Introduction
Esophageal squamous cell carcinoma (ESCC) is a
common type of cancer in China and other parts of the world, which
is associated with a poor prognosis for the affected patients
(1). ESCC is the fourth most common
type of cancer in China, accounting for ~375,000 deaths in 2015,
which represented ~13% of all cancer-related deaths (2). ESCC is a frequently recurrent cancer
with a high mortality rate and an average 5-year survival rate of
>20% (3), which highlights the
importance of discovering novel therapeutic strategies to treat the
disease.
Tyrosine-protein kinase receptor UFO (AXL) is a
member of the TYRO3, AXL and MERTK (TAM) family of receptor
tyrosine kinases (RTKs), which is activated by its ligand, growth
arrest-specific protein 6 (GAS6), leading to the activation of
several downstream signaling pathways, which is dependent on the
cell type, but predominantly includes the PI3K/AKT and
mitogen-activated protein kinase (MEK)/ERK signaling pathways. AXL
has been previously implicated in the pathophysiology of numerous
types of cancer, including breast, gastric, prostate, ovarian and
lung cancer (4,5), where it was discovered to promote
cancer cell survival, angiogenesis, metastasis and drug-resistance
(6-8).
AXL was revealed to be overexpressed in several ESCC
tumors, which was associated with an adverse prognosis (9,10) and
drug resistance (11). In addition,
our previous study demonstrated that AXL was associated with ESCC
epithelial-mesenchymal transition (EMT) by analyzing online ESCC
gene sets using Gene Set Enrichment Analysis (12). The present study revealed that
targeting AXL with R428, the most selective AXL inhibitor currently
available (13), suppressed ESCC
tumor cell proliferation and invasion through inhibiting matrix
metalloproteinase (MMP)2 and MMP9 activity.
Materials and methods
Cell lines and reagents
TE1 and KYSE150 ESCC cell lines were purchased from
The Cell Bank of Type Culture Collection of the Chinese Academy of
Sciences. TE1 cells were cultured in RPMI-1640 medium (Gibco;
Thermo Fisher Scientific, Inc.), supplemented with 10% FBS
(Zhejiang Tianhang Biotechnology Co., Ltd.), whereas KYSE150 cells
were cultured in a 1:1 mixture of Ham's F12 medium (Gibco; Thermo
Fisher Scientific, Inc.) and RPMI-1640 medium, supplemented with 5%
FBS. Both cell lines were also supplemented with 100 U/ml
penicillin and 0.1 mg/ml streptomycin (Beyotime Institute of
Biotechnology) and maintained in a humidified atmosphere at 37˚C
with 5% CO2. Cell line authentication was achieved by
genetic profiling using the polymorphic short tandem repeat (STR)
method at the Forensic Science Center, Jining Medical University
(Shandong, China). The results (Table
SI) revealed that these cells were identical to the original
cells, whereas the STR profile of KYSE150 cells was identical to
that reported by Wu et al (14). R428 was purchased from
MedChemExpress and the concentration used was the lowest it could
be to suppress the activation of AXL and its downstream signaling
pathways (≤5 µM in cell proliferation assay and ≤3 µM in other
assays), these concentrations have also been widely used in other
studies involving R428 (15-17).
The MTT reagent, dimethylformamide (DMF), DMSO and sodium dodecyl
sulfate (SDS) were purchased from Sigma-Aldrich (Merck KGaA).
MTT assay
The MTT assay was performed according to the method
described by Hansen et al (18). Briefly, 3x103 cells/well
were plated into 96-well plates and incubated overnight.
Subsequently, the cells were treated with R428 or vehicle (DMSO)
and incubated for 24, 48 or 72 h at 37˚C. Then, 25 µl MTT (5 mg/ml
MTT in sterile PBS) was added to the medium and cells were
incubated for 2 h at 37˚C. The reaction was stopped, and the cells
were lysed with the addition of 100 µl lysis buffer consisting of
20% SDS in a water/dimethylformamide (1:1) solution (pH 4.7). Cell
lysates were incubated at 37˚C overnight to allow for cell lysis
and dye solubilization. The absorbance (OD) was measured at 570 and
630 nm, as a reference, using a Multiskan Mk3 microplate reader
(Thermo Fisher Scientific, Inc.). The absorbance of each well was
calculated as OD570-OD630. MTT assays were
performed in triplicate, with three technical repeats.
Colony formation assay
A total of 1x103 cells/well were seeded
into six-well plates and incubated overnight. Then, the cells were
treated with R428 or vehicle (DMSO) for 24 h at 37˚C before the
medium was changed to normal growth medium. Following 8 days of
incubation for KYSE150 cells, and 14 days for TE1, the cells were
washed with PBS twice and stained with crystal violet staining
solution at room temperature for 5 min. The number of colonies
formed (>50 cells) was counted under a light microscope at x40
magnification.
