Introduction
Chronic obstructive pulmonary disease (COPD) occurs
in >380 million individuals globally, especially amongst adults
aged >30 years (1). The global
prevalence of COPD is expected to increase continuously during the
next few decades due to an ageing population, uncontrolled smoking
prevalence in developing countries and additional environmental
exposures such as air pollution and biomass fuel exposure (2). It is estimated that in 2030, more than
4.5 million deaths annually will be attributable to COPD and
related conditions worldwide (2),
and by 2060 there may be over 5.4 million deaths annually from COPD
and related conditions globally (3). COPD is a progressive lung disease
characterized by persistent airway inflammation, accompanied by
irreversible airflow obstruction, including chronic bronchitis and
emphysema (4,5). Despite the rising the incidence of
COPD, there are currently no specific drugs to treat and protect
against COPD (5-7).
Therefore, the identification of novel agents for targeting this
disease is urgently needed.
Elevated levels of pathogen-associated molecular
patterns (PAMPs) are often associated with progressive inflammation
in COPD (8,9). Lipopolysaccharide (LPS) is one of the
most common PAMPs, which is produced from the cell walls of
Gram-negative bacteria (4). LPS has
been recognized as an essential contributor to lung inflammation
and injury in COPD (10,11). Moreover, LPS can activate
inflammatory cells, such as monocytes and macrophages, to produce
tumor necrosis factor-α (TNF-α) and other related inflammatory
cytokines (8,9). The levels of TNF-α are significantly
increased in the sputum, bronchoalveolar lavage fluid, plasma and
lung tissue of patients with COPD, which may serve as a therapeutic
target for chronic lung inflammation (4). Strategies to reduce the secretion of
TNF-α may effectively alleviate the inflammation in patients with
COPD (11-13).
AMP-activated protein kinase (AMPK) plays a vital
role in maintaining cellular energy homeostasis (14). Studies have suggested that AMPK also
exerts potent anti-inflammatory effects (15-18).
A plethora of studies have demonstrated that AMPK inhibits the
inflammatory response by indirect inhibition of NF-κB activation
(19-22).
In addition, a study suggested that AMPK exerted anti-inflammatory
effects in immune cells by switching metabolic activity from a
glycolysis driven process to a mitochondrial oxidative metabolic
process, such as fatty acid oxidation (FAO) (23,24).
Furthermore, an additional study indicated that transformation of
pro-inflammatory M1 macrophages to anti-inflammatory M2 macrophages
is dependent on AMPK and FAO (25).
This evidence indicates that AMPK has a functional role in
attenuating the inflammatory response (15,24,26).
The well-established AMPK activators 5-Aminoimidazole-4-carboxamide
ribonucleotide (AICAR) and A769662 have been reported to suppress
LPS-induced cytokine production and NF-κB activation (27,28).
An additional classic AMPK activator and widely used antidiabetic
agent, metformin, has been indicated to decrease the expression
levels of pro-inflammation and adhesion molecules by activating the
AMPK signaling pathway (29).
Furthermore, it has been reported that perifosine and cordycepin
activate the AMPK signaling pathway, and subsequently inhibit
LPS-induced TNF-α expression (27,30).
Therefore, AMPK activation may serve as an effective and novel
therapeutic strategy for inhibiting LPS-induced inflammatory
responses (31).
Zicao is a commonly used herbal medicine in China
(32). Zicao is believed to possess
detoxification properties and has been used for the treatment of
macular eruptions, measles, sore-throat, carbuncles and burns
(33). Shikonin, a naphthoquinone
compound, is the primary active constituent of Zicao, which can be
produced from the dried root of Lithospermum erythrorhizon
(34). Shikonin has been reported
to exert multiple pharmacological effects, including both
anti-cancer and anti-inflammatory properties (35,36).
However, the strong non-selective cytotoxicity of shikonin
restricts its clinical application (36). Thus, it is essential to investigate
the pharmacological effects of shikonin at low cytotoxic doses. The
aims of the present study were to examine the effects of shikonin
at non-cytotoxic doses on the pro-inflammation functions of
monocytes and macrophages, as well as identifying the underlying
molecular mechanisms.
Materials and methods
Reagents
Shikonin was obtained from Yuanye Biotechnology Co.,
Ltd. (cat. no. B21682). LPS (cat. no. L6143), compound C (an AMPK
inhibitor; cat. no. 171261) and D-galactosamine (cat. no. G0500-5G)
were obtained from Sigma-Aldrich (Merck KGaA). MTT was purchased
from Beyotime Institute of Biotechnology (cat. no. C0009). Cell
culture reagents, including: DMEM (Dulbecco's modified Eagle's
medium; cat. no. 11965084), FBS (fetal bovine serum; cat. no.
16140071), glutamine (cat. no. 25030081) and
penicillin-streptomycin (cat. no. 15070063) were supplied by Gibco
(Thermo Fisher Scientific, Inc.).
Culture of RAW 264.7 cells
The mouse macrophage cell line RAW 264.7 was
obtained from the Cell Bank of Type Culture Collection of the
Chinese Academy of Sciences (cat. no. TCM13). The cells were
cultured in DMEM with 10% FBS, 100 U/ml penicillin and 100 µg/ml
streptomycin, and 2 mM glutamine at 37˚C in a 5% CO2
humidified incubator as described previously (30).
