NSCLC.
Another oncogene in several types of tumor and a hub
gene identified in the present study was CDC20(36). CDC20 is an important regulator of the
cell cycle and altered expression or functional impairment may
induce mitotic arrest to prevent activation of adenomatous
polyposis coli and hence, increase premature anaphase manifesting
as aneuploidy in daughter cells (37). CDC20 was observed to be upregulated
at mRNA and protein levels in NSCLC, and was significantly
correlated with tumor size, pleural invasion and histological
classification (38). Of note,
knockdown of CDC20 caused inhibition of growth, migration ability
and formation of colonies in lung cancer cells, as well as cell
cycle arrest in G2/M phase and induction of apoptosis (39), making this oncogene a potential
target molecule to address NSCLC therapy. Upregulation of CDC20 has
been associated with shorter OS in patients with LUAD, but not in
patients with LUSC (40), which is
consistent with the results of the present study.
TOP2A encodes for a DNA topoisomerase involved in
torsional dynamics during replication and transcription (41), which is also associated with cell
proliferation (42). TOP2A has been
indicated to be upregulated in numerous types of tumor, including
breast, nasopharyngeal and renal cell carcinomas, and is associated
with poor prognosis; therefore, TOP2A has important roles in cancer
(43-45).
The ability of NSCLC cells to proliferate and invade tissues is
associated with elevated TOP2A expression. Several anti-cancer
agents have been developed to target this gene (46), and the development of drug resistance
has been associated with mutation of TOP2A (47).
AURKA and AURKB are highly conserved
serine/threonine kinases, the former of which is associated with
regulating centrosome duplication and spindle formation (48), and the latter of which is important
for regulating chromatin modifications and suppressing cytokinesis
(49). NSCLC prognosis is known to
be associated with elevated expression of these 2 genes (50,51). In
the present study, TCGA dataset analysis suggested that the
prognosis of patients with LUAD was associated with AURKA and
AURKB, which require further clinical trial validation.
CCNB2 is a cyclin gene that activates
cyclin-dependent kinase 1 to drive the G2/M cell cycle transition,
and inhibition of CCNB2 leads to cell cycle arrest (52). It has been previously confirmed that
CCNB2 is upregulated in tissue and serum samples from patients with
NSCLC (53,54). Elevated CCNB2 mRNA levels are known
to be closely associated with tumor differentiation grade and
histological type, and upregulation of CCNB2 at the protein level
has been significantly associated with the degree of
differentiation, tumor size, lymph node metastasis, distant
metastasis and clinical stage (54,55).
Previous studies of NSCLC have suggested that there was no
statistically significant correlation between the levels of CCNB2
protein and mRNA in NSCLC (55). The
results of the present study indicated that upregulation of CCNB2
mRNA was a poor prognostic biomarker in patients with LUAD, while a
previous study suggested that the protein levels of CCNB2 may serve
as an independent prognostic marker in NSCLC (54). Therefore, the role of CCNB2 in NSCLC
should be further elucidated.
MELK is a serine/threonine kinase that has been
indicated to be highly expressed in several human cancer types
(prostate, breast, brain, colorectal and gastric cancer) and
glioblastoma multiforme stem cells (56). Elevated expression of MELK is
associated with the degree of tumor malignancy and with poor
survival in cervical cancer (57),
breast cancer (58) and gastric
cancer (59). Furthermore, the
present study suggested that upregulation of MELK is associated
with the progression of NSCLC.
miRNAs regulate a wide array of target mRNAs via
3'-UTR binding and subsequent translational repression. As a
result, complex miRNA-mRNA networks may govern a wide range of
biological pathways, making miRNAs critical for the progression of
numerous types of cancer (60). In
the present study, a total of 46 DEMs were identified, and
miRWalk-mediated predictive analyses were performed to identify
those DEMs that were predicted to interact with DEGs, yielding 6
hub miRNAs and associated mRNAs, including miR-127-5p, miR134-5p,
miR-130b-3p, miR-1181, miR-145-3p, miR-153-3p, CDO1, SLIT3 and
PGM5. Survival analyses revealed that dysregulation of miR-127-5p,
miR-130b-3p, miR-1181, CDO1, SLIT3, PGM5, EMCN and DPYSL2 were
significantly associated with the prognosis of patients with
LUAD.
