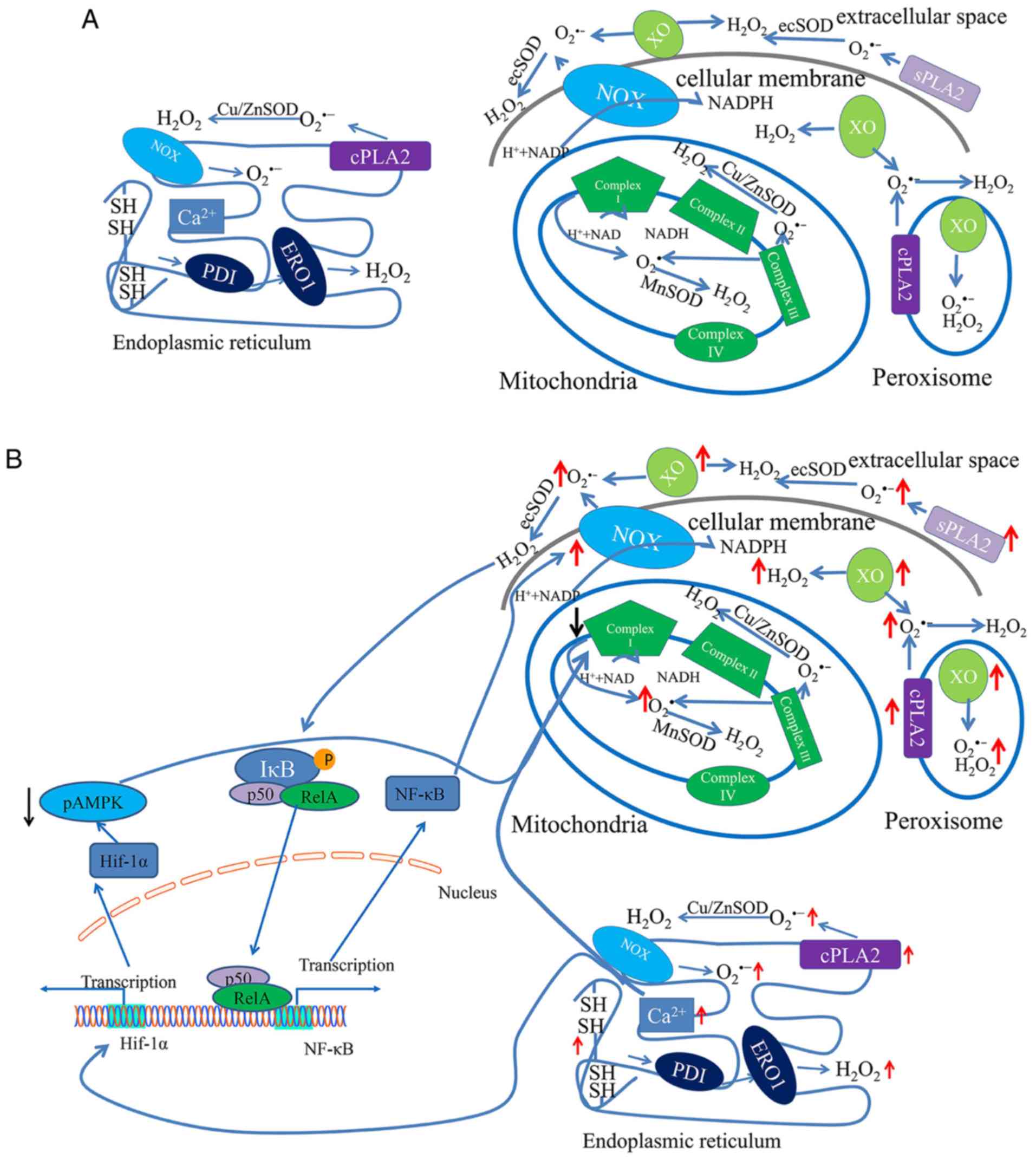
Obstructive sleep apnea (OSA) is a common breathing
and sleeping disorder, the primary cause of which is aspiratory
collapse of the pharyngeal airway, and leads to intermittent
hypoxia (IH) (1). Previous studies
have shown that OSA is associated with behavioral and
neuropsychological deficits, including impaired spatial learning
memory and cognition (2,3). However, the specific mechanisms
underlying the chain of events from the development of IH to
cognitive impairment remain elusive. It has been suggested that
reactive oxygen species (ROS), which are produced in excess in
cases of IH, are strongly associated with the presence of
IH-induced cognitive impairment (4,5).
ROS are essential for many biological processes and
these molecules are constantly formed in cells and removed by
antioxidant defenses (6). Moreover,
previous studies have demonstrated that increased oxidative stress
is present in OSA (7-10).
It has been shown that there is increased ROS production in
stimulated neutrophils from patients with OSA, while the levels of
ROS are attenuated after continuous positive airway pressure (CPAP)
treatment (7,8). Furthermore, lipid peroxidation and
protein carbonylation, both of which are markers of oxidative
stress, are observed in patients with OSA (9,10).
Moreover, these processes damage biomolecules such as lipids,
proteins and DNA (6).
The aims of the present review were to examine the
production of ROS and its role in the pathobiology of cognitive
impairment in an OSA model and to investigate the underlying
mechanism of ROS-induced cognitive deficiencies.
As described above, the majority of ROS are
generated from mitochondria during ATP synthesis, which supplies
energy for physiological functions (Fig. 1A) (16). During this process, electrons flow
along the mitochondrial electron transport chain (ETC) and protons
are translocated from the mitochondrial matrix into the
intermembrane space, thus creating a proton gradient (4). During normal aerobic respiration,
approximately 2% of electrons ‘leak’ from the flow, resulting in the
formation of O2•- (17). O2•- is easily
detoxified by superoxide dismutase, which leads to the formation of
H2O2 (18).
While H2O2 is further reduced to
H2O by glutathione peroxidase.
H2O2 can also form highly reactive OH• by
reacting with metal ions, which may be responsible for oxidative
stress-induced cellular damage (19).
During mitochondrial respiration, some
oxidoreductases affect ROS production, including complex I via
complex IV (4). Under physiological
conditions, a low concentration of ROS is maintained by the action
of these oxidoreductases. However, increased ROS production can be
caused by the inhibition of complex I activity in the mitochondrial
ETC, under IH conditions (20).
Furthermore, complex I, the first oxidoreductase in the ETC, is
dependent on NADH-producing substrates to produce
O2•- (21). A
previous study demonstrated that IH induced complex I inhibition
via the upregulation of NADPH oxidase (NOX) in PC12 cell cultures
(22). Moreover, when complex I
activity is decreased, ROS levels are increased in mitochondria due
to the inability of electrons to be transported, which results in
the formation of superoxide via a one-electron reduction of oxygen
(23). However, complex III,
another major producer of superoxide and ROS within the
mitochondrial ETC, is unaffected (24). These results suggest that complex I,
and not complex III, may be involved in increasing ROS levels
(Fig. 1B).
The ER is a large organelle that participates in the
correct assembly and folding of nascent proteins and is also the
site of post-translational protein modifications in ATP-dependent
chaperone-mediated processes (25).
Furthermore, approximately 25% of ROS are derived from the ER and
are primarily required for oxidative protein folding (26). Correct protein folding involves the
formation of disulfide bonds in a process driven by protein
disulfide isomerase (PDI) and ER oxidoreductin (ERO-1) (27). In this process, electrons are
transferred from PDI to O2, resulting in
H2O2 formation (Fig. 1A) (27).