Preparation of bone marrow-derived
macrophages (BMDMs)
All animal experiments were approved by the Animal
Ethics Committee of The Fourth Military Medical University (Xi'an,
Shaanxi, China). The present study used 2-month-old male C57BL/6J
mice (supplied by Beijing Vital River Laboratory, Beijing, China;
n=10; 20-25 g). Mice were maintained under specific pathogen-free
conditions. All mice were provided free access to food and water
and were maintained at a temperature of 23±2˚C, humidity of 40-80%
and on a 12 h light/dark cycle. Mice were euthanized with sodium
pentobarbital (250 mg/kg; cat. no. P3761; Sigma-Aldrich; Merck
KGaA), and their femurs and tibias were isolated and rinsed in 75%
(vol/vol) ethanol. Bone marrow cells were flushed out with PBS and
passed through 40 µM filters, then centrifuged at 450 x g for 5 min
at room temperature. Cell pellets were suspended in
ammonium-chloride-potassium (ACK) hypotonic buffer (cat. no. C3702;
Beyotime Institute of Biotechnology) at a volume ratio of cell
pellet to ACK hypotonic buffer of 1:5 at room temperature for 1
min, then washed with serum-free DMEM. After centrifugation at 450
x g for 10 min at room temperature, the isolated bone marrow cells
were resuspended and cultured in DMEM containing 10% FBS and 30%
L929-conditioned media at 37˚C in a 5% CO2 incubator to
allow for differentiation. The L929-conditioned media was obtained
from L929 fibroblasts (cat. no. GNM28; Cell Bank of Type Culture
Collection of the Chinese Academy of Sciences) that were grown in
DMEM (cat. no. 11965084; Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (cat. no. 16140071; Gibco; Thermo Fisher
Scientific, Inc.) at 37˚C in a 5% CO2 incubator for 3
days following confluency. The medium was filtered through a
0.22-µm filter (cat. no. FF362-100pcs; Beyotime Institute of
Biotechnology) and kept at 4˚C for the subsequent applications, as
previously described (37). After
culturing for 7 days, the adherent macrophages were trypsinized and
sub-cultured in DMEM supplemented with 10% FBS and 15%
L929-conditioned media, 2 mM glutamine, 100 U/ml penicillin and 100
µg/ml streptomycin at 37˚C in a 5% CO2 incubator for
subsequent experiments. The animal experimental procedures were
carried out strictly in accordance with the guidelines approved by
The Animal Ethics Committee of The Fourth Military Medical
University.
Ex vivo culture of human peripheral
blood mononuclear cells (PBMCs)
After obtaining their written informed consent,
PBMCs were collected from patients with COPD. A total of 10 COPD
patients were recruited at the Xijing Hospital of Fourth Military
Medical University between October 2017 and March 2018. The
patients were male, and aged 52-65 years old (mean age, 61.1;
median age, 62.5). Cells were collected using lymphocyte separation
medium (cat. no. 10771; Sigma-Aldrich; Merck KGaA) as previously
described (4,28). The collected PBMCs were cultured in
DMEM containing 10% FBS and other essential nutrients (38). The study protocol involving clinical
specimens was approved by The Institutional Review Board of Xijing
Hospital Affiliated to The Fourth Military Medical University.
Assessment of cytotoxicity
The survival rates of RAW 264.7 cells, BMDMs and
PBMCs were assessed by MTT assay. Cells were cultured in 96-well
plates at a density of 1x104 cells/well and incubated
with 5% CO2 at 37˚C for 24 h, Subsequently, the cells
were treated with gradually increasing doses of shikonin (0, 0.5,
1.0, 1.5, and 2.0 µM) at 37˚C for a further 24 h. After shikonin
treatment, 20 µl/well of MTT (5 mg/ml) was added into each well,
and then incubated at 37˚C for 4 h. Then, the culture supernatant
was removed from each well, and DMSO (150 µl/well) was added to
dissolve the formazan crystals (30). The optical density (OD) values were
determined at 570 nm using a microplate reader (ELX 800; BioTek
Instruments, Inc.).
The percentage of cell death was determined by
trypan blue staining (4,39). After shikonin treatment, the cells
were collected, centrifuged at 1,000 x g for 1 min at room
temperature and resuspended in 0.4% trypan blue (cat. no. 15250061;
Gibco; Thermo Fisher Scientific, Inc.), stained for 3 min at room
temperature and then manually counted using a hemocytometer. The
numbers of stained cells were presented as a ratio of the total
(stained and unstained) cells.
Detection of apoptosis
The level of apoptosis in RAW 264.7 cells was
evaluated using a TUNEL assay kit (cat. no. C1088; Beyotime
Institute of Biotechnology) according to the manufacturer's
protocol. Briefly, RAW 264.7 cells were cultured in 24-well plates
at a density of 1.5x105 cells/well and incubated with 5%
CO2 at 37˚C for 24 h, subsequently, the cells were
treated with gradually increasing doses of shikonin (0, 0.5, 1.0,
1.5 and 2.0 µM) at 37˚C for a further 24 h. After treatment, the
cells were fixed in 4% paraformaldehyde for 20 min at room
temperature. Subsequently, the cells were washed with PBS three
times and permeabilized with 0.3% Triton X-100 for 5 min at room
temperature before the cells were incubated with TUNEL working
solution for 60 min in a humidified atmosphere at 37˚C in the dark.
The cells were then counterstained with 5 µg/ml DAPI (cat. no.
D8417; Sigma-Aldrich; Merck KGaA) for 5 min at room temperature and
mounted using antifade mounting medium (cat. no. P0126; Beyotime
Institute of Biotechnology). The cells were observed using a
fluorescence microscope (magnification, x100), in which five fields
were randomly selected, and the TUNEL and DAPI stained nuclei in
the cells were counted manually (40). The percentage of positive cells was
calculated using the following equation: TUNEL-positive cells
(%)=(number of TUNEL positive cells/total number of cells) x 100%,
as previously described (41). The
apoptosis of RAW 264.7 cells was also assessed using a Dead Cell
Annexin-V-FITC Propidium iodide (PI) apoptosis detection kit (cat.
no. V13242; Invitrogen; Thermo Fisher Scientific, Inc.) according
to the manufacturer's protocol. The rate of apoptotic cells was
evaluated using a standard EPICS Elite flow cytometer (Beckman
Coulter, Inc.) as previously described (42), and data were analyzed using CXP
Analysis Software version 1.0 (Beckman Coulter, Inc.).
ELISA assay
Cells (RAW 264.7 cells, BMDMs and PBMCs) were
cultured in 6-well plates at a density of 1x106
cells/well and incubated with 5% CO2 at 37˚C for 24 h.