Wound healing assay
TE1 and KYSE150 cells were harvested using trypsin
and cell suspensions were seeded into six-well plates at a density
of 1x106 cells/well. Upon the cells reaching 90%
confluence, a 10-µl pipette tip was used to scratch a wound on the
surface of the cell monolayer. After washing with pre-warmed PBS,
the cells were incubated with their respective culture medium
containing 1% FBS and R428 or vehicle (DMSO). Images of the wells
were obtained at 0 h, and then again at 14 h for TE1 cells and 24 h
for KYSE150 cells, in the same position as that at 0 h under a
light microscope at x40 magnification. The images were analyzed
using ImageJ software (version 1.52a; National Institutes of
Health) to obtain the area. The migration of cells towards the open
gap was determined as the area at 0 h-the area at 14 h for TE1
cells and the area at 0 h-the area at 24 h for KYSE150 cells. The
assay was performed in triplicate, with three technical repeats.
The migratory rate of the control cells was set at 100% and the
migratory rate of the other plates was normalized to the control
cells.
Matrigel assay
To investigate cell invasion, 1x106 TE1
or KYSE150 cells in normal medium were pretreated with R428 for 3 h
at 37˚C, then plated into the upper chambers of Transwell plates
(8-µm pore size; 6.5 mm diameter; Corning Life Sciences) precoated
with 500 µg/ml Matrigel (Corning Life Sciences) for 1 h at 37˚C, in
medium supplemented with 0.3% FBS. Medium supplemented with 10% FBS
was plated into the lower chambers. Following incubation for 16 h
(for TE1 cells) or 24 h (for KYSE150 cells), the invasive cells
were fixed with 100% methanol for 10 min at room temperature,
stained with 0.5% crystal violet for 30 min at room temperature and
washed with water to remove the excess dye. Stained cells were
visualized using an optical microscope under x100 magnification in
five randomly selected fields of view. The invasive rate of the
control cells was set at 100% and the invasive rate of the other
groups was normalized to the control cells.
Western blotting
Western blotting was performed as previously
described (19). Briefly, total
cellular protein was extracted from cells using RIPA lysis buffer
containing protease inhibitors and phosphatase inhibitors.
Extracted protein (20 µg per lane) was separated via 10% SDS-PAGE
and transferred onto PVDF membranes. The membrane was blocked for 1
h in 5% non-fat milk solution at room temperature and then
incubated with the indicated primary antibodies overnight at
4°C. Following the primary antibody incubation, the
membranes were incubated with horseradish peroxidase-conjugated
secondary antibodies at room temperature for 1 h. Protein bands
were visualized using an ECL reagent (cat. no. CW0049; CoWin
Biosciences). GAPDH was used as the endogenous loading control.
Anti-AXL (cat. no. 8661; 1:1,000), anti-phosphorylated (p)-AKT
(cat. no. 4060; 1:1,000) and anti-AKT (cat. no. 4691; 1:1,000)
primary antibodies were purchased from Cell Signaling Technology,
Inc. The anti-p-AXL antibody (cat. no. AF2228; 1:200) was obtained
from R&D Systems China Co., Ltd. Anti-GAPDH antibody (Clone
OT12DG; cat. no. TA-08; 1:1,000) was purchased from OriGene
Technologies, Inc. Anti-ERK1+ERK2 (cat. no. ab184699; 1:10,000) and
anti-p-ERK1+ERK2 (cat. no. ab76299; 1:10,000) primary antibodies
were purchased from Abcam.
Gelatin zymography to determine the
activity of MMP2 and MMP9
MMP2 and MMP9 activity from the collected samples
was assayed using 0.05% gelatin zymography, as previously described
(20). Briefly, 5x105
cells were cultured in six-well plates overnight, and then R428 was
added to the medium and incubated for 24 h at 37˚C. Following
incubation, the medium was changed to 700 µl serum-free medium
containing R428. Following incubation for 18 h, the medium was
collected for analysis. The protein concentration of each sample
was measured using a bicinchoninic acid protein assay kit (cat. no.
P0010; Beyotime Institute of Biotechnology) and equal amounts of
total protein were separated via 10% SDS-PAGE gels co-polymerized
with 0.05% gelatin. After electrophoresis, the gels were washed in
2.5% Triton X-100 three times for 1.5 h at room temperature to
remove the SDS and regain activity. The washed gels were then
bathed in proteolysis buffer (50 mM CaCl2, 0.5 M NaCl,
50 mM Tris; pH 7.8) and incubated at 37˚C for 15 h. Following
incubation, the gels were rinsed in 2.5% Triton X-100 solution and
stained at room temperature with Coomassie blue (45% methanol,
44.75% H2O, 10% acetic acid, 0.25% Coomassie blue R-250)
for 1 h on a rotator. Gels were de-stained with a solution
containing 40% methanol, 7.5% acetic acid and 52.5% H2O
for a few hours until the bands became clear. The gelatin inside
the gel bound to the Coomassie blue, dyeing the gel blue and
leaving the MMP-digested regions white in color. Bands were scanned
using a Bio-Rad ChemiDoc™ imager (Bio-Rad Laboratories, Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cells using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocol. First-strand cDNA was synthesized from
0.5 µg RNA using a PrimeScript™ RT Master mix (cat. no. RR036A,
Takara Biotechnology Co., Ltd.), by incubation at 37˚C for 15 min
and then 85˚C for 5 sec, according to the manufacturer's
instructions. qPCR was subsequently performed in triplicate using
the SYBR Green PCR Master mix (CoWin Biosciences) on a QuantStudio™
5 Real-Time PCR system (Thermo Fisher Scientific, Inc.). The
following primer pairs were used: MMP2 forward,
5'-TACAGGATCATTGGCTACACACC-3' and reverse,
5'-GGTCACATCGCTCCAGACT-3'; MMP9 forward, 5'-GGGACGCAGACATCGTCATC-3'
and reverse, 5'-TCGTCATCGTCGAAATGGGC-3'; and GAPDH forward,
5'-GGAGCGAGATCCCTCCAAAAT-3' and reverse,
5'-GGCTGTTGTCATACTTCTCATGG-3'. Each qPCR reaction was performed in
a final volume of 20 µl, containing 10 µl SYBR Green Master mix
(2X), 0.5 µl forward and reverse primers (10 µM), 8 µl DEPC treated
water and 1 µl cDNA. The following thermocycling conditions were
used for the qPCR: 95˚C for 10 min, 40 cycles of 95˚C for 15 sec
and 60˚C for 1 min. Data were normalized to GAPDH expression
levels, the housekeeping gene. The relative expression of target
genes was calculated by the comparative 2-ΔΔCq method
(21).