To perform these functions, the ER lumen possesses a
unique environment composed of molecular chaperones, folding
enzymes and high concentrations of ATP and calcium (28). In addition, the oxidative
environment favors protein folding and, in particular, the
formation of intra- and intermolecular disulfide bonds (29). However, increased ROS production may
lead to a loss of ER homeostasis and the subsequent accumulation of
misfolded proteins, a process known as ER stress (30). Under conditions of ER stress,
additional misfolded or unfolded proteins are synthesized, leading
to the depletion of glutathione (GSH) (31). After GSH is utilized, the oxidizing
environment facilitates the reoxidation of protein thiols through
interaction with ERO-1/PDI (27).
These steps produce repetitive cycles of disulfide bond breakage
and formation, with each cycle generating additional ROS as a
byproduct (25). Furthermore, the
accumulation of unfolded proteins in the ER elicits Ca2+
leakage into the cytosol, thus causing increased ROS production in
the mitochondria (Fig. 1B)
(29).
NOX is a multisubunit enzymatic complex that is
localized to both cellular and subcellular membranes (32). Furthermore, NOX catalyzes
O2•-production via the one-electron reduction
of O2 using NADPH (33).
There have been ≥7 NOX isoforms, including NOX1-5 and DUOX1-2,
identified in this enzymatic system (34). Moreover, NOX can regulate electron
flow and transfer to the outer heme, where the electron accepts
O2 to form O2•- (35).
As a complex of major ROS-generating enzymes, NOX
plays an important role in the OSA model (36). NOX2 expression and ROS production
have been shown to be simultaneously increased in patients with OSA
(37). Furthermore, pharmacological
inhibition of NOX by apocynin and NOX2 deficiency can attenuate
arterial hypertension in an OSA model by suppressing ROS production
(38). In addition to
cardiovascular risk, the cognitive deficits induced by oxidative
stress are partially mediated by excessive NOX activity during IH
(39). Furthermore, mice lacking
NOX activity present with a learning ability comparable to that of
IH-exposed wild-type littermates, which exhibit spatial learning
deficits in a water maze test (40). Collectively, these studies indicated
that NOX may be involved in ROS production in a model of OSA.
However, the specific mechanistic involvement of NOX in IH injury
requires further investigation.
In addition to mitochondria and the ER, superoxide
and other ROS are generated in subcellular organelles peroxisomes
(Fig. 1). The primary function of
these particles is the oxidative degradation of long-chain fatty
acids (41). Peroxisomes are
spherical or oval shaped particles with a diameter of 0.2-1 mm that
are surrounded by a single membrane (42). Moreover, peroxisomes in mammals
harbor >100 enzymes and other proteins, some of which are
associated with OSA or IH (43).
XO is localized to the outer surface of the cellular
membrane, as well as in the cytosol and peroxisomes (44). XO catalyzes the oxidation of
xanthine to uric acid at the site of flavin adenine dinucleotide,
together with the reduction of NAD+ and O2
(45). In this process, the affinity
for O2 (44) is
significantly enhanced, resulting in univalent and divalent electron
transfer to O2 to generate O2•-
and H2O2, respectively (46).
XO is a critical source of ROS in
ischemia/reperfusion injury, which is similar to chronic cycles of
hypoxia and reoxygenation (47).
Moreover, IH processes are associated with elevated XO activity and
promotion of ROS formation by increasing the proteolytic conversion
of xanthine dehydrogenase to XO (48). XO has been identified to be involved
in ROS production in OSA, as lipid peroxidation, a marker of
oxidative stress, is reduced after the application of the XO
inhibitor allopurinol (49).
Furthermore, allopurinol reduces the level of ROS by decreasing
free radical generation via inhibition of the XO system (50). These findings suggest that XO plays
a key role in increased ROS production under IH conditions.
NO is known to play a critical role in the
regulation of cellular processes that include catalyzation by
nitric oxide synthase (NOS) (51).
Although excess NO can induce apoptosis via the aggravation of
oxidative stress (52), the
specific mechanism is not fully understood.
Moreover, two NOS enzymes, the constitutive
calcium/calmodulin-dependent neuronal and endothelial isoforms
(53), show sustained expression in
the central nervous system. However, under certain pathological
conditions, including ischemia, hypoxia and other pathological
stimuli, another NOS, inducible calcium-independent isoform (iNOS),
is activated (54). Furthermore, IH
has been shown to augment NO generation by activating iNOS
(55), which induces apoptosis via
the aggravation of oxidative stress. In addition, overexpression of
O2•-in the endothelial tissue of patients
with OSA is reduced by the NOS inhibitor l-nitroarginine
methyl-ester (56). NOS has also
been revealed to play a critical role in enhancing oxidative stress
in OSA (56).
PLA2s belong to a family of enzymes that hydrolyze
the acyl bond at the sn-2 position of phospholipids to generate
free fatty acids and lysophospholipids (57). PLA2s can be classified into three
families based on their calcium requirement for catalytic activity,
including calcium-dependent cytosolic PLA2 (cPLA2),
calcium-independent PLA2 and secretory PLA2 (sPLA2) (58). It has been shown that all these
PLA2sare involved in ROS production via arachidonic acid and
lipoxygenase, which produce O2•- (59).
While there is no direct evidence to establish a
connection between PLA2 expression and IH treatment, PLA2 activity
is significantly increased after ischemia/reperfusion, which is
dependent on Ca2+ concentration (60). Previous studies have revealed that
cPLA2 immunoreactivity is selectively higher in the hippocampal CA1
region compared with other regions in a hypoxia-ischemia rat model
(61). Furthermore, inhibition of
cPLA2 attenuates oxygen-glucose deprivation-induced neuronal death
in the hippocampus, which suggests that cPLA2 is involved in
ischemic injury (62). In addition,
a biphasic increase in them RNA expression of sPLA2 in the cortex
of a rat brain after 20 min of transient forebrain occlusion has
been shown (63). These results
suggest that enhanced PLA2 activity results in the increased
production of ROS in ischemia/reperfusion. Thus, it is hypothesized
that the excessive ROS levels under IH conditions may also be due
to increased PLA2 activity.
Misregulation of ROS-sensitive transcription factors
and their downstream genes, such as hypoxia-inducible factor
(HIF)-1 α and NF-κB (10), also
aggravates oxidative stress (Figs.
1B and 2).
Although many organelles and peroxisomes are
involved in ROS production during IH treatment, there is no direct
evidence that demonstrates the underlying mechanism responsible for
ROS production (64). Therefore, on
the basis of previous studies, it is hypothesized that
mitochondrial dysfunction may be the main source of excessive ROS
production in OSA (20,22,65).
Thus, attenuation of mitochondrial dysfunction may be an effective
strategy for future clinical therapeutics.
OSA is commonly associated with cognitive
impairment, particularly memory, verbal fluency, attention and
perception impairments (66,67),
but the cause of this cognitive dysfunction is not fully
understood.
Various different mechanisms that link OSA to
cognitive dysfunction have been suggested (4,5,68-70).