Subsequently, the cells were treated with LPS alone (100 ng/ml) or
10-250 ng/ml in combination with different doses of shikonin (0.5
and 1.0 µM) at 37˚C for 24 h, with or without 1 h pretreatment with
10 µM compound C. After treatment, the extracellular levels of
TNF-α in DMEM were detected using commercial ELISA kits (RAW 264.7
cells, cat. no. MTA00B, R&D Systems, Inc.; BMDMs, cat. no.
PT512; Beyotime Institute of Biotechnology; PBMCs, cat. no. PT518;
Beyotime Institute of Biotechnology) following each manufacturer's
protocol. The general procedure was as follows: The culture medium
was collected by centrifuging at 500 x g for 10 min at room
temperature. The culture supernatant was added into anti-TNF-α
antibody-coated wells (in the corresponding kit) and incubated for
2 h at room temperature. Then, the corresponding biotinylated
antibody was added into each well and incubated for a further 1 h
at room temperature. Horseradish peroxidase (HRP)-streptavidin was
added and incubated in the dark for 20 min at room temperature,
then TMB substrate was added into each well and incubated in the
dark for another 20 min at room temperature. Stop solution was
added and gently mixed in the dark for 2 min at room temperature.
The absorbance values at 450 nm were detected using a microplate
reader (ELX 800; BioTek Instruments, Inc.), and the concentration
of TNF-α was calculated by referring to the standard curve.
Reverse transcription-quantitative PCR
(RT-qPCR)
The cells (RAW 264.7 cells, BMDMs and PBMCs) were
cultured in 6-well plates at a density of 1x106
cells/well and incubated with 5% CO2 at 37˚C for 24 h.
Subsequently, the cells were treated with LPS (100 ng/ml alone or
10-250 ng/ml), or in combination with different doses of shikonin
(0.5 and 1.0 µM) at 37˚C for 24 h, with or without 1 h pretreatment
of 10 µM compound C. After treatment, RNA isolation and RT-qPCR
assay were carried out according to previously published methods
(4,43). Briefly, total RNA was extracted from
the cells using TRIzol® reagent (cat. no. 15596018;
Invitrogen; Thermo Fisher Scientific, Inc.). Then, the extracted
RNA (2 µg; 10 µl) was reverse transcribed using AMV reverse
transcriptase (cat. no. M5101; Promega Corporation) in a 25 µl
final reaction volume containing AMV Reverse Transcriptase (3 µl),
AMV Reverse Transcriptase 5X Reaction Buffer (5 µl; cat. no. M5101;
Promega Corporation), 10 mM dNTP (2.5 µl; cat. no. U1330; Promega
Corporation), RNasin® Ribonuclease Inhibitor (1 µl; cat.
no. N2511; Promega Corporation), 500 µg/ml oligo(dT)15
primer (2 µl; cat. no. C1101; Promega Corporation) and
nuclease-free water (1.5 µl; cat. no. P1193; Promega Corporation),
which were incubated at 42˚C for 60 min. qPCR reactions were
carried out on an ABI Prism 7500 RT PCR instrument (Applied
Biosystems; Thermo Fisher Scientific, Inc.) in triplicate using
SYBR Premix Ex Taq (cat. no. RR420A; Takara Bio Inc.). The
thermocycling conditions were as follows: 95˚C for 5 min, followed
by 40 cycles of 95˚C for 15 sec and 60˚C for 1 min. The relative
changes in TNF-α mRNA expression levels were calculated using the
2-ΔΔCq method after normalization to GAPDH (44). The primer sequences for TNF-α and
GAPDH were as follows: Mouse TNF-α (forward,
5'-CATCTTCTCAAAATTCGAGTGAC-3' and reverse,
5'-TGGGAGTAGACAAGGTACAACCC-3'), Mouse GAPDH (forward,
5'-GGCCTTCCGTGTTCCTAC-3' and reverse, 5'-TGTCATCATATCTGGCAGGTT-3'),
Human TNF-α (forward, 5'-CGAGTGACAAGCCTGTAGCC-3' and reverse,
5'-TTGAAGAGGACCTGGGAGTAG-3'), Human GAPDH (forward,
5'-AACGGATTTGGTCGTATTG-3' and reverse, 5'-GGAAGATGGTGATGGGATT-3').
All the primers were synthesized by Sangon Biotech (Shanghai) Co.,
Ltd.
Western blotting analysis
Total protein was extracted from the cells with RIPA
buffer (cat. no. P0013; Beyotime Institute of Biotechnology),
followed by quantification using a bicinchoninic acid protein assay
kit (cat. no. P0012; Beyotime Institute of Biotechnology). Equal
amounts of protein (30 µg protein per lane) from each sample were
separated by 10% SDS-PAGE and transferred to nitrocellulose filter
membrane at 100 mV for 75 min. After blocking with 5% non-fat skim
milk [diluted with Tris-buffered saline containing 0.1% Tween-20
(TBST)] for 1 h at room temperature, the membrane was incubated
overnight with primary antibody [diluted with 2% bovine serum
albumin (cat. no. ST023; Beyotime Institute of Biotechnology) in
TBST] at 4˚C (45). The following
primary antibodies were used: Anti-acetyl-CoA carboxylase (ACC;
1:1,000; cat. no. AF1867), anti-phosphorylated (p)-ACC (1:1,000;
cat. no. AA110), anti-AMPK (1:2,000; cat. no. AF1627), anti-p-AMPK
(1:1,000; cat. no. AA393), anti-p-IκB kinase α/β (1:1,000; IKKα/β;
cat. no. AI139), anti-IKKα/β (1:1,000; cat. no. AF2221), and
anti-β-actin (1:1,000; cat. no. AF0003), all of the above primary
antibodies were purchased from Beyotime Institute of Biotechnology.
On the next day, blots were washed and incubated with anti-rabbit
(1:5,000; cat. no. SA00001-2; ProteinTech Group) or anti-mouse
(1:5,000; cat. no. SA00001-1, ProteinTech Group)
horseradish-peroxidase-conjugated secondary antibody for 1 h at
room temperature. The protein bands were visualized with an
enhanced chemiluminescence western blot detection kit (Pierce;
Thermo Fisher Scientific, Inc.), and the protein expression levels
were semi quantified by densitometry using ImageJ version 1.46r
(National Institutes of Health) (45).