Statistical analysis
Statistical differences between experimental and
control groups were analyzed using an unpaired two-tailed Student's
t-test (2 groups) or a one-way ANOVA (>2 groups) followed by a
multiple post hoc comparisions test (Dunnett's test) using GraphPad
Prism 8 software (GraphPad Software, Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
AXL is activated in ESCC cells and
R428 suppresses AXL activation
To determine the effect of R428 on ESCC tumor cells,
the expression levels of AXL in TE1 and KYSE150 cells were
determined using western blotting. Both cell lines were found to
express AXL and activated AXL (AXL phosphorylated at tyrosine
residue 779; Fig. 1A). It was also
confirmed that R428 treatment suppressed AXL activation in both TE1
and KYSE150 cells in a dose-dependent manner (Fig. 1B-E). AKT and ERK signaling are two
major branches of AXL signaling (4,22,23),
thus their responses to R428 were determined; it was identified
that R428 treatment suppressed AKT and ERK signaling activation in
a dose-dependent manner (Fig.
1B-E).
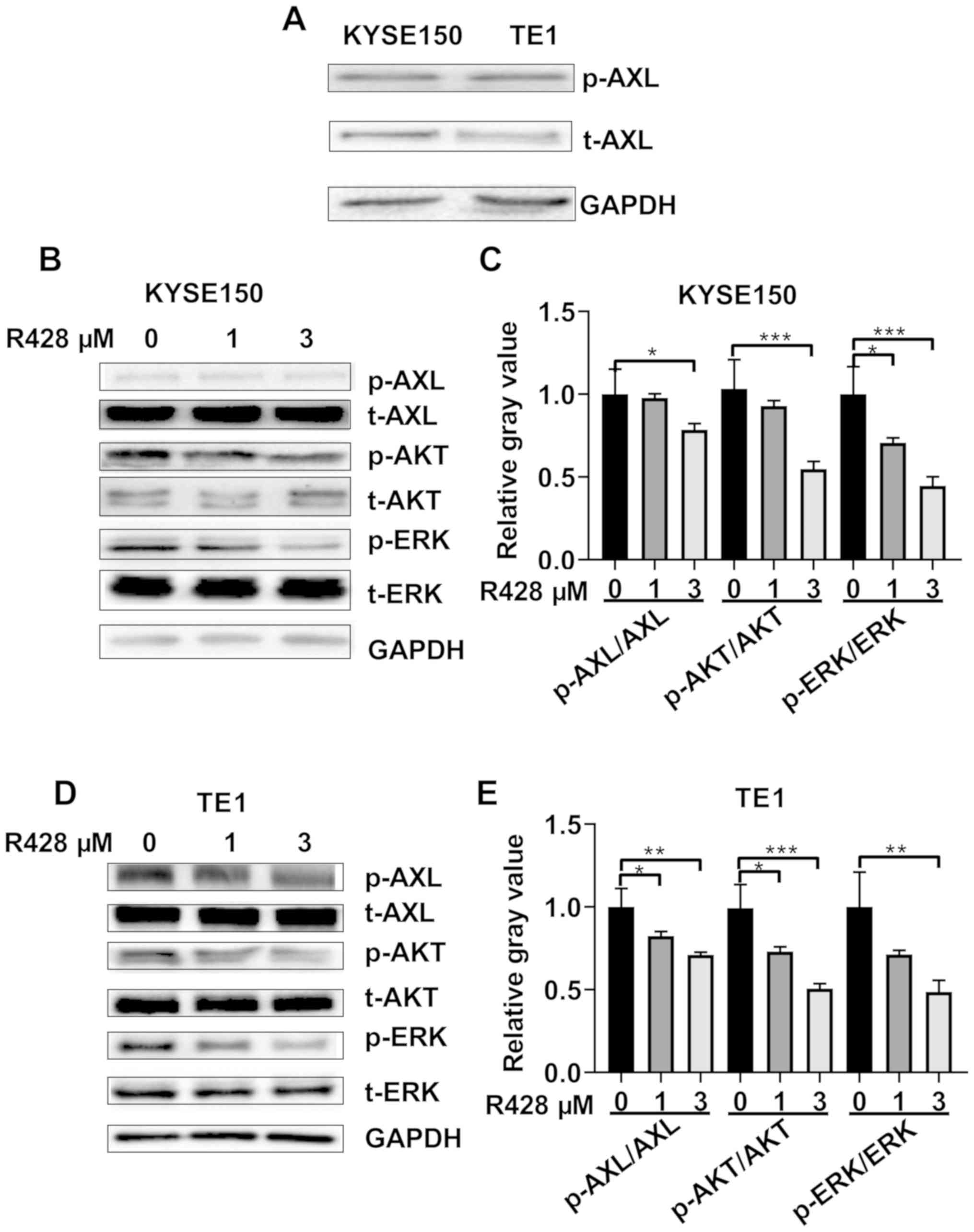 | Figure 1R428 suppresses the activation of AXL