miR-127 has previously been indicated to function as
either a promoter or suppressor of cancer development depending on
the specific context (61,62). Based on the NSCLC network established
in the present study, miR-127 was among the most prominent
regulatory miRNAs, suggesting it serves complex regulatory
functions in the context of NSCLC. In a previous study, miR-127
expression was indicated to be elevated in LUAD and associated with
poor prognosis (63), consistent
with the results of the present study. High levels of miR-127
induce epithelial-to-mesenchymal transition, rendering tumor cells
with stem cell-like properties, and propagate tumor resistance to
epidermal growth factor receptor inhibitor (63). The aggressiveness of the cancer was
associated with a circuit involving miR-127, NF-κB and tumor
necrosis factor α-induced protein 3, which are markers of
inflammation (63).
miR-130b has also been documented in several other
types of tumor, with upregulation observed in prostate cancer
(64), while downregulation was
identified in thyroid carcinomas (65). In the present study, miR-130b-3p
expression was determined to be significantly increased in NSCLC
and associated with poor survival, although this was specifically
restricted to patients with LUAD in the TCGA dataset, warranting
further investigation.
miR-134 has been indicated to be differentially
regulated in lung cancer and other types of cancer (including
gastric cancer, breast cancer and oral cancer), with certain
studies reporting increased expression in lung cancer (66,67),
while other studies observed that it was downregulated (68,69).
miR-134 may function to either promote or suppress tumor
progression (69,70), highlighting complex mechanisms
warranting further investigation.
miR-1181 has been observed to be downregulated in
nasopharyngeal carcinoma, ovarian cancer and pancreatic cancer
(71-73).
In addition, miR-1181 inhibited invasion and proliferation via
STAT3 in pancreatic cancer cells and inhibited metastasis by
modulating the WNT/β-catenin pathway in nasopharyngeal carcinoma
(71,73). Thus, the role of miR-1181 in NSCLC
requires to be further investigated in the future.
In summary, the present study identified 782 DEGs
and 46 DEMs between NSCLC tumor and normal tissues, and a
miRNA-mRNA regulatory network was established. Certain hub genes
were screened out from the PPI network and miRNA-mRNA regulatory
network, including UBE2C, CDC20, TOP2A, AURKA, AURKB, CCNB2, MELK,
SLIT3, PGM5, EMCN, CDO1, DPYSL2, miR-130b, miR-1181 and miR-127.
These analyses suggested a comprehensive overview of the
mechanistic basis of NSCLC, potentially highlighting future avenues
for treatment. However, analysis of TCGA datasets indicated that
the expression of certain hub genes was only associated with the
prognosis in patients with LUAD, which requires further validation.
The present results remain to be verified by further clinical
investigation in the future.
Acknowledgements
Not applicable.
Funding
The present study was supported by Beijing Municipal
Science and Technology Commission Fund (grant no. 2018-A20).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
LP and WW designed the study; WW and SW analyzed the
microarray datasets and interpreted the results; WW wrote the
manuscript; LP and SW revised the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Osmani L, Askin F, Gabrielson E and Li QK:
Current WHO guidelines and the critical role of immunohistochemical
markers in the subclassification of non-small cell lung carcinoma
(NSCLC): Moving from targeted therapy to immunotherapy. Semin
Cancer Biol. 52:103–109. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Tang J, Kong D, Cui Q, Wang K, Zhang D,
Yuan Q, Liao X, Gong Y and Wu G: Bioinformatic analysis and
identification of potential prognostic microRNAs and mRNAs in
thyroid cancer. PeerJ. 6(e4674)2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297.
2004.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Volinia S, Calin GA, Liu CG, Ambs S,
Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et
al: A microRNA expression signature of human solid tumors defines
cancer gene targets. Proc Natl Acad Sci USA. 103:2257–2261.