Reactive oxygen species (ROS)
determination
The levels of ROS in RAW 264.7 cells, BMDMs and
PBMCs were assessed using a ROS detection kit (cat. no. S0033;
Beyotime Institute of Biotechnology) as described previously
(43). Cells were harvested by
trypsinization and then resuspended into serum-free DMEM. The cell
suspension was incubated with 10 µM Dichloro-dihydro-fluorescein
diacetate solution at 37˚C for 20 min in the dark, and mixed by
repeatedly inverting the tube for 5 min. After being washed three
times with serum-free culture medium, the cell samples were
analyzed by a flow cytometry (FACScan; Becton, Dickinson and
Company). Fluorescence intensity values of the treatment group were
normalized as fold changes relative to the control group.
Thiobarbituric acid reactive
substances (TBARS) assay
TBARS assay was used for the assessment of lipid
peroxidation (28,46). RAW 264.7 cells were treated with 100
ng/ml of LPS, or in combination with different doses of shikonin
(0.5 and 1.0 µM) for 2 h, with or without 1 h pretreatment of 10 µM
compound C. After treatment, the cells were harvested by
trypsinization, and the cell extracts were prepared by sonication
(at 200 W four times, for 5 sec each time, with a 2 sec interval
between pulses) at 4˚C in ice-cold RIPA buffer (cat. no. P0013;
Beyotime Institute of Biotechnology). To remove debris, the lysed
cells were centrifuged at 10,000 x g, at 4˚C for 20 min. After
centrifugation, the malondialdehyde (MDA) levels in the supernatant
were measured following the manufacturer's protocol of the TBARS
assay kit (cat. no. S0131; Beyotime Institute of Biotechnology)
(47). A protein assay kit (cat.
no. P0012; Beyotime Institute of Biotechnology) was used to
quantify the total protein concentration, and the MDA levels were
then normalized to mg protein. The values of MDA in treatment
groups were presented as fold changes relative to the control
group.
Measurement of NF-κB p65 subunit DNA
binding activity
The DNA binding activity of NF-κB p65 subunit in RAW
264.7 cells, BMDMs and PBMCs was assessed as described previously
(4,31). The cells were treated with 100 ng/ml
of LPS, or in combination with different doses of shikonin (0.5 and
1.0 µM) for 2vh, with or without 1 h pretreatment of 10 µM compound
C. After treatment, 1 µg of nuclear extracts per treatment was
analyzed by the TransAM ELISA kit (cat. no. 40098; Active Motif,
Inc.) according to the manufacturer's instructions. To determine
the relative changes in NF-κB p65 subunit DNA binding activities,
the OD values of the treatment group were compared with the
untreated group.
LPS-induced endotoxin shock
The animal experiments were approved by the Animal
Ethics Committee of The Fourth Military Medical University (Xi'an,
Shaanxi, China). BALB/c mice (supplied by Beijing Vital River
Laboratory; male; 4-6 weeks old; weighing 18-20 g; n=30) had ad
libitum access to food and water, and were maintained under a
12 h light/ dark cycle at 24±1˚C with 40-80% relative humidity. The
experiment was divided into three groups (n=10 per group): Control
group, D-galactosamine/LPS group, and D-galactosamine/LPS +
Shikonin group. The mice were intraperitoneally injected with 30
mg/kg body weight (BW) LPS, and 300 mg/kg BW D-galactosamine or 2.5
mg/kg BW shikonin as described previously (4,28).
Blood samples of ~50-150 µl were collected from the tail vein of
mice 8 h after LPS induction. The blood samples were incubated at
4˚C for 2 h to allow clotting and then centrifuged at 8,000 x g to
obtain serum (~15-50 µl/per mouse), as previously described
(48). The levels of TNF-α in serum
(diluted with the dilution buffer of the corresponding kit) were
detected using ELISA (mouse TNF-α ELISA kit; cat. no. PT512;
Beyotime Institute of Biotechnology) following the manufacturer's
protocol (38). The survival of
mice was recorded within 72 h after LPS injection, and the animal
suffering was minimized based on the humane endpoints. Clinical
signs of the humane endpoints included a significant decrease in
locomotion, severe diarrhea, piloerection and a 20% reduction in
body weight (4). Animals were
weighed and monitored every hour after the initial administration
of LPS/D-galactosamine. Once the animals reached the endpoints,
they were euthanized with sodium pentobarbital (50 mg/kg BW, i.p;
cat. no. P3761; Sigma-Aldrich; Merck KGaA), followed by cervical
dislocation. The procedures of animal experimentation were carried
out strictly in accordance with the guidelines approved by The
Animal Ethics Committee of The Fourth Military Medical
University.
Statistical analysis
Data were obtained from ≥3 independent experiments.