in esophageal squamous cell carcinoma cells. (A) Expression levels
of t-AXL and p-AXL in KYSE150 and TE1 cells. (B) R428 treatment
inhibited AXL activation, in addition to AKT and ERK activation, in
a dose-dependent manner in KYSE150 cells. (C) Semi-quantitative
analysis of the expression levels of p-AXL, p-AKT and p-ERK. (D)
R428 inhibited AXL activation, in addition to AKT and ERK
activation in a dose-dependent manner in TE1 cells. (E)
Semi-quantitative analysis of the expression levels of p-AXL, p-AKT
and p-ERK. *P<0.05, **P<0.01,
***P<0.001. AXL, tyrosine-protein kinase receptor
UFO; t, total; p, phosphorylated. |
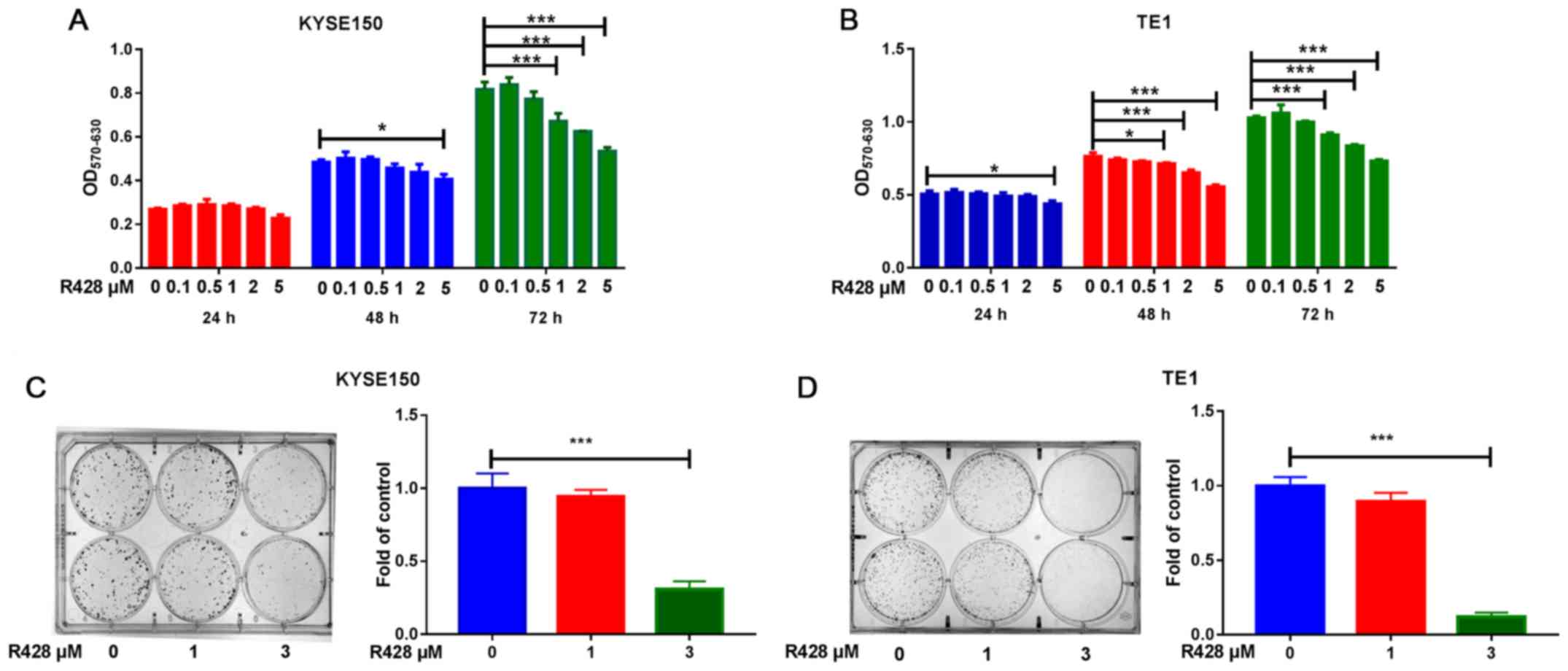
R428 treatment reduces cell
proliferation in ESCC cells in vitro
The effects of R428 treatment on cell proliferation
were subsequently investigated. The results of the MTT assay
revealed that R428 treatment reduced cell viability in a time- and
dose-dependent manner in both cell lines (Fig. 2A and B). Furthermore, the colony formation assay
demonstrated that R428 treatment suppressed cell colony formation
in a dose-dependent manner (Fig. 2C
and D), suggesting that AXL
signaling may enhance ESCC proliferation.