A major proposed mechanism is IH, which induces a reduction in
memory performance in patients with OSA via oxidative stress injury
(71). Moreover, increased levels
of ROS have been confirmed to contribute to learning and memory
impairments (72). In addition,
increased ROS production induced by IH can result in deficits in
spatial learning and memory in rodent models, which are largely
dependent on the hippocampus. Neuroimaging studies have been
performed to investigate the effects of OSA (73-75).
Regional gray matter (GM), which is closely associated with memory
formation, is observed to be lost in multiple brain regions of
patients with OSA via MRI of the cortex (75). In another study, voxel-based
morphometry was performed to measure cortical GM volume and
indicated that, compared with healthy control individuals, patients
with OSA exhibited significant cognitive impairment and had a
smaller right hippocampus (76).
Therefore, the cortex and hippocampus are the two major regions
associated with cognitive impairment in OSA models. Although the
underlying mechanism is not fully understood, it is hypothesized
that the density of capillaries and large-diameter vessels is
abundant in the two regions and that these are sensitive to
oxidative stress (77).
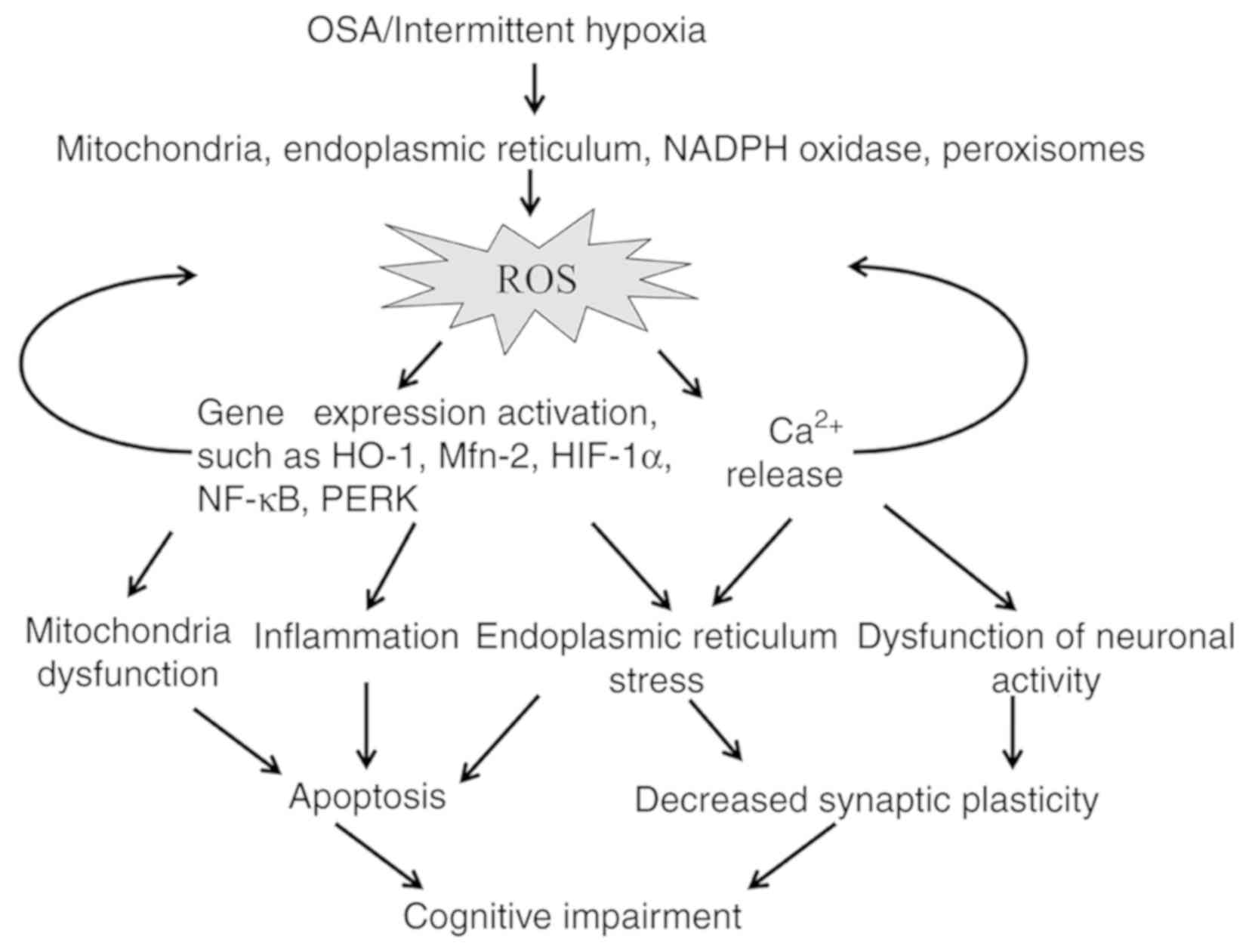
Many pathways, including mitochondrial dysfunction,
inflammation, apoptosis, ER stress and neuronal activity
disturbance, are proposed to be associated with cognitive
impairment induced by oxidative stress (Fig. 2).
Mitochondrial dysfunction is a common feature of
patients with OSA, and the function of oxidoreductases in the
mitochondrial ETC is disturbed under IH conditions (4). Furthermore, mitochondrial fusion and
fission, which play important roles in mitochondrial function, have
been observed in an IH model (78).
Under IH conditions, mitochondrial fission occurs more frequently
than fusion and leads to apoptosis through regulation of the
expression of mitochondrial fusion protein-2 (Mfn2) (76). Thus, ROS may control the expression
of Mfn2. While the full mechanism has not been elucidated, it is
hypothesized that the mechanism could be associated with the
overexpression of heme oxygenase-1, a gene that responds to
oxidative stress (79). In
addition, a significant correlation is found between OSA severity
and decreased mitochondrial DNA (mtDNA) copy number, suggesting
that patients with a high apnea-hypopnea index are exposed to
greater systemic blood oxidative stress (65). Despite the lack of direct evidence
to show that mitochondrial dysfunction is a cause of cognitive
impairment in OSA, clinical research has identified that the number
of copies of mtDNA are biomarkers for assessing cognitive status in
neurodegenerative diseases, such as Alzheimer's and Huntington's
disease (80,81). The main mechanism of this effect is
that mtDNA depletion induced by persistent oxidative stress leads
to cognitive decline via cell apoptosis (82).
When NF-κB is activated, spatial learning and memory
are impaired, which may be associated with declining hippocampal
long-term potentiation (LTP) and dendritic branching (93). On the other hand, inflammation
decreases the efficiency of the capillary system and oxygen supply
to the brain, thus reducing metabolic function and oxygen intake in
neurons (94). Consequently,
individuals with neuroinflammation and OSA may present with
cognitive deterioration.
A parallel increase in the expression of HIF-1α, a
key regulator of cell adaptation to hypoxia, is often observed
alongside the formation of ROS (98). Under IH, the
Ca2+-dependent activation of calcium-calmodulin protein
kinase stimulates HIF-1α transcriptional activity and protein
expression (99). Then, in turn,
the stabilization of HIF-1α promotes the synthesis of ROS in
mitochondria to induce cell death via the suppression of
AMP-activated protein kinase (100,101). HIF-1α stabilization, along with a
decline in Bcl-2 and substantial caspase-3 expression, has been
observed after IH exposure (102).