Statistical analyses were carried out using SPSS ver. 16.0 software
(SPSS, Inc.). Data are presented as the mean ± SD. Statistical
differences between groups were compared using Student's t-test or
one-way ANOVA with Bonferroni's post hoc test. Intergroup survival
rates were analyzed using Kaplan-Meier curves with log-rank test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Cytotoxic effects of shikonin on RAW
264.7 mouse macrophages
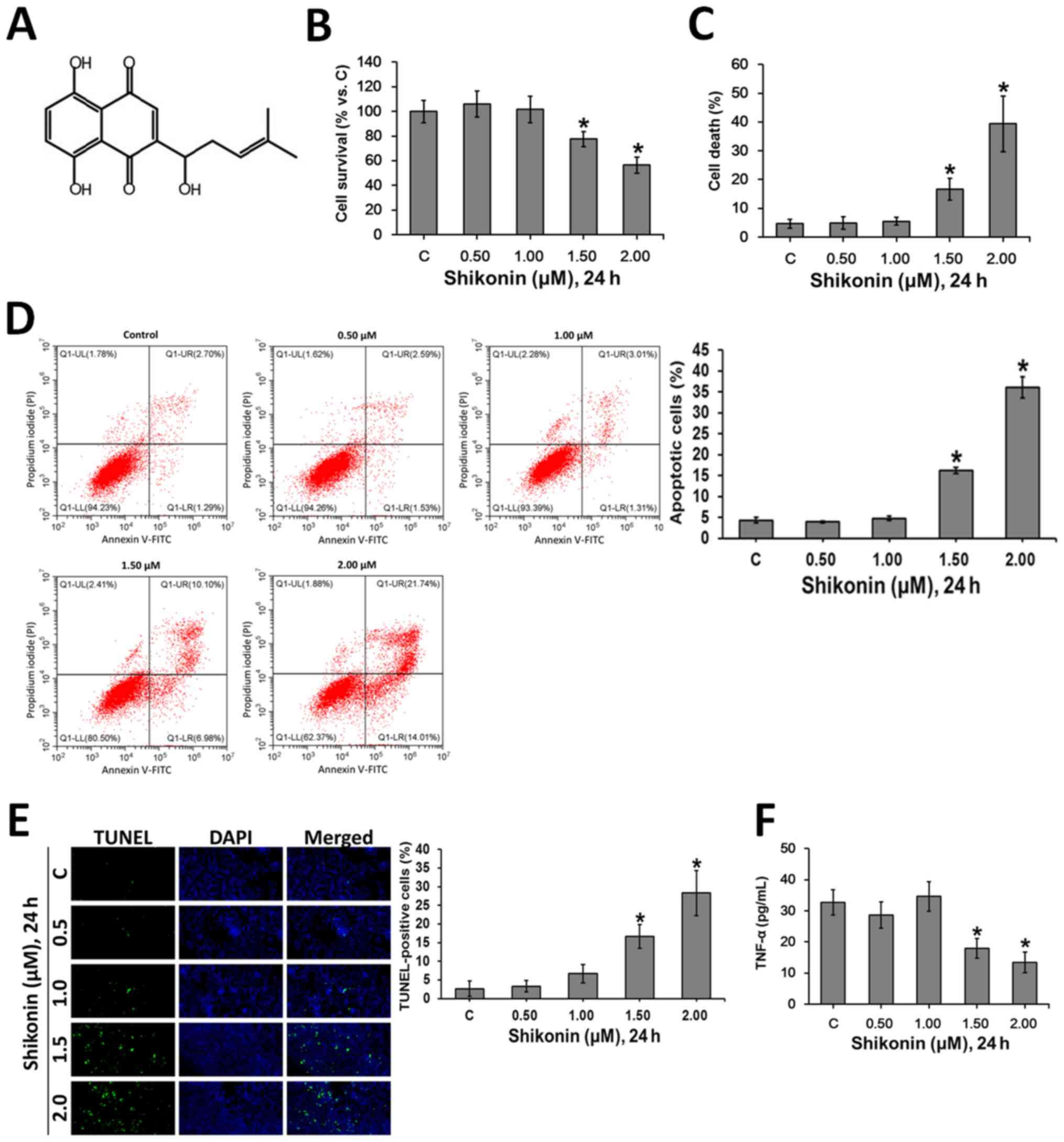
Shikonin, a naphthoquinone compound, is the primary
active constituent of Zicao (32).
Shikonin (Fig. 1A) has been
reported to have a wide variety of medical functions, but its
non-selectivity and strong cytotoxicity limits its clinical use
(36). The present study
investigated the cytotoxic profile of shikonin in RAW 264.7
macrophages. The cultured macrophages were treated with gradually
increasing doses of shikonin for 24 h. Then, MTT (Fig. 1B) and trypan blue exclusion
(Fig. 1C) assays were performed to
assess the viability of cells. It was found that shikonin (1.5-2.0
µM) was cytotoxic to RAW 264.7 macrophages, however, there was no
obvious cytotoxicity with 0.5 and 1.0 µM shikonin. The flow
cytometry analysis (Fig. 1D) and
TUNEL staining (Fig. 1E) results
suggested that shikonin induced apoptosis at the concentrations of
1.5 and 2.0 µM, but did not have these effects at 0.5 and 1.0 µM.
In addition, at non-cytotoxic concentrations (0.5 and 1.0 µM),
shikonin did not affect the levels of TNF-α in RAW 264.7
macrophages (Fig. 1F). However, at
1.5 and 2.0 µM, shikonin significantly inhibited the production of
TNF-α (Fig. 1F), which may
attribute to shikonin-induced apoptosis (Fig. 1D and E).