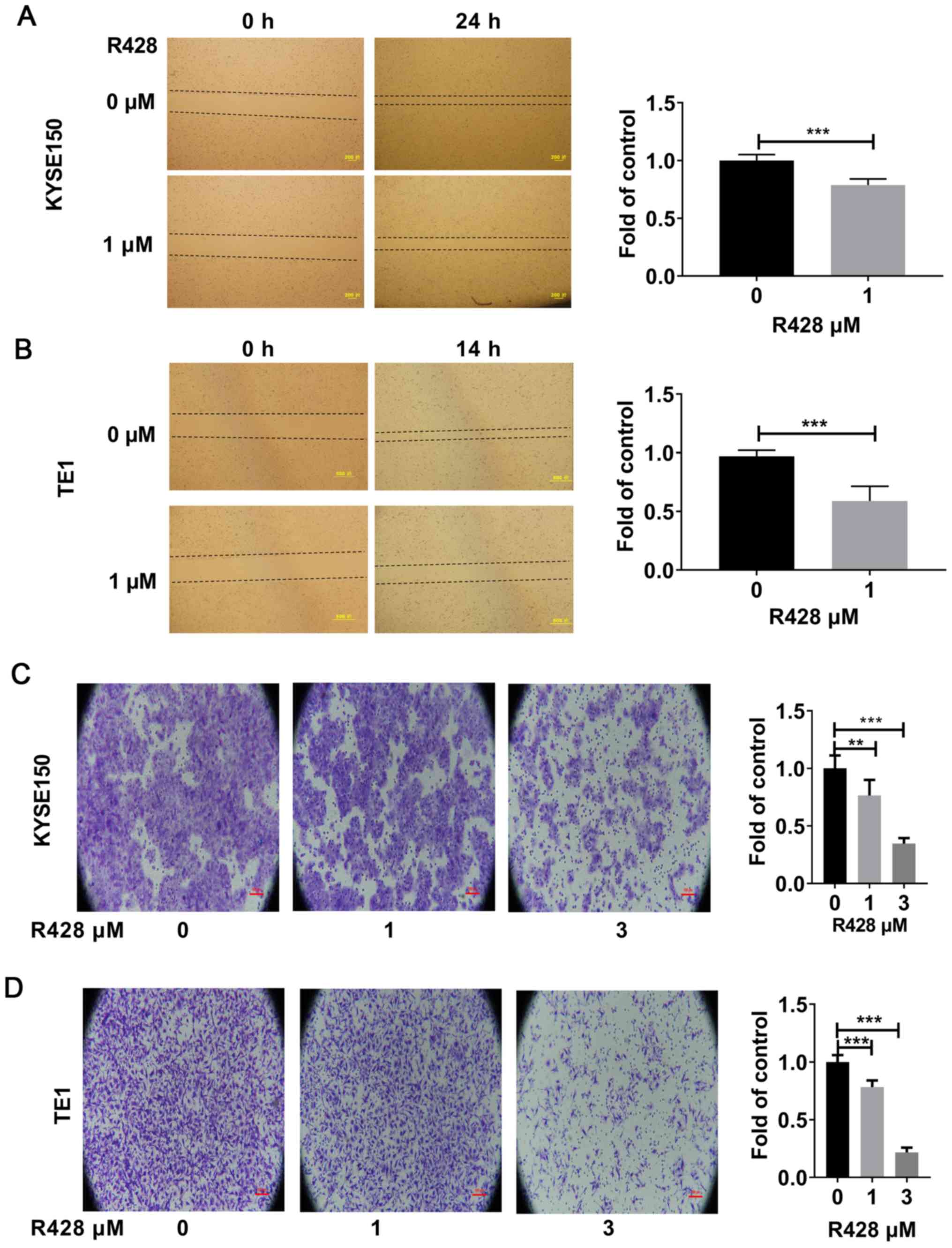
R428 treatment inhibits ESCC cell
migration and invasion
The effects of R428 treatment on ESCC cell migration
and invasion were subsequently investigated. The results of the
wound healing assay revealed that R428 treatment inhibited the
migration of both KYSE150 and TE1 cells (Fig. 3A and B). In addition, the Matrigel assay also
demonstrated that both 1 and 3 µM R428 treatment suppressed tumor
cell invasion (Fig. 3C and D). These results suggested that R428
treatment may significantly inhibit tumor migration and
invasion.