2006.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Sempere LF: Integrating contextual miRNA
and protein signatures for diagnostic and treatment decisions in
cancer. Expert Rev Mol Diagn. 11:813–827. 2011.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Lv K, Yang J, Sun J and Guan J:
Identification of key candidate genes for pancreatic cancer by
bioinformatics analysis. Exp Ther Med. 18:451–458. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Edgar R, Domrachev M and Lash AE: Gene
Expression Omnibus: NCBI gene expression and hybridization array
data repository. Nucleic Acids Res. 30:207–210. 2002.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Sanchez-Palencia A, Gomez-Morales M,
Gomez-Capilla JA, Pedraza V, Boyero L, Rosell R and Fárez-Vidal ME:
Gene expression profiling reveals novel biomarkers in nonsmall cell
lung cancer. Int J Cancer. 129:355–364. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Selamat SA, Chung BS, Girard L, Zhang W,
Zhang Y, Campan M, Siegmund KD, Koss MN, Hagen JA, Lam WL, et al:
Genome-scale analysis of DNA methylation in lung adenocarcinoma and
integration with mRNA expression. Genome Res. 22:1197–1211.
2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ma L, Huang Y, Zhu W, Zhou S, Zhou J, Zeng
F, Liu X, Zhang Y and Yu J: An integrated analysis of miRNA and
mRNA expressions in non-small cell lung cancers. PLoS One.
6(e26502)2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Diboun I, Wernisch L, Orengo CA and
Koltzenburg M: Microarray analysis after RNA amplification can
detect pronounced differences in gene expression using limma. BMC
Genomics. 7(252)2006.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: The Gene Ontology Consortium: Gene ontology: Tool for the
unification of biology. Nat Genet. 25:25–29. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Scardoni G, Petterlini M and Laudanna C:
Analyzing biological network parameters with CentiScaPe.
Bioinformatics. 25:2857–2859. 2009.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Cellai D, Lawlor A, Dawson KA and Gleeson
JP: Tricritical point in heterogeneous k-core percolation. Phys Rev
Lett. 107(175703)2011.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Han JDJ, Bertin N, Hao T, Goldberg DS,
Berriz GF, Zhang LV, Dupuy D, Walhout AJ, Cusick ME, Roth FP, et
al: Evidence for dynamically organized modularity in the yeast
protein-protein interaction network. Nature. 430:88–93.
2004.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Wong N and Wang X: miRDB: An online
resource for microRNA target prediction and functional annotations.
Nucleic Acids Res. 43 (D1):D146–D152. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Tomczak K, Czerwińska P and Wiznerowicz M:
The Cancer Genome Atlas (TCGA): An immeasurable source of
knowledge. Contemp Oncol (Pozn). 19 (1A):A68–A77. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Li B and Dewey CN: RSEM: Accurate
transcript quantification from RNA-Seq data with or without a
reference genome. BMC Bioinformatics. 12(323)2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Li B, Ruotti V, Stewart RM, Thomson JA and
Dewey CN: RNA-Seq gene expression estimation with read mapping
uncertainty. Bioinformatics. 26:493–500. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Carter SL, Eklund AC, Kohane IS, Harris LN
and Szallasi Z: A signature of chromosomal instability inferred
from gene expression profiles predicts clinical outcome in multiple
human cancers. Nat Genet. 38:1043–1048. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
23
|
Choi YL, Park SH, Jang JJ and Park CK:
Expression of the G1-S modulators in hepatitis B virus-related
hepatocellular carcinoma and dysplastic nodule: Association of
cyclin D1 and p53 proteins with the progression of hepatocellular
carcinoma. J Korean Med Sci. 16:424–432. 2001.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Tripathi V, Shen Z, Chakraborty A, Giri S,
Freier SM, Wu X, Zhang Y, Gorospe M, Prasanth SG, Lal A, et al:
Long noncoding RNA MALAT1 controls cell cycle progression by
regulating the expression of oncogenic transcription factor B-MYB.
PLoS Genet. 9(e1003368)2013.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Bumbaca B and Li W: Taxane resistance in
castration-resistant prostate cancer: Mechanisms and therapeutic
strategies. Acta Pharm Sin B. 8:518–529. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhong Z, Pannu V, Rosenow M, Stark A and
Spetzler D: KIAA0100 modulates cancer cell aggression behavior of
MDA-MB-231 through microtubule and heat shock proteins. Cancers
(Basel). 10(10)2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Guglietta S and Rescigno M:
Hypercoagulation and complement: Connected players in tumor
development and metastases. Semin Immunol. 28:578–586.