Non-cytotoxic doses of shikonin
suppress LPS-induced TNF-α expression in RAW 264.7 mouse
macrophages
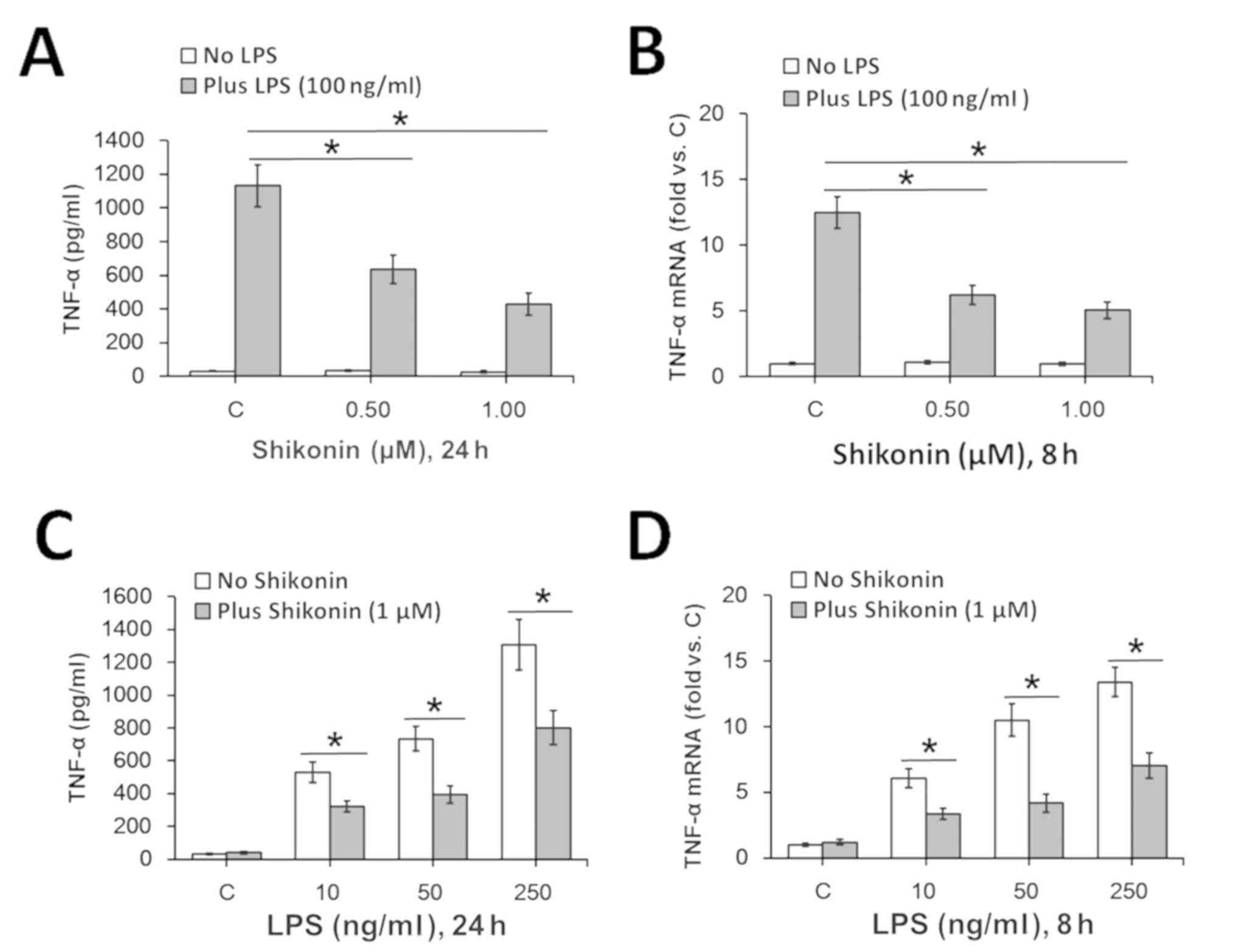
Therefore, based on the present results the
non-cytotoxic doses (0.5 and 1.0 µM) of shikonin were chosen for
subsequent experimentation. In line with previous studies (4,28), 100
ng/ml of LPS treatment significantly increased TNF-α expression in
RAW 264.7 macrophages at both the protein (Fig. 2A) and mRNA (Fig. 2B) levels. However, co-treatment with
0.5 and 1.0 µM of shikonin significantly attenuated LPS-induced
TNF-α expression levels (Fig. 2A
and B). Furthermore, 1.0 µM
shikonin-mediated inhibition of TNF-α expression was identified in
RAW 264.7 macrophages in response to different concentrations of
LPS (Fig. 2C and D). Collectively, the present results
suggested that non-cytotoxic doses of shikonin inhibit LPS-induced
TNF-α expression in RAW 264.7 macrophages.
Shikonin suppresses LPS-induced TNF-α
expression via AMPK activation
AMPK activation is involved in the suppression of
inflammatory responses (31).
Previous studies showed that the activation of AMPK inhibits
LPS-induced TNF-α expression in mouse macrophages (28,30).
However, it remains unknown whether the AMPK signaling pathway is
activated in shikonin-treated RAW 264.7 macrophages. The present
results indicated that the phosphorylation levels of ACC and AMPKα
were significantly increased after treatment with non-cytotoxic
doses of shikonin (0.5 and 1.0 µM; Fig.
3A). However, pretreatment with compound C, an AMPK inhibitor,
not only reversed the AMPK activation induced by shikonin (Fig. 3A), but also attenuated the
inhibitory effects of shikonin on LPS-induced TNF-α expression at
both protein and mRNA levels (Fig.
3B and C). Consistent with
previous results (49), in the
present study compound C alone showed no obvious effect on TNF-α
expression under LPS stimulation (Fig.
3D and E). Therefore, the
present results suggested that the AMPK signaling pathway may play
a key role in the inhibition of LPS-induced TNF-α expression after
treatment with shikonin at non-cytotoxic doses.