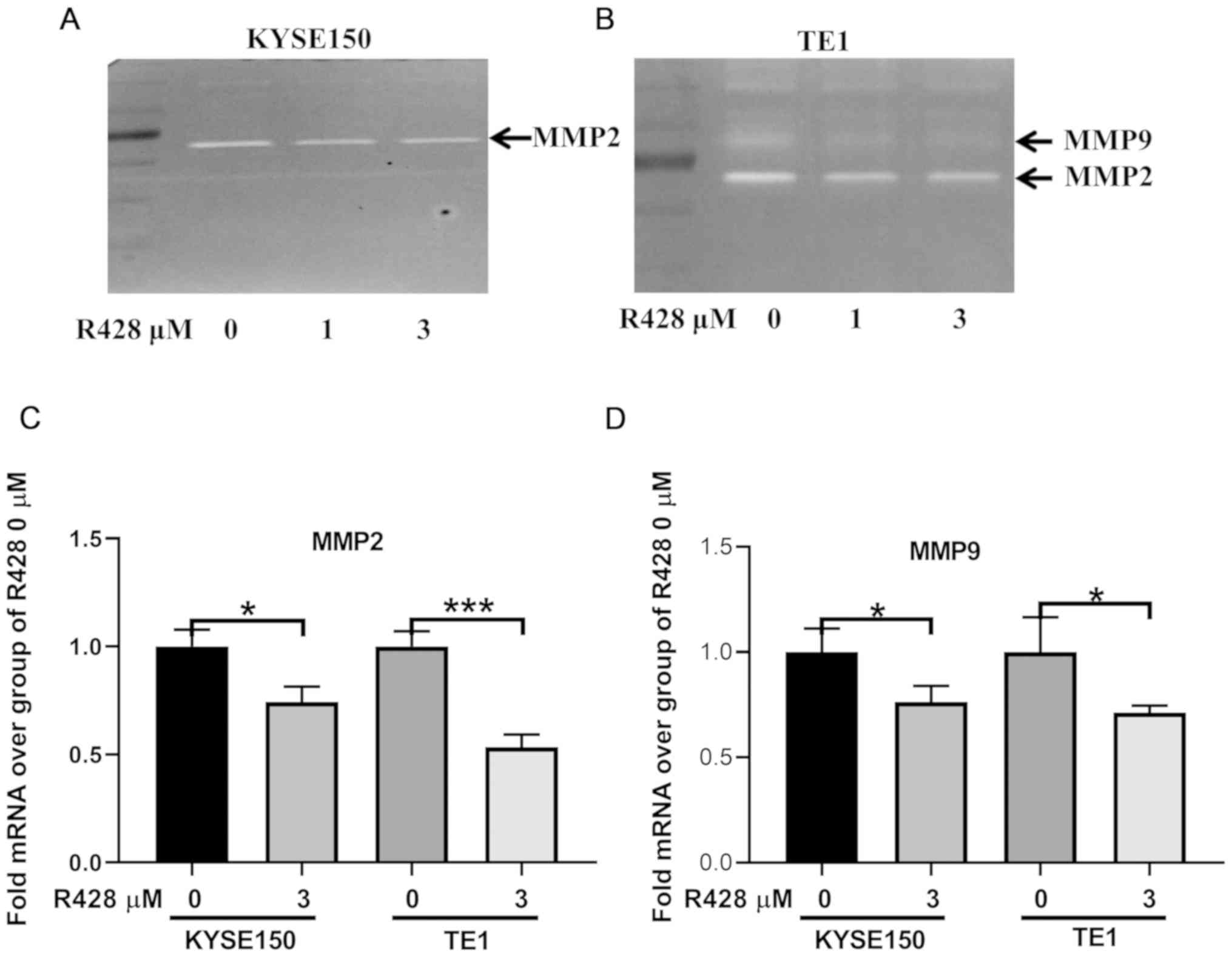
R428 treatment downregulates MMP2 and
MMP9 expression levels
MMP2 and MMP9 are important regulators of tumor
invasion, and both have been previously revealed to be involved in
AXL-mediated tumor cell invasion (24-27).
Thus, the present study aimed to determine whether MMP2 and MMP9
were also involved in the R428-induced suppression of ESCC
invasion. Gelatin zymography demonstrated that R428 treatment
suppressed both MMP2 and MMP9 activity in TE1 cells. In KYSE150
cells, MMP2 activity was also inhibited, whereas MMP9 activity was
not detected (Fig. 4A and B). Furthermore, the mRNA expression levels
of MMP2 and MMP9 were analyzed in both cell lines; R428 treatment
downregulated the expression levels of both MMP2 and MMP9 (Fig. 4C and D).
Discussion
ESCC is a cancer with high mortality rates, for
which no targeted therapies have been approved. R428 treatment is
currently under phase I or II clinical trials in several types of
cancer (28). Previous
immunohistochemistry studies have revealed that AXL was
overexpressed in ESCC tumors (9,10).
Thus, the aim of the present study was to investigate the effect of
R428 treatment on ESCC cancer cells in vitro.
The present study revealed that the two ESCC cell
lines, KYSE150 and TE1, expressed AXL in the activated form. AXL
can be activated by its ligand GAS6 or by other molecules via
atypical activation (23,29). For example, epidermal growth factor
(EGF), which activates the EGF receptor, was discovered to
subsequently transactivate AXL (17). In addition, hepatocyte growth factor
(HGF) was reported to activate AXL by promoting the formation of
HGF receptor MET and AXL complexes (30). However, the pathway involved in the
activation of AXL in the two cell lines requires further
investigations. AXL has been elucidated to be involved in the
proliferation of cancer cells (4,16,31,32).