2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
van Ree JH, Jeganathan KB, Malureanu L and
van Deursen JM: Overexpression of the E2 ubiquitin-conjugating
enzyme UbcH10 causes chromosome missegregation and tumor formation.
J Cell Biol. 188:83–100. 2010.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Berlingieri MT, Pallante P, Guida M, Nappi
C, Masciullo V, Scambia G, Ferraro A, Leone V, Sboner A,
Barbareschi M, et al: UbcH10 expression may be a useful tool in the
prognosis of ovarian carcinomas. Oncogene. 26:2136–2140.
2007.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Parris TZ, Kovács A, Aziz L, Hajizadeh S,
Nemes S, Semaan M, Forssell-Aronsson E, Karlsson P and Helou K:
Additive effect of the AZGP1, PIP, S100A8 and UBE2C molecular
biomarkers improves outcome prediction in breast carcinoma. Int J
Cancer. 134:1617–1629. 2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Zhang HQ, Zhao G, Ke B, Ma G, Liu GL,
Liang H, Liu LR and Hao XS: Overexpression of UBE2C correlates with
poor prognosis in gastric cancer patients. Eur Rev Med Pharmacol
Sci. 22:1665–1671. 2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Okamoto Y, Ozaki T, Miyazaki K, Aoyama M,
Miyazaki M and Nakagawara A: UbcH10 is the cancer-related E2
ubiquitin-conjugating enzyme. Cancer Res. 63:4167–4173.
2003.PubMed/NCBI
|
|
33
|
Guo J, Wu Y, Du J, Yang L, Chen W, Gong K,
Dai J, Miao S, Jin D and Xi S: Deregulation of UBE2C-mediated
autophagy repression aggravates NSCLC progression. Oncogenesis.
7(49)2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Wu Y, Jin D, Wang X, Du J, Di W, An J,
Shao C and Guo J: UBE2C induces cisplatin resistance via
ZEB1/2-dependent upregulation of ABCG2 and ERCC1 in NSCLC cells. J
Oncol. 2019(8607859)2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Wang C, Pan YH, Shan M, Xu M, Bao JL and
Zhao LM: Knockdown of UbcH10 enhances the chemosensitivity of dual
drug resistant breast cancer cells to epirubicin and docetaxel. Int
J Mol Sci. 16:4698–4712. 2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wang L, Zhang J, Wan L, Zhou X, Wang Z and
Wei W: Targeting Cdc20 as a novel cancer therapeutic strategy.
Pharmacol Ther. 151:141–151. 2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Mondal G, Sengupta S, Panda CK, Gollin SM,
Saunders WS and Roychoudhury S: Overexpression of Cdc20 leads to
impairment of the spindle assembly checkpoint and aneuploidization
in oral cancer. Carcinogenesis. 28:81–92. 2007.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kato T, Daigo Y, Aragaki M, Ishikawa K,
Sato M and Kaji M: Overexpression of CDC20 predicts poor prognosis
in primary non-small cell lung cancer patients. J Surg Oncol.
106:423–430. 2012.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Kidokoro T, Tanikawa C, Furukawa Y,
Katagiri T, Nakamura Y and Matsuda K: CDC20, a potential cancer
therapeutic target, is negatively regulated by p53. Oncogene.
27:1562–1571. 2008.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Wu F, Lin Y, Cui P, Li H, Zhang L, Sun Z,
Huang S, Li S, Huang S, Zhao Q, et al: Cdc20/p55 mediates the
resistance to docetaxel in castration-resistant prostate cancer in
a Bim-dependent manner. Cancer Chemother Pharmacol. 81:999–1006.
2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Pommier Y, Sun Y, Huang SN and Nitiss JL:
Roles of eukaryotic topoisomerases in transcription, replication
and genomic stability. Nat Rev Mol Cell Biol. 17:703–721.
2016.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Liu T, Zhang H, Yi S, Gu L and Zhou M:
Mutual regulation of MDM4 and TOP2A in cancer cell proliferation.
Mol Oncol. 13:1047–1058. 2019.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Chen D, Maruschke M, Hakenberg O,
Zimmermann W, Stief CG and Buchner A: TOP2A, HELLS, ATAD2, and TET3
are novel prognostic markers in renal cell carcinoma. Urology.