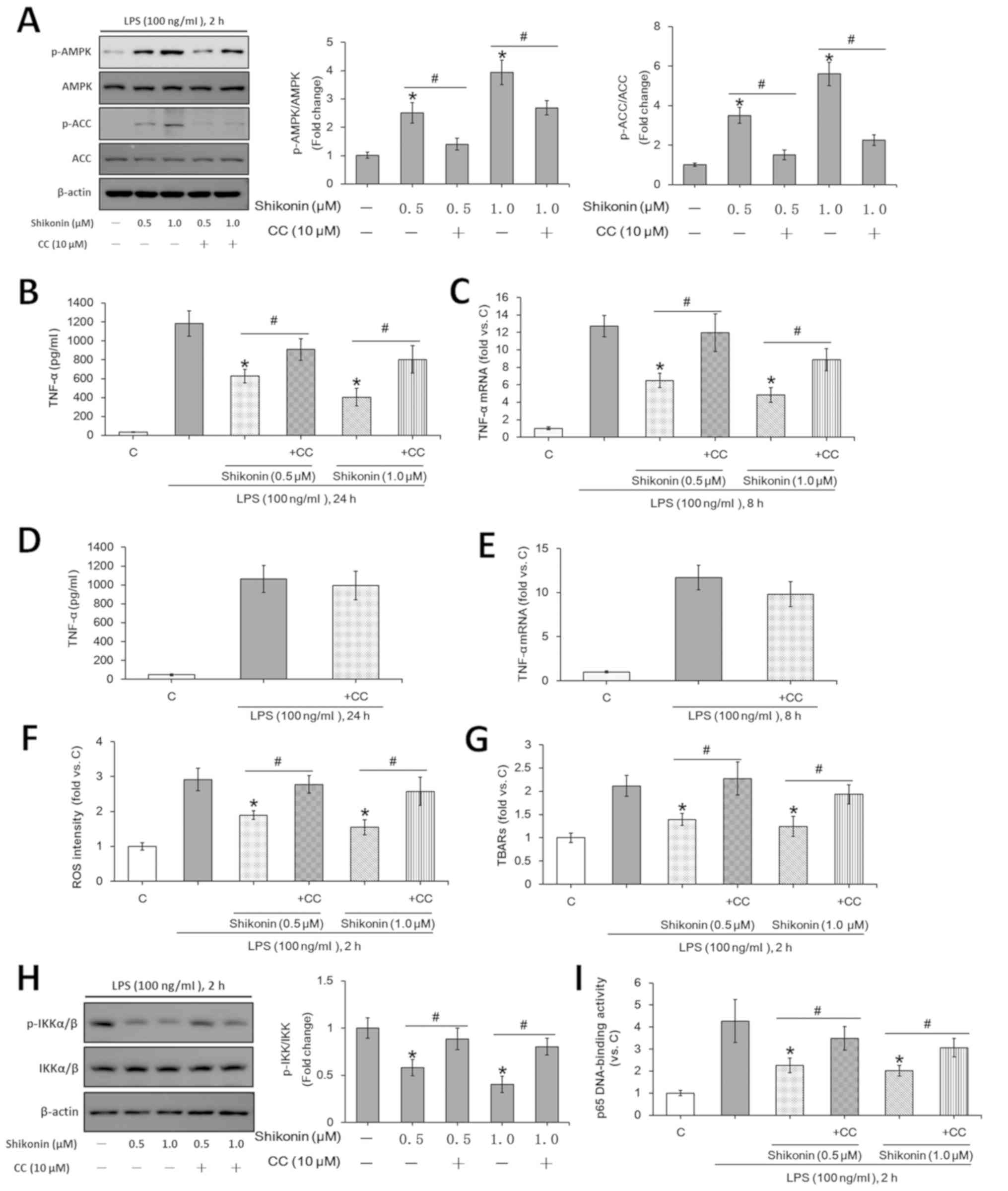 | Figure 3Non-cytotoxic doses of shikonin
suppress LPS-induced TNF-α expression via activating AMPK pathway.
RAW 264.7 cells were treated with 100 ng/ml of LPS, or in
combination with the indicated doses of shikonin with and without 1
h pretreatment of 10 µM compound C. (A) Protein expression levels
of ACC, p-ACC, AMPKα and p-AMPKα were determined by western
blotting and then semi-quantified by densitometric analysis. (B)
Extracellular contents of TNF-α were examined using ELISA assay.
(C) Relative mRNA levels of TNF-α were examined using RT-qPCR
assay. RAW 264.7 cells were treated with 100 ng/ml of LPS with or
without 1 h pretreatment of 10 µM compound C for (D) 24 h or (E) 8
h. Extracellular concentration of TNF-α was examined using ELISA
assay. Relative mRNA levels of TNF-α were examined using RT-qPCR
assay. RAW 264.7 cells were treated with 100 ng/ml of LPS for 2 h,
or in combination with the indicated doses of shikonin with or
without 1 h pretreatment with 10 µM compound C. (F) Relative ROS
intensity and (G) TBARs content were detected by flow cytometry and
TBARs production assay, respectively. Activation of NF-κB was
evaluated by detecting (H) p-IKKα/β and IKKα/β levels, (I) and p65
DNA-binding activity. *P<0.05 vs. LPS alone group.
#P<0.05 vs. corresponding shikonin combined with LPS
group. C, control; LPS, lipopolysaccharide; TNF-α, tumor necrosis
factor α; p-, phosphorylated; ACC, acetyl-CoA carboxylase; AMPK,
AMP-activated protein kinase; IKKα/β, IκB kinase α/β; CC, Compound
C; ROS, reactive oxygen species; TBARs, thiobarbituric acid
reactive substances. |
Previous studies have reported that LPS-induced ROS
is required for subsequent TNF-α expression and NF-κB activation
(28,30). AMPK activation suppresses ROS
production under different stress conditions (14,43).
The present results indicated that shikonin treatment at
non-cytotoxic doses (0.5 and 1.0 µM) significantly attenuated
LPS-induced ROS generation (Fig.
3F), lipid peroxidation (TBARs production; Fig. 3G) and NF-κB activation (Fig. 3H and I) in RAW 264.7 macrophages. Moreover,
pretreatment with compound C reversed the effects of shikonin
(Fig. 3F-I). However, the
non-cytotoxic doses of shikonin exerted no significant effects on
both NF-κB activation and ROS generation in the absence of LPS
stimulation (data not shown). Therefore, shikonin, via AMPK
signaling activation, inhibited LPS-induced ROS generation and
NF-κB activation, which suppressed TNF-α expression.
Non-cytotoxic doses shikonin inhibit
LPS-induced TNF-α expression in murine BMDMs via AMPK
activation
The potential role of shikonin in LPS-induced TNF-α
production was investigated using the primary murine BMDMs. Similar
to the results from RAW 264.7 macrophages, the indicated doses of
shikonin (0.5 and 1.0 µM) were also non-cytotoxic to BMDMs
(Fig. 4A and B). Moreover, the non-cytotoxic
concentrations of shikonin activated AMPK signaling (Fig. 4C), suppressed LPS-induced ROS
generation (Fig. 4D) and NF-κB
activation (Fig. 4E), whilst
downregulating TNF-α expression at both the protein (Fig. 4F) and mRNA (Fig. 4G) levels. However, pretreatment with
compound C significantly attenuated such effects of shikonin
(Fig. 4C-G). Collectively, the
present results suggested that the non-cytotoxic doses of shikonin
suppressed LPS-induced TNF-α expression in murine BMDMs via AMPK
activation.