Furthermore, a large number of apoptotic events have been
identified in rat myocardium exposed to IH (103), and the inhibition of HIF-1α
expression has been shown to decrease neuronal apoptosis (104). Additionally, >50 oxygen
sensitive genes have been identified as direct targets of
HIF-1α-mediated transactivation, such as the enzymeβ-secretase 1
(BACE1) (105). It was reported
that 3 days of IH treatment upregulated BACE and generated amyloid
β (Aβ) via HIF-1α, thus compared with age-matched control
individuals, patients with Alzheimer's disease have a 5-fold
increased risk of presenting with OSA (106). These data suggest that inhibition
of HIF-1α not only suppresses apoptosis, but also reduces Aβ
generation. Thus, HIF-1α may be a potential drug target for future
OSA therapy.
ER stress induced by increased ROS expression is
initiated when unfolded or misfolded proteins accumulate in the ER
(30). Furthermore, ER stress
induces the coordinated adaptive program known as the ‘unfolded
protein response’ (UPR), which involves the degradation of unfolded
proteins (107). The UPR consists
of three independent signaling pathways, pancreatic ER kinase
signaling, activating transcription factor 6 signaling and
inositol-requiring enzyme 1 signaling (108). If the accumulation of toxic
unfolded and misfolded proteins results in prolonged ER stress, the
perturbed and overloaded ER-folding environment persists and is
associated with increased cell death (109). Previous studies have demonstrated
that ER stress is present in the brain in a mice model of OSA,
accompanied by an increase in the expression of cleaved caspase-3,
which is a protein marker of apoptosis (69). In addition, the expression of growth
arrest and DNA damage-inducible gene 153 (GADD153), a proapoptotic
protein activated by ER stress, is increased in the cortex and
hippocampus of the OSA model, which is accompanied by cognitive
impairment (69). Moreover,
GADD153-/- mice exhibit resistance to oxidative stress
(110). In contrast, synaptic
plasticity impairment is prevented by incubation with phenylbutyric
acid (PBA), an inhibitor of ER stress (111). Furthermore, synapse degeneration
is associated with elevatedGADD153 expression (112). Therefore, these studies suggest
that ER stress also affects synaptic formation. Our previous study
using a OSA model found that ER stress is involved in cognitive
deficits due to LTP impairment in the hippocampus, as application
of tauroursodeoxycholic acid, an inhibitor of ER stress, rescued
LTP impairment by decreasing the number of apoptotic neurons and
promoting the formation of synapses (69).
Neuronal firing, especially robust persistent
activity of neurons in the cortex and hippocampus, is critical in
memory formation (113,114). In a previous study, it was found
that hypoxia could affect membrane excitability and may involve
acute modulation of ion channels, including K+-channels,
Na+-channels and Ca2+-channels, which results
in the depolarization or hyperpolarization of neurons (115). Moreover, hypoxia/hypoxemia
underlies several pathological processes, including neuronal
activity, in these regions (116).
ROS have been implicated in LTP of neural activity
as they are associated with a number of K+ channels and
lead to the Ca2+-dependent release of the
neurotransmitter glutamate and excitotoxicity (117). ROS promote the activation of
inositol 1,4,5-trisphosphate receptors in the ER, leading to
Ca2+ mobilization into the cytosol, thus enhancing
membrane permeability and promoting the release of glutamate
(118). Furthermore, it was
observed that a higher concentration of glutamate is found in the
cortex of patients with OSA (119). Thus, it is hypothesized that the
release of glutamate could increase neuronal excitability, and
result in neuron dysfunction and apoptosis due to excitotoxicity.
In addition, motor-evoked potentials, which are indicative of
neuronal excitability, are higher in patients with OSA compared
with healthy controls (120).
Moreover, increased expression of c-Fos is observed in the cortex
after IH treatment and is accompanied by increased apoptosis
(102). Collectively, these
previous studies have suggested that these neurons are under
excitotoxic conditions, which leads to cognitive dysfunction, as
measured by the Morris water maze (121). Furthermore, our previous study
implanted multiple electrodes into the CA1 region of the
hippocampus to monitor spontaneous discharges after chronic IH
treatment. It was found that the frequency of pyramidal neuron
firing in the CA1 region increased after 1-2 days of IH exposure.
However, this firing decreased after 14 days of IH treatment
(unpublished data). These results indicated that cognitive
malfunction in the OSA model may be associated with marked cellular
changes over time.
This review has focused on the mechanism of ROS
overexpression and cognitive impairment induced by oxidative stress
in IH or OSA models. While the role of ROS in cognitive impairment
in OSA models is not fully understood, ROS are known to activate
multiple pathways to induce neuronal dysfunction, mitochondrial
dysfunction, inflammation, apoptosis, ER stress and neuronal
activity disturbance (69,80,82,117).
These factors ultimately lead to cognitive dysfunction in the OSA
model. Currently, the most commonly used methods in the clinical
treatment of OSA are surgery and CPAP (122,123). However, each of these methods has
limitations and neither fully reverses the cognitive impairment
seen in patients with OSA (124,125). It is therefore important to
investigate the molecular mechanisms underlying neurological
impairments in patients with OSA. Moreover, identification of these
molecular mechanisms will facilitate the development of targeted
drug-based therapy to rescue cognitive impairment in OSA.
Not applicable.
The present study was supported by the National
Natural Science Foundation of China (grant no. 81601972) and the
Natural Science Foundation of Zhejiang Province (grant no.
LY19H010002).
Not applicable.
LX conceived the idea for this review article and
wrote the manuscript. YY was responsible for the manuscript
revision. The manuscript was critically revised for important
intellectual content by JC. All authors were involved in the
writing of the manuscript. All authors have read and approved the
final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Dewan NA, Nieto FJ and Somers VK:
Intermittent hypoxemia and OSA: Implications for comorbidities.
Chest. 147:266–274. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Vaessen TJ, Overeem S and Sitskoorn MM:
Cognitive complaints in obstructive sleep apnea. Sleep Med Rev.
19:51–58. 2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lal C, Strange C and Bachman D:
Neurocognitive impairment in obstructive sleep apnea. Chest.
141:1601–1610. 2012.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wang Y, Zhang SX and Gozal D: Reactive
oxygen species and the brain in sleep apnea. Respir Physiol
Neurobiol. 174:307–316. 2010.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Dayyat EA, Zhang SX, Wang Y, Cheng ZJ and
Gozal D: Exogenous erythropoietin administration attenuates
intermittent hypoxia-induced cognitive deficits in a murine model
of sleep apnea. BMC Neurosci. 13(77)2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Roy J, Galano JM, Durand T, Le Guennec JY
and Lee JC: Physiological role of reactive oxygen species as
promoters of natural defenses. FASEB J. 31:3729–3745.
2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Pilkauskaite G, Miliauskas S and
Sakalauskas R: Reactive oxygen species production in peripheral
blood neutrophils of obstructive sleep apnea patients.
ScientificWorldJournal. 2013(421763)2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Schulz R, Mahmoudi S, Hattar K, Sibelius
U, Olschewski H, Mayer K, Seeger W and Grimminger F: Enhanced
release of superoxide from polymorphonuclear neutrophils in
obstructive sleep apnea. Impact of continuous positive airway
pressure therapy. Am J Respir Crit Care Med. 162:566–570.