102:265.e1–265.e7. 2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Lan J, Huang HY, Lee SW, Chen TJ, Tai HC,
Hsu HP, Chang KY and Li CF: TOP2A overexpression as a poor
prognostic factor in patients with nasopharyngeal carcinoma. Tumour
Biol. 35:179–187. 2014.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Zheng H, Li X, Chen C, Chen J, Sun J, Sun
S, Jin L, Li J, Sun S and Wu X: Quantum dot-based immunofluorescent
imaging and quantitative detection of TOP2A and prognostic value in
triple-negative breast cancer. Int J Nanomedicine. 11:5519–5529.
2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Węsierska-Gądek J and Składanowski A:
Therapeutic intervention by the simultaneous inhibition of DNA
repair and type I or type II DNA topoisomerases: One strategy, many
outcomes. Future Med Chem. 4:51–72. 2012.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Wang TL, Ren YW, Wang HT, Yu H and Zhao
YX: Association of topoisomerase II (TOP2A) and dual-specificity
phosphatase 6 (DUSP6) single nucleotide polymorphisms with
radiation treatment response and prognosis of lung cancer in Han
Chinese. Med Sci Monit. 23:984–993. 2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Kovarikova V, Burkus J, Rehak P, Brzakova
A, Solc P and Baran V: Aurora kinase A is essential for correct
chromosome segregation in mouse zygote. Zygote. 24:326–337.
2016.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Xu Z, Ogawa H, Vagnarelli P, Bergmann JH,
Hudson DF, Ruchaud S, Fukagawa T, Earnshaw WC and Samejima K:
INCENP-aurora B interactions modulate kinase activity and
chromosome passenger complex localization. J Cell Biol.
187:637–653. 2009.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Takeshita M, Koga T, Takayama K, Ijichi K,
Yano T, Maehara Y, Nakanishi Y and Sueishi K: Aurora-B
overexpression is correlated with aneuploidy and poor prognosis in
non-small cell lung cancer. Lung Cancer. 80:85–90. 2013.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Schneider MA, Christopoulos P, Muley T,
Warth A, Klingmueller U, Thomas M, Herth FJ, Dienemann H, Mueller
NS, Theis F, et al: AURKA, DLGAP5, TPX2, KIF11 and CKAP5: Five
specific mitosis-associated genes correlate with poor prognosis for
non-small cell lung cancer patients. Int J Oncol. 50:365–372.
2017.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Wu T, Zhang X, Huang X, Yang Y and Hua X:
Regulation of cyclin B2 expression and cell cycle G2/m transition
by menin. J Biol Chem. 285:18291–18300. 2010.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Mo ML, Chen Z, Li J, Li HL, Sheng Q, Ma
HY, Zhang FX, Hua YW, Zhang X, Sun DQ, et al: Use of serum
circulating CCNB2 in cancer surveillance. Int J Biol Markers.
25:236–242. 2010.PubMed/NCBI
|
|
54
|
Qian X, Song X, He Y, Yang Z, Sun T, Wang
J, Zhu G, Xing W and You C: CCNB2 overexpression is a poor
prognostic biomarker in Chinese NSCLC patients. Biomed
Pharmacother. 74:222–227. 2015.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Takashima S, Saito H, Takahashi N, Imai K,
Kudo S, Atari M, Saito Y, Motoyama S and Minamiya Y: Strong
expression of cyclin B2 mRNA correlates with a poor prognosis in
patients with non-small cell lung cancer. Tumour Biol.
35:4257–4265. 2014.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Ganguly R, Mohyeldin A, Thiel J, Kornblum
HI, Beullens M and Nakano I: MELK-a conserved kinase: Functions,
signaling, cancer, and controversy. Clin Transl Med.
4(11)2015.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Wang J, Wang Y, Shen F, Xu Y, Zhang Y, Zou
X, Zhou J and Chen Y: Maternal embryonic leucine zipper kinase: A
novel biomarker and a potential therapeutic target of cervical
cancer. Cancer Med. 7:5665–5678. 2018.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Speers C, Zhao SG, Kothari V, Santola A,
Liu M, Wilder-Romans K, Evans J, Batra N, Bartelink H, Hayes DF, et
al: Maternal embryonic leucine zipper kinase (MELK) as a novel
mediator and biomarker of radioresistance in human breast cancer.