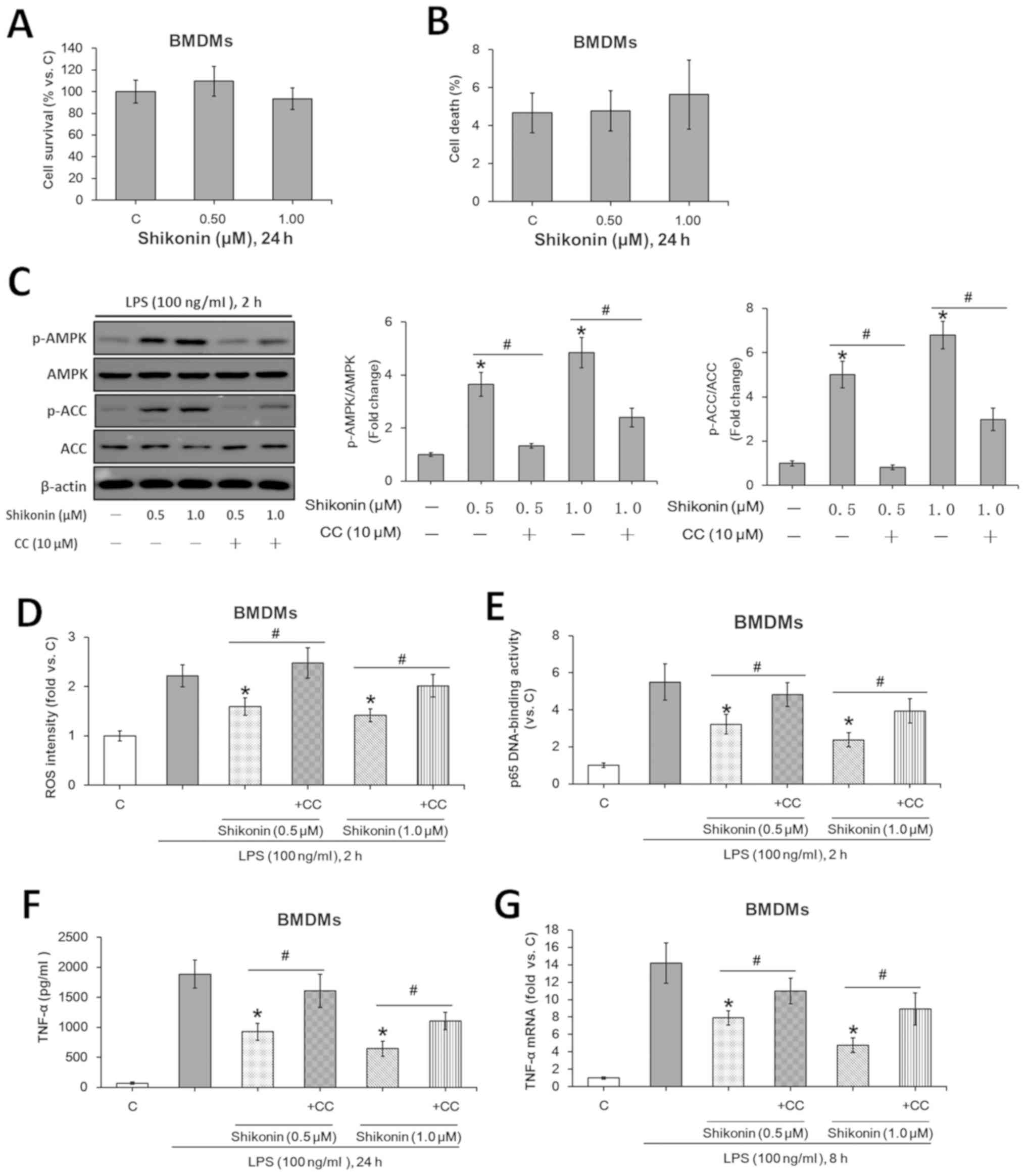 | Figure 4Non-cytotoxic doses of shikonin
suppress LPS-induced TNF-α expression in primary murine BMDMs via
AMPK activation. After treating with the indicated doses of
shikonin for 24 h, the survival and death rates of primary murine
BMDMs were evaluated by (A) MTT and (B) trypan blue exclusion
assays, respectively. (C) Expression levels of ACC, p-ACC, AMPKα
and p-AMPKα were detected by western blotting. Relative ROS
intensity and NF-κB activation were examined by (D) flow cytometry
and (E) p65 DNA-binding activity assay, respectively. Extracellular
contents and relative mRNA levels of TNF-α were examined by (F)
ELISA and (G) reverse transcription-quantitative PCR, respectively.
*P<0.05 vs. LPS alone group. #P<0.05
vs. corresponding shikonin combined with LPS group. C, control;
LPS, lipopolysaccharide; TNF-α, tumor necrosis factor α; p-,
phosphorylated; ACC, acetyl-CoA carboxylase; AMPK, AMP-activated
protein kinase; IKKα/β, IκB kinase α/β; CC, Compound C; ROS,
reactive oxygen species; BMDMs, bone marrow-derived
macrophages. |
Non-cytotoxic doses of shikonin
inhibit LPS-induced TNF-α expression in PBMCs from patients with
COPD via AMPK activation
The potential role of shikonin in LPS-induced TNF-α
production was assessed using ex vivo cultured primary human
PBMCs collected from patients with COPD. Based on the MTT (Fig. 5A) and trypan blue exclusion
(Fig. 5B) assay results, 0.5 µM was
selected as the non-cytotoxic dose for shikonin. Consistent with
the data of murine macrophages, the non-cytotxoic concentration of
shikonin (0.5 µM) activated the AMPK signaling pathway (Fig. 5C), suppressed LPS-induced ROS
generation (Fig. 5D) and NF-κB
activation (Fig. 5E) in PBMCs.
Moreover, 0.5 µM shikonin downregulated TNF-α expression at both
protein (Fig. 5F) and mRNA
(Fig. 5G) levels in the monocytes
derived from patients with COPD.