Consistent with these results, the present study also revealed that
R428 treatment suppressed ESCC proliferation. Of note, Hsieh et
al (9) revealed that ESCC cells
were more sensitive to the AXL inhibitor foretinib compared with
the HER2 inhibitor lapatinib, suggesting that AXL seems a better
target in the treatment of ESCC than HER2.
Patients with solid tumors primarily succumb to
mortality due to metastatic lesions, rather than from the primary
tumor (33). AXL has been
identified to be highly associated with cancer EMT and invasion
(22). Our previous study also
revealed that ESCC tumors expressing high levels of AXL displayed
enhanced EMT properties and the invasive ESCC cells expressed
increased levels of AXL (12).
Thus, it is not surprising that targeting AXL with R428 suppressed
ESCC cancer cell migration and invasion in the present study, which
is also consistent with the study of Yang et al (16). However, in the wound healing assay
1% FBS was used, which may be a limitation. Mechanistically, MMP9
and MMP2 have been reported to mediate, at least partially, the
effect of AXL on cell invasion. In previous studies, AXL was
discovered to upregulate the expression levels of MMP9 and MMP2
(24-27),
which promoted tissue remodeling and cancer invasion. The present
study confirmed the association between MMP2, MMP9 and AXL by
observing that R428 decreased the expression of MMP2 and MMP9. MMP9
was detected by real-time PCR, but not detected by gelatin
zymography in KYSE150 cells, this may be because gelatin zymography
is not a sensitive enough technique for the detection of MMPs. It
would be useful to study whether GAS6, the ligand of AXL, could
promote the proliferation and invasion of ESCC, and whether R428
could suppress the effects of GAS6, which will further confirm the
role of AXL in ESCC progression. However, these experiments were
not performed, which is a limitation in the present study.
In conclusion, ESCC is a common type of cancer with
a poor prognosis, of which there are currently no targeted
therapies approved for the treatment of this disease. The findings
of the present study suggested that R428 treatment may suppress
ESCC tumor cell proliferation and invasion, therefore representing
a potential therapeutic strategy for the treatment of ESCC.
Supplementary Material
STR profiling of TE1 and KYSE150
cells.
Acknowledgements
Not applicable.
Funding
This study was supported by innovation and
entrepreneurship training program for college students of Jining
Medical University (grant nos. cx2016034 and cx2019014); Supporting
Fund for Teachers' Research of Jining Medical University (grant
nos. JY2017KJ040 and JYFC2018KJ010); Research Fund for Lin He's
Academician Workstation of New Medicine and Clinical Translation in
Jining Medical University (grant nos. JYHL2018MS13 and
JYHL2018MS14); Jining Science and Technology Boost the Old and New
Kinetic Energy Conversion Project (grant no. 2018SMNS001), and
Shandong Provincial Natural Science Foundation (grant no.
ZR2019PH039).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
GZ, WC and NL designed the study. SH, YW, CG, MG,
XW, FW and LS performed the experiments. SL, TD and ZD analyzed the
data. GZ and NL prepared the manuscript. All authors read and
reviewed the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Allum WH, Stenning SP, Bancewicz J, Clark
PI and Langley RE: Long-term results of a randomized trial of
surgery with or without preoperative chemotherapy in esophageal
cancer. J Clin Oncol. 27:5062–5067. 2009.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Paccez JD, Vogelsang M, Parker MI and
Zerbini LF: The receptor tyrosine kinase Axl in cancer: Biological
functions and therapeutic implications. Int J Cancer.
134:1024–1033. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Qu X, Liu G, Zhong X, Li X and Zhang Q:
Role of AXL expression in non-small cell lung cancer. Oncol Lett.
12:5085–5091. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Graham DK, DeRyckere D, Davies KD and Earp
HS: The TAM family: Phosphatidylserine sensing receptor tyrosine
kinases gone awry in cancer. Nat Rev Cancer. 14:769–785.
2014.PubMed/NCBI View
Article : Google Scholar
|
|
7
|
Zhang Z, Lee JC, Lin L, Olivas V, Au V,
LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, et al:
Activation of the AXL kinase causes resistance to EGFR-targeted
therapy in lung cancer. Nat Genet. 44:852–860. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
8
|
Duan Y, Hu B, Qiao C, Luo L, Li X, Wang J,
Liu H, Zhou T, Shen B, Lv M and Feng J: Engineered AXL-ECD-Fc
variants that abolish the AXL/Gas6 interaction suppress tumor cell
migration. Oncol Lett. 17:5784–5792. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hsieh MS, Yang PW, Wong LF and Lee JM: The
AXL receptor tyrosine kinase is associated with adverse prognosis
and distant metastasis in esophageal squamous cell carcinoma.
Oncotarget. 7:36956–36970. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Paccez JD, Duncan K, Vava A, Correa RG,
Libermann TA, Parker MI and Zerbini LF: Inactivation of GSK3β and
activation of NF-κB pathway via Axl represents an important
mediator of tumorigenesis in esophageal squamous cell carcinoma.