2000.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Vatansever E, Surmen-Gur E, Ursavas A and
Karadag M: Obstructive sleep apnea causes oxidative damage to
plasma lipids and proteins and decreases adiponectin levels. Sleep
Breath. 15:275–282. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Lavie L: Oxidative stress inflammation and
endothelial dysfunction in obstructive sleep apnea. Front Biosci
(Elite Ed). 4:1391–1403. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
11
|
Zou Z, Chang H, Li H and Wang S: Induction
of reactive oxygen species: An emerging approach for cancer
therapy. Apoptosis. 22:1321–1335. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Bonekamp NA, Völkl A, Fahimi HD and
Schrader M: Reactive oxygen species and peroxisomes: Struggling for
balance. Biofactors. 35:346–355. 2009.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Fransen M, Nordgren M, Wang B and
Apanasets O: Role of peroxisomes in ROS/RNS-metabolism:
Implications for human disease. Biochimica et biophysica Biochim
Biophys Acta. 1822:1363–1373. 2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kubota C, Torii S, Hou N, Saito N,
Yoshimoto Y, Imai H and Takeuchi T: Constitutive reactive oxygen
species generation from autophagosome/lysosome in neuronal
oxidative toxicity. J Biol Chem. 285:667–674. 2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Santos CX, Tanaka LY, Wosniak J and
Laurindo FR: Mechanisms and implications of reactive oxygen species
generation during the unfolded protein response: Roles of
endoplasmic reticulum oxidoreductases, mitochondrial electron
transport, and NADPH oxidase. Antioxid Redox Signal. 11:2409–2427.
2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Angelova PR and Abramov AY: Role of
mitochondrial ROS in the brain: From physiology to
neurodegeneration. FEBS Lett. 592:692–702. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Oyewole AO and Birch-Machin MA:
Mitochondria-targeted antioxidants. FASEB J. 29:4766–4771.
2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Halliwell B: Oxidative stress and
neurodegeneration: Where are we now? J Neurochem. 97:1634–1658.
2006.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Ohsawa I, Ishikawa M, Takahashi K,
Watanabe M, Nishimaki K, Yamagata K, Katsura K, Katayama Y, Asoh S
and Ohta S: Hydrogen acts as a therapeutic antioxidant by
selectively reducing cytotoxic oxygen radicals. Nat Med.
13:688–694. 2007.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Prabhakar NR: Sensory plasticity of the
carotid body: Role of reactive oxygen species and physiological
significance. Respir Physiol Neurobiol. 178:375–380.
2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhao RZ, Jiang S, Zhang L and Yu ZB:
Mitochondrial electron transport chain, ROS generation and
uncoupling (Review). Int J Mol Med. 44:3–15. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Khan SA, Nanduri J, Yuan G, Kinsman B,
Kumar GK, Joseph J, Kalyanaraman B and Prabhakar R: NADPH oxidase 2
mediates intermittent hypoxia-induced mitochondrial complex I
inhibition: Relevance to blood pressure changes in rats. Antioxid
Redox Signal. 14:533–542. 2011.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Kang PT, Chen CL, Lin P, Zhang L, Zweier
JL and Chen YR: Mitochondrial complex I in the post-ischemic heart:
Reperfusion-mediated oxidative injury and protein cysteine
sulfonation. J Mol Cell Cardiol. 121:190–204. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Yuan G, Adhikary G, McCormick AA, Holcroft
JJ, Kumar GK and Prabhakar NR: Role of oxidative stress in
intermittent hypoxia-induced immediate early gene activation in rat
PC12 cells. J Physiol. 557:773–783. 2004.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Higa A and Chevet E: Redox signaling loops
in the unfolded protein response. Cell Signal. 24:1548–1555.
2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Görlach A, Bertram K, Hudecova S and
Krizanova O: Calcium and ROS: A mutual interplay. Redox Biol.
6:260–271. 2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Bhandary B, Marahatta A, Kim HR and Chae
HJ: An involvement of oxidative stress in endoplasmic reticulum
stress and its associated diseases. Int J Mol Sci. 14:434–456.
2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Yong J, Bischof H, Burgstaller S, Siirin
M, Murphy A, Malli R and Kaufman RJ: Mitochondria supply ATP to the
ER through a mechanism antagonized by cytosolic Ca2.
Elife. 8: pii(e49682)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Malhotra JD and Kaufman RJ: Endoplasmic
reticulum stress and oxidative stress: A vicious cycle or a
double-edged sword? Antioxid Redox Signal. 9:2277–2293.
2007.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Mello T, Zanieri F, Ceni E and Galli A:
Oxidative stress in the healthy and wounded hepatocyte: A cellular
organelles perspective. Oxid Med Cell Longev.
2016(8327410)2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Tu BP and Weissman JS: The FAD- and
O(2)-dependent reaction cycle of Ero1-mediated oxidative protein
folding in the endoplasmic reticulum. Mol Cell. 10:983–994.
2002.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Panday A, Sahoo MK, Osorio D and Batra S:
NADPH oxidases: An overview from structure to innate
immunity-associated pathologies. Cell Mol Immunol. 12:5–23.
2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Kaur G, Sharma A, Guruprasad K and Pati
PK: Versatile roles of plant NADPH oxidases and emerging concepts.
Biotechnol Adv. 32:551–563. 2014.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Konior A, Schramm A, Czesnikiewicz-Guzik M
and Guzik TJ: NADPH oxidases in vascular pathology. Antioxid Redox
Signal. 20:2794–2814. 2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Nisimoto Y, Motalebi S, Han CH and Lambeth
JD: The p67(phox) activation domain regulates electron flow from
NADPH to flavin in flavocytochrome b(558). J Biol Chem.
274:22999–23005. 1999.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Li L, Ren F, Qi C, Xu L, Fang Y, Liang M,
Feng J, Chen B, Ning W and Cao J: Intermittent hypoxia promotes
melanoma lung metastasis via oxidative stress and inflammation
responses in a mouse model of obstructive sleep apnea. Respir Res.
19(28)2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Del Ben M, Fabiani M, Loffredo L, Polimeni
L, Carnevale R, Baratta F, Brunori M, Albanese F, Augelletti T,
Violi F and Angelico F: Oxidative stress mediated arterial
dysfunction in patients with obstructive sleep apnoea and the
effect of continuous positive airway pressure treatment. BMC Pulm
Med. 12(36)2012.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Schulz R, Murzabekova G, Egemnazarov B,
Kraut S, Eisele HJ, Dumitrascu R, Heitmann J, Seimetz M, Witzenrath
M, Ghofrani HA, et al: Arterial hypertension in a murine model of
sleep apnea: Role of NADPH oxidase 2. J Hypertens. 32:300–305.
2014.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Nair D, Dayyat EA, Zhang SX, Wang Y and
Gozal D: Intermittent hypoxia-induced cognitive deficits are
mediated by NADPH oxidase activity in a murine model of sleep
apnea. PLoS One. 6(e19847)2011.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Nair D, Ramesh V and Gozal D: Adverse
cognitive effects of high-fat diet in a murine model of sleep apnea
are mediated by NADPH oxidase activity. Neuroscience. 227:361–369.
2012.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Reddy JK and Mannaerts GP: Peroxisomal
lipid metabolism. Annu Rev Nutr. 14:343–370. 1994.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Lodhi IJ and Semenkovich CF: Peroxisomes:
A nexus for lipid metabolism and cellular signaling. Cell Metab.