Clin Cancer Res. 22:5864–5875. 2016.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Li S, Li Z, Guo T, Xing XF, Cheng X, Du H,
Wen XZ and Ji JF: Maternal embryonic leucine zipper kinase serves
as a poor prognosis marker and therapeutic target in gastric
cancer. Oncotarget. 7:6266–6280. 2016.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Lou W, Ding B, Xu L and Fan W:
Construction of Potential Glioblastoma Multiforme-Related
miRNA-mRNA Regulatory Network. Front Mol Neurosci.
12(66)2019.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Gao X, Wang X, Cai K, Wang W, Ju Q, Yang
X, Wang H and Wu H: MicroRNA-127 is a tumor suppressor in human
esophageal squamous cell carcinoma through the regulation of
oncogene FMNL3. Eur J Pharmacol. 791:603–610. 2016.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Goswami RS, Atenafu EG, Xuan Y, Waldron L,
Reis PP, Sun T, Datti A, Xu W, Kuruvilla J, Good DJ, et al:
MicroRNA signature obtained from the comparison of aggressive with
indolent non-Hodgkin lymphomas: Potential prognostic value in
mantle-cell lymphoma. J Clin Oncol. 31:2903–2911. 2013.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Shi L, Wang Y, Lu Z, Zhang H, Zhuang N,
Wang B, Song Z, Chen G, Huang C, Xu D, et al: miR-127 promotes EMT
and stem-like traits in lung cancer through a feed-forward
regulatory loop. Oncogene. 36:1631–1643. 2017.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Fort RS, Mathó C, Oliveira-Rizzo C, Garat
B, Sotelo-Silveira JR and Duhagon MA: An integrated view of the
role of miR-130b/301b miRNA cluster in prostate cancer. Exp Hematol
Oncol. 7(10)2018.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Dettmer MS, Perren A, Moch H, Komminoth P,
Nikiforov YE and Nikiforova MN: MicroRNA profile of poorly
differentiated thyroid carcinomas: New diagnostic and prognostic
insights. J Mol Endocrinol. 52:181–189. 2014.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Ruiz-Martinez M, Navarro A, Marrades RM,
Viñolas N, Santasusagna S, Muñoz C, Ramírez J, Molins L and Monzo
M: YKT6 expression, exosome release, and survival in non-small cell
lung cancer. Oncotarget. 7:51515–51524. 2016.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Mirzadeh Azad F, Naeli P, Malakootian M,
Baradaran A, Tavallaei M, Ghanei M and Mowla SJ: Two lung
development-related microRNAs, miR-134 and miR-187, are
differentially expressed in lung tumors. Gene. 577:221–226.
2016.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Sun CC, Li SJ and Li DJ: Hsa-miR-134
suppresses non-small cell lung cancer (NSCLC) development through
down-regulation of CCND1. Oncotarget. 7:35960–35978.
2016.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Qin Q, Wei F, Zhang J, Wang X and Li B:
miR-134 inhibits non-small cell lung cancer growth by targeting the
epidermal growth factor receptor. J Cell Mol Med. 20:1974–1983.
2016.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Pan JY, Zhang F, Sun CC, Li SJ, Li G, Gong
FY, Bo T, He J, Hua RX, Hu WD, et al: miR-134: A human cancer
suppressor? Mol Ther Nucleic Acids. 6:140–149. 2017.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Hua X and Fan KC: Down-regulation of
miR-1181 indicates a dismal prognosis for nasopharyngeal carcinoma
and promoted cell proliferation and metastasis by modulating
Wnt/β-catenin signaling. Eur Rev Med Pharmacol Sci. 23:1077–1086.
2019.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Nam EJ, Lee M, Yim GW, Kim JH, Kim S, Kim
SW and Kim YT: MicroRNA profiling of a CD133(+) spheroid-forming
subpopulation of the OVCAR3 human ovarian cancer cell line. BMC Med
Genomics. 5(18)2012.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Wang J, Guo XJ, Ding YM and Jiang JX:
miR-1181 inhibits invasion and proliferation via STAT3 in
pancreatic cancer. World J Gastroenterol. 23:1594–1601.
2017.PubMed/NCBI View Article : Google Scholar
|