Mol Biol Cell. 26:821–831. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Elkabets M, Pazarentzos E, Juric D, Sheng
Q, Pelossof RA, Brook S, Benzaken AO, Rodon J, Morse N, Yan JJ, et
al: AXL mediates resistance to PI3Kα inhibition by activating the
EGFR/PKC/mTOR Axis in head and neck and esophageal squamous cell
carcinomas. Cancer Cell. 27:533–546. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhang G, Kong X, Wang M, et al: AXL is a
marker for epithelial-mesenchymal transition in esophageal squamous
cell carcinoma. Oncol Lett. 15:1900–1906. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Myers SH, Brunton VG and Unciti-Broceta A:
AXL inhibitors in cancer: A medicinal chemistry perspective. J Med
Chem. 59:3593–3608. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wu Y, Hu L, Liang Y, Li J, Wang K, Chen X,
Meng H, Guan X, Yang K and Bai Y: Up-regulation of lncRNA CASC9
promotes esophageal squamous cell carcinoma growth by negatively
regulating PDCD4 expression through EZH2. Mol Cancer.
16(150)2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Holland SJ, Pan A, Franci C, Hu Y, Chang
B, Li W, Duan M, Torneros A, Yu J, Heckrodt TJ, et al: R428, a
selective small molecule inhibitor of Axl kinase, blocks tumor
spread and prolongs survival in models of metastatic breast cancer.
Cancer Res. 70:1544–1554. 2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Yang PW, Liu YC, Chang YH, Lin CC, Huang
PM, Hua KT, Lee JM and Hsieh MS: Cabozantinib (XL184) and R428
(BGB324) inhibit the growth of esophageal squamous cell carcinoma
(ESCC). Front Oncol. 9(1138)2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Meyer AS, Miller MA, Gertler FB and
Lauffenburger DA: The receptor AXL diversifies EGFR signaling and
limits the response to EGFR-targeted inhibitors in Triple-negative
breast cancer cells. Sci Signal. 6(ra66)2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Hansen MB, Nielsen SE and Berg K:
Re-examination and further development of a precise and rapid dye
method for measuring cell growth/cell kill. J Immunol Methods.
119:203–210. 1989.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Kong LL, Man DM, Wang T, Zhang GA and Cui
W: siRNA targeting RBP2 inhibits expression, proliferation,
tumorigenicity and invasion in thyroid carcinoma cells. Oncol Lett.
10:3393–3398. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Tang Z, Yang L, Wang Y, Xue R, Zhang J,
Huang W, Chen PC and Sung KL: Contributions of different
intraarticular tissues to the acute phase elevation of synovial
fluid MMP-2 following rat ACL rupture. J Orthop Res. 27:243–248.
2009.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Antony J and Huang RY: AXL-Driven EMT
state as a targetable conduit in cancer. Cancer Res. 77:3725–3732.
2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zhang G, Wang M, Zhao H and Cui W:
Function of Axl receptor tyrosine kinase in non-small cell lung
cancer. Oncol Lett. 15:2726–2734. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Tai KY, Shieh YS, Lee CS, Shiah SG and Wu
CW: Axl promotes cell invasion by inducing MMP-9 activity through
activation of NF-kappaB and Brg-1. Oncogene. 27:4044–4055.
2008.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Reichl P, Dengler M, van Zijl F, Huber H,
Führlinger G, Reichel C, Sieghart W, Peck-Radosavljevic M,
Grubinger M and Mikulits W: Axl activates autocrine transforming
growth factor-β signaling in hepatocellular carcinoma. Hepatology.
61:930–941. 2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Chiu KC, Lee CH, Liu SY, Yeh CT, Huang RY,
Yuh DY, Cheng JC, Chou YT and Shieh YS: Protumoral effect of
macrophage through Axl activation on mucoepidermoid carcinoma. J
Oral Pathol Med. 43:538–544. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Han J, Tian R, Yong B, Luo C, Tan P, Shen
J and Peng T: Gas6/Axl mediates tumor cell apoptosis, migration and
invasion and predicts the clinical outcome of osteosarcoma
patients. Biochem Biophys Res Commun. 435:493–500. 2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wu G, Ma Z, Hu W, Wang D, Gong B, Fan C,
Jiang S, Li T, Gao J and Yang Y: Molecular insights of Gas6/TAM in
cancer development and therapy. Cell Death Dis.
8(e2700)2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Linger RM, Keating AK, Earp HS and Graham
DK: TAM receptor tyrosine kinases: Biologic functions, signaling,
and potential therapeutic targeting in human cancer. Adv Cancer
Res. 100:35–83. 2008.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhang G, Wang M, Zhao H and Cui W:
Function of Axl receptor tyrosine kinase in non-small cell lung
cancer. Oncol Lett. 15:2726–2734. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Verma A, Warner SL, Vankayalapati H,
Bearss DJ and Sharma S: Targeting Axl and Mer kinases in cancer.
Mol Cancer Ther. 10:1763–1773. 2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Scaltriti M, Elkabets M and Baselga J:
Molecular pathways: AXL, a membrane receptor mediator of resistance
to therapy. Clin Cancer Res. 22:1313–1317. 2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000.PubMed/NCBI View Article : Google Scholar
|