19:380–392. 2014.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Suzuki J: Short-duration intermittent
hypoxia enhances endurance capacity by improving muscle fatty acid
metabolism in mice. Physiol Rep. 4: pii(e12744)2016.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Cantu-Medellin N and Kelley EE: Xanthine
oxidoreductase-catalyzed reactive species generation: A process in
critical need of reevaluation. Redox Biol. 1:353–358.
2013.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Harris CM and Massey V: The reaction of
reduced xanthine dehydrogenase with molecular oxygen. Reaction
kinetics and measurement of superoxide radical. J Biol Chem.
272:8370–8379. 1997.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Harris CM and Massey V: The oxidative
half-reaction of xanthine dehydrogenase with NAD; Reaction kinetics
and steady-state mechanism. J Biol Chem. 272:28335–28341.
1997.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Wang S, Li Y, Song X, Wang X, Zhao C, Chen
A and Yang P: Febuxostat pretreatment attenuates myocardial
ischemia/reperfusion injury via mitochondrial apoptosis. J Transl
Med. 13(209)2015.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Nanduri J, Vaddi DR, Khan SA, Wang N,
Makerenko V and Prabhakar NR: Xanthine oxidase mediates
hypoxia-inducible factor-2α degradation by intermittent hypoxia.
PLoS One. 8(e75838)2013.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Morgan BJ, Bates ML, Rio RD, Wang Z and
Dopp JM: Oxidative stress augments chemoreflex sensitivity in rats
exposed to chronic intermittent hypoxia. Respir Physiol Neurobiol.
234:47–59. 2016.PubMed/NCBI View Article : Google Scholar
|
|
50
|
El Solh AA, Saliba R, Bosinski T, Grant
BJ, Berbary E and Miller N: Allopurinol improves endothelial
function in sleep apnoea: A randomised controlled study. Eur Respir
J. 27:997–1002. 2006.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Griscavage JM, Rogers NE, Sherman MP and
Ignarro LJ: Inducible nitric oxide synthase from a rat alveolar
macrophage cell line is inhibited by nitric oxide. J Immunol.
151:6329–6337. 1993.PubMed/NCBI
|
|
52
|
Kumar A, Chen SH, Kadiiska MB, Hong JS,
Zielonka J, Kalyanaraman B and Mason RP: Inducible nitric oxide
synthase is key to peroxynitrite-mediated, LPS-induced protein
radical formation in murine microglial BV2 cells. Free Radic Biol
Med. 73:51–59. 2014.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Contestabile A: Role of nitric oxide in
cerebellar development and function: Focus on granule neurons.
Cerebellum. 11:50–61. 2012.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Li S, Hafeez A, Noorulla F, Geng X, Shao
G, Ren C, Lu G, Zhao H, Ding Y and Ji X: Preconditioning in
neuroprotection: From hypoxia to ischemia. Prog Neurobiol.
157:79–91. 2017.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Yuan X, Guo X, Deng Y, Zhu D, Shang J and
Liu H: Chronic intermittent hypoxia-induced neuronal apoptosis in
the hippocampus is attenuated by telmisartan through suppression of
iNOS/NO and inhibition of lipid peroxidation and inflammatory
responses. Brain Res. 1596:48–57. 2015.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Varadharaj S, Porter K, Pleister A,
Wannemacher J, Sow A, Jarjoura D, Zweier JL and Khayat RN:
Endothelial nitric oxide synthase uncoupling: A novel pathway in
OSA induced vascular endothelial dysfunction. Respir Physiol
Neurobiol. 207:40–47. 2015.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Sun GY, Shelat PB, Jensen MB, He Y, Sun AY
and Simonyi A: Phospholipases A2 and inflammatory responses in the
central nervous system. Neuromolecular Med. 12:133–148.
2010.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Akiba S and Sato T: Cellular function of
calcium-independent phospholipase A2. Biol Pharm Bull.
27:1174–1178. 2004.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Radogna F, Sestili P, Martinelli C,
Paolillo M, Paternoster L, Albertini MC, Accorsi A, Gualandi G and
Ghibelli L: Lipoxygenase-mediated pro-radical effect of melatonin
via stimulation of arachidonic acid metabolism. Toxicol Appl
Pharmacol. 238:170–177. 2009.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Rordorf G, Uemura Y and Bonventre JV:
Characterization of phospholipase A2 (PLA2) activity in gerbil
brain: Enhanced activities of cytosolic, mitochondrial, and
microsomal forms after ischemia and reperfusion. J Neurosci.
11:1829–1836. 1991.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Stephenson D, Rash K, Smalstig B, Roberts
E, Johnstone E, Sharp J, Panetta J, Little S, Kramer R and Clemens
J: Cytosolic phospholipase A2 is induced in reactive glia following
different forms of neurodegeneration. Glia. 27:110–128.
1999.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Arai K, Ikegaya Y, Nakatani Y, Kudo I,
Nishiyama N and Matsuki N: Phospholipase A2 mediates ischemic
injury in the hippocampus: A regional difference of neuronal
vulnerability. Eur J Neurosci. 13:2319–2323. 2001.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Lauritzen I, Heurteaux C and Lazdunski M:
Expression of group II phospholipase A2 in rat brain after severe
forebrain ischemia and in endotoxic shock. Brain Res. 651:353–356.
1994.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Elliot-Portal E, Laouafa S, Arias-Reyes C,
Janes TA, Joseph V and Soliz J: Brain-derived erythropoietin
protects from intermittent hypoxia-induced cardiorespiratory
dysfunction and oxidative stress in mice. Sleep: 41, 2018 doi:
10.1093/sleep/zsy072.
|
|
65
|
Kim YS, Kwak JW, Lee KE, Cho HS, Lim SJ,
Kim KS, Yang HS and Kim HJ: Can mitochondrial dysfunction be a
predictive factor for oxidative stress in patients with obstructive
sleep apnea? Antioxid Redox Signal. 21:1285–1288. 2014.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Slonkova J, Bar M, Nilius P, Berankova D,
Salounova D and Sonka K: Spontaneous improvement in both
obstructive sleep apnea and cognitive impairment after stroke.
Sleep Med. 32:137–142. 2017.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Quan SF, Chan CS, Dement WC, Gevins A,
Goodwin JL, Gottlieb DJ, Green S, Guilleminault C, Hirshkowitz M,
Hyde PR, et al: The association between obstructive sleep apnea and
neurocognitive performance-the Apnea Positive Pressure Long-term
Efficacy Study (APPLES). Sleep. 34:303–314B. 2011.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Gagnon K, Baril AA, Gagnon JF, Fortin M,
Décary A, Lafond C, Desautels A, Montplaisir J and Gosselin N:
Cognitive impairment in obstructive sleep apnea. Pathol Biol
(Paris). 62:233–240. 2014.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Xu LH, Xie H, Shi ZH, Du LD, Wing YK, Li
AM, Ke Y and Yung WH: Critical role of endoplasmic reticulum stress
in chronic intermittent Hypoxia-induced deficits in synaptic
plasticity and Long-term memory. Antioxid Redox Signal. 23:695–710.
2015.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Xie H, Leung KL, Chen L, Chan YS, Ng PC,
Fok TF, Wing YK, Ke Y, Li AM and Yung WH: Brain-derived
neurotrophic factor rescues and prevents chronic intermittent
hypoxia-induced impairment of hippocampal long-term synaptic
plasticity. Neurobiol Dis. 40:155–162. 2010.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Feng J, Wu Q, Zhang D and Chen BY:
Hippocampal impairments are associated with intermittent hypoxia of
obstructive sleep apnea. Chin Med J (Engl). 125:696–701.
2012.PubMed/NCBI
|
|
72
|
Milton VJ and Sweeney ST: Oxidative stress
in synapse development and function. Dev Neurobiol. 72:100–110.
2012.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Lin WC, Huang CC, Chen HL, Chou KH, Chen
PC, Tsai NW, Chen MH, Friedman M, Lin HC and Lu CH: Longitudinal
brain structural alterations and systemic inflammation in
obstructive sleep apnea before and after surgical treatment. J
Transl Med. 14(139)2016.PubMed/NCBI View Article : Google Scholar
|
|
74
|
O'Donoghue FJ, Wellard RM, Rochford PD,
Dawson A, Barnes M, Ruehland WR, Jackson ML, Howard ME, Pierce RJ
and Jackson GD: Magnetic resonance spectroscopy and neurocognitive
dysfunction in obstructive sleep apnea before and after CPAP
treatment. Sleep. 35:41–48. 2012.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Canessa N, Castronovo V, Cappa SF, Aloia
MS, Marelli S, Falini A, Alemanno F and Ferini-Strambi L:
Obstructive sleep apnea: Brain structural changes and
neurocognitive function before and after treatment. Am J Respir
Crit Care Med. 183:1419–1426. 2011.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Torelli F, Moscufo N, Garreffa G, Placidi
F, Romigi A, Zannino S, Bozzali M, Fasano F, Giulietti G, Djonlagic
I, et al: Cognitive profile and brain morphological changes in
obstructive sleep apnea. NeuroImage. 54:787–793. 2011.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Nizari S, Carare RO, Romero IA and Hawkes
CA: 3D reconstruction of the neurovascular unit reveals
differential loss of cholinergic innervation in the cortex and
hippocampus of the adult mouse brain. Front Aging Neurosci.
11(172)2019.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Han Q, Li G, Ip MS, Zhang Y, Zhen Z, Mak
JC and Zhang N: Haemin attenuates intermittent hypoxia-induced
cardiac injury via inhibiting mitochondrial fission. J Cell Mol
Med. 22:2717–2726. 2018.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Hull TD, Boddu R, Guo L, Tisher CC,
Traylor AM, Patel B, Joseph R, Prabhu SD, Suliman HB, Piantadosi
CA, et al: Heme oxygenase-1 regulates mitochondrial quality control
in the heart. JCI Insight. 1(e85817)2016.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Delbarba A, Abate G, Prandelli C, Marziano
M, Buizza L, Arce Varas N, Novelli A, Cuetos F, Martinez C, Lanni
C, et al: Mitochondrial alterations in peripheral mononuclear blood
cells from Alzheimer's disease and mild cognitive impairment
patients. Oxid Med Cell Longev. 2016(5923938)2016.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Petersen MH, Budtz-Jørgensen E, Sørensen
SA, Nielsen JE, Hjermind LE, Vinther-Jensen T, Nielsen SM and
Nørremølle A: Reduction in mitochondrial DNA copy number in
peripheral leukocytes after onset of Huntington's disease.
Mitochondrion. 17:14–21. 2014.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Leuner K, Pantel J, Frey C, Schindowski K,
Schulz K, Wegat T, Maurer K, Eckert A and Müller WE: Enhanced
apoptosis, oxidative stress and mitochondrial dysfunction in
lymphocytes as potential biomarkers for Alzheimer's disease. J
Neural Transm. Suppl:207–215. 2007.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Ciccone MM, Scicchitano P, Zito A, Cortese
F, Boninfante B, Falcone VA, Quaranta VN, Ventura VA, Zucano A, Di
Serio F, et al: Correlation between inflammatory markers of
atherosclerosis and carotid intima-media thickness in Obstructive
Sleep Apnea. Molecules. 19:1651–1662. 2014.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Yang Q, Wang Y, Feng J, Cao J and Chen B:
Intermittent hypoxia from obstructive sleep apnea may cause
neuronal impairment and dysfunction in central nervous system: The
potential roles played by microglia. Neuropsychiatr Dis Treat.
9:1077–1086. 2013.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Lee EJ, Heo W, Kim JY, Kim H, Kang MJ, Kim
BR, Kim JH, Park DY, Kim CH, Yoon JH and Cho HJ: Alteration of
inflammatory mediators in the upper and lower airways under chronic
intermittent hypoxia: Preliminary animal study. Mediators Inflamm.
2017(4327237)2017.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Ryan S, Taylor CT and McNicholas WT:
Selective activation of inflammatory pathways by intermittent
hypoxia in obstructive sleep apnea syndrome. Circulation.
112:2660–2667. 2005.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Morgan MJ and Liu ZG: Crosstalk of
reactive oxygen species and NF-κB signaling. Cell Res. 21:103–115.
2011.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Schoonbroodt S, Ferreira V, Best-Belpomme
M, Boelaert JR, Legrand-Poels S, Korner M and Piette J: Crucial
role of the amino-terminal tyrosine residue 42 and the
carboxyl-terminal PEST domain of I kappa B alpha in NF-kappa B
activation by an oxidative stress. J Immunol. 164:4292–4300.
2000.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. Cell. 132:344–362. 2008.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Fang L, Choudhary S, Zhao Y, Edeh CB, Yang
C, Boldogh I and Brasier AR: ATM regulates NF-κB-dependent
immediate-early genes via RelA Ser 276 phosphorylation coupled to
CDK9 promoter recruitment. Nucleic Acids Res. 42:8416–8432.
2014.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Williams A and Scharf SM: Obstructive
sleep apnea, cardiovascular disease, and inflammation-is NF-kappaB
the key? Sleep Breath. 11:69–76. 2007.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Anrather J, Racchumi G and Iadecola C:
NF-kappaB regulates phagocytic NADPH oxidase by inducing the
expression of gp91phox. J Biol Chem. 281:5657–5667. 2006.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Vasconcelos AR, Yshii LM, Viel TA, Buck
HS, Mattson MP, Scavone C and Kawamoto EM: Intermittent fasting
attenuates lipopolysaccharide-induced neuroinflammation and memory
impairment. J Neuroinflammation. 11(85)2014.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Do K, Laing BT, Landry T, Bunner W,
Mersaud N, Matsubara T, Li P, Yuan Y, Lu Q and Huang H: The effects
of exercise on hypothalamic neurodegeneration of Alzheimer's
disease mouse model. PLoS One. 13(e0190205)2018.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Wang J, Ming H, Chen R, Ju JM, Peng WD,
Zhang GX and Liu CF: CIH-induced neurocognitive impairments are
associated with hippocampal Ca(2+) overload, apoptosis, and
dephosphorylation of ERK1/2 and CREB that are mediated by
overactivation of NMDARs. Brain Res. 1625:64–72. 2015.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Qi G, Mi Y, Wang Y, Li R, Huang S, Li X
and Liu X: Neuroprotective action of tea polyphenols on oxidative
stress-induced apoptosis through the activation of the
TrkB/CREB/BDNF pathway and Keap1/Nrf2 signaling pathway in SH-SY5Y
cells and mice brain. Food Funct. 8:4421–4432. 2017.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Yin X, Zhang X, Lv C, Li C, Yu Y, Wang X
and Han F: Protocatechuic acid ameliorates neurocognitive functions
impairment induced by chronic intermittent hypoxia. Sci Rep.
5(14507)2015.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Cervellati F, Cervellati C, Romani A,
Cremonini E, Sticozzi C, Belmonte G, Pessina F and Valacchi G:
Hypoxia induces cell damage via oxidative stress in retinal
epithelial cells. Free Radic Res. 48:303–312. 2014.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Semenza GL and Prabhakar NR:
HIF-1-dependent respiratory, cardiovascular, and redox responses to
chronic intermittent hypoxia. Antioxid Redox Signal. 9:1391–1396.
2007.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Jung SN, Yang WK, Kim J, Kim HS, Kim EJ,
Yun H, Park H, Kim SS, Choe W, Kang I and Ha J: Reactive oxygen
species stabilize hypoxia-inducible factor-1 alpha protein and
stimulate transcriptional activity via AMP-activated protein kinase
in DU145 human prostate cancer cells. Carcinogenesis. 29:713–721.
2008.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Rabinovitch RC, Samborska B, Faubert B, Ma
EH, Gravel SP, Andrzejewski S, Raissi TC, Pause A, St-Pierre J and
Jones RG: AMPK maintains cellular metabolic homeostasis through
regulation of mitochondrial reactive oxygen species. Cell Rep.
21:1–9. 2017.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Guo H, Cao J, Li J, Yang X, Jiang J, Feng
J, Li S, Zhang J and Chen B: Lymphocytes from intermittent
hypoxia-exposed rats increase the apoptotic signals in endothelial
cells via oxidative and inflammatory injury in vitro. Sleep Breath.
19:969–976. 2015.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Bianchi G, Di Giulio C, Rapino C, Rapino
M, Antonucci A and Cataldi A: p53 and p66 proteins compete for
hypoxia-inducible factor 1 alpha stabilization in young and old rat
hearts exposed to intermittent hypoxia. Gerontology. 52:17–23.
2006.PubMed/NCBI View Article : Google Scholar
|
|
104
|
da Rosa DP, Forgiarini LF, e Silva MB,
Fiori CZ, Andrade CF, Martinez D and Marroni NP: Antioxidants
inhibit the inflammatory and apoptotic processes in an intermittent
hypoxia model of sleep apnea. Inflamm Res. 64:21–29.
2015.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Pan W and Kastin AJ: Can sleep apnea cause
Alzheimer's disease? Neurosci Biobehav Rev. 47:656–669.
2014.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Andrade AG, Bubu OM, Varga AW and Osorio
RS: The relationship between obstructive sleep apnea and
Alzheimer's disease. J Alzheimers Dis. 64 (Suppl 1):S255–S270.
2018.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Casagrande R, Stern P, Diehn M, Shamu C,
Osario M, Zúñiga M, Brown PO and Ploegh H: Degradation of proteins
from the ER of S. cerevisiae requires an intact unfolded protein
response pathway. Mol cell. 5:729–735. 2000.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Walter P and Ron D: The unfolded protein
response: From stress pathway to homeostatic regulation. Science.
334:1081–1086. 2011.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Chou YT, Zhan G, Zhu Y, Fenik P, Panossian
L, Li Y, Zhang J and Veasey S: C/EBP homologous binding protein
(CHOP) underlies neural injury in sleep apnea model. Sleep.
36:481–492. 2013.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Yao ZH, Kang X, Yang L, Niu Y, Lu Y, Gong
CX, Tian Q and Wang JZ: Phenylbutyric acid protects against spatial
memory deficits in a model of repeated electroconvulsive therapy.
Curr Neurovasc Res. 11:156–167. 2014.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Nosyreva E and Kavalali ET:
Activity-dependent augmentation of spontaneous neurotransmission
during endoplasmic reticulum stress. J Neurosci. 30:7358–7368.
2010.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Archbold KH, Borghesani PR, Mahurin RK,
Kapur VK and Landis CA: Neural activation patterns during working
memory tasks and OSA disease severity: Preliminary findings. J Clin
Sleep Med. 5:21–27. 2009.PubMed/NCBI
|
|
114
|
Miller JF, Neufang M, Solway A, Brandt A,
Trippel M, Mader I, Hefft S, Merkow M, Polyn SM, Jacobs J, et al:
Neural activity in human hippocampal formation reveals the spatial
context of retrieved memories. Science. 342:1111–1114.
2013.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Pena F and Ramirez JM: Hypoxia-induced
changes in neuronal network properties. Mol Neurobiol. 32:251–283.
2005.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Clark RS, Kochanek PM, Dixon CE, Chen M,
Marion DW, Heineman S, DeKosky ST and Graham SH: Early
neuropathologic effects of mild or moderate hypoxemia after
controlled cortical impact injury in rats. J Neurotrauma.
14:179–189. 1997.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Fung SJ, Xi MC, Zhang JH, Sampogna S,
Yamuy J, Morales FR and Chase MH: Apnea promotes glutamate-induced
excitotoxicity in hippocampal neurons. Brain Res. 1179:42–50.
2007.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Socodato R, Portugal CC, Rodrigues A,
Henriques J, Rodrigues C, Figueira C and Relvas JB: Redox tuning of
Ca2+ signaling in microglia drives glutamate release
during hypoxia. Free Radic Biol Med. 118:137–149. 2018.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Macey PM, Sarma MK, Nagarajan R, Aysola R,
Siegel JM, Harper RM and Thomas MA: Obstructive sleep apnea is
associated with low GABA and high glutamate in the insular cortex.
J Sleep Res. 25:390–394. 2016.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Opie GM, Catcheside PG, Usmani ZA, Ridding
MC and Semmler JG: Motor cortex plasticity induced by theta burst
stimulation is impaired in patients with obstructive sleep apnoea.
Eur J Neurosci. 37:1844–1852. 2013.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Gozal D, Nair D and Goldbart AD: Physical
activity attenuates intermittent hypoxia-induced spatial learning
deficits and oxidative stress. Am J Respir Crit Care Med.
182:104–112. 2010.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Toraldo DM, Di Michele L, Ralli M,
Arigliani M, Passali GC, De Benedetto M and Passali D: Obstructive
sleep apnea syndrome in the pediatric age: The role of the
pneumologist. Eur Rev Med Pharmacol Sci. 23 (1 Suppl):S15–S18.
2019.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Li Y, Ye J, Han D, Zhao D, Cao X, Orr J,
Jen R, Deacon-Diaz N, Sands SA, Owens R and Malhotra A: The effect
of upper airway surgery on loop gain in obstructive sleep apnea. J
Clin Sleep Med. 15:907–913. 2019.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Epstein LJ, Kristo D, Strollo PJ Jr,
Friedman N, Malhotra A, Patil SP, Ramar K, Rogers R, Schwab RJ,
Weaver EM, et al: Clinical guideline for the evaluation, management
and long-term care of obstructive sleep apnea in adults. J Clin
Sleep Med. 5:263–276. 2009.PubMed/NCBI
|
|
125
|
Carlucci A, Ceriana P, Mancini M, Cirio S,
Pierucci P, D'Artavilla Lupo N, Gadaleta F, Morrone E and Fanfulla
F: Efficacy of Bilevel-auto treatment in patients with obstructive
sleep apnea not responsive to or intolerant of continuous positive
airway pressure ventilation. J Clin Sleep Med. 11:981–985.
2015.PubMed/NCBI View Article : Google Scholar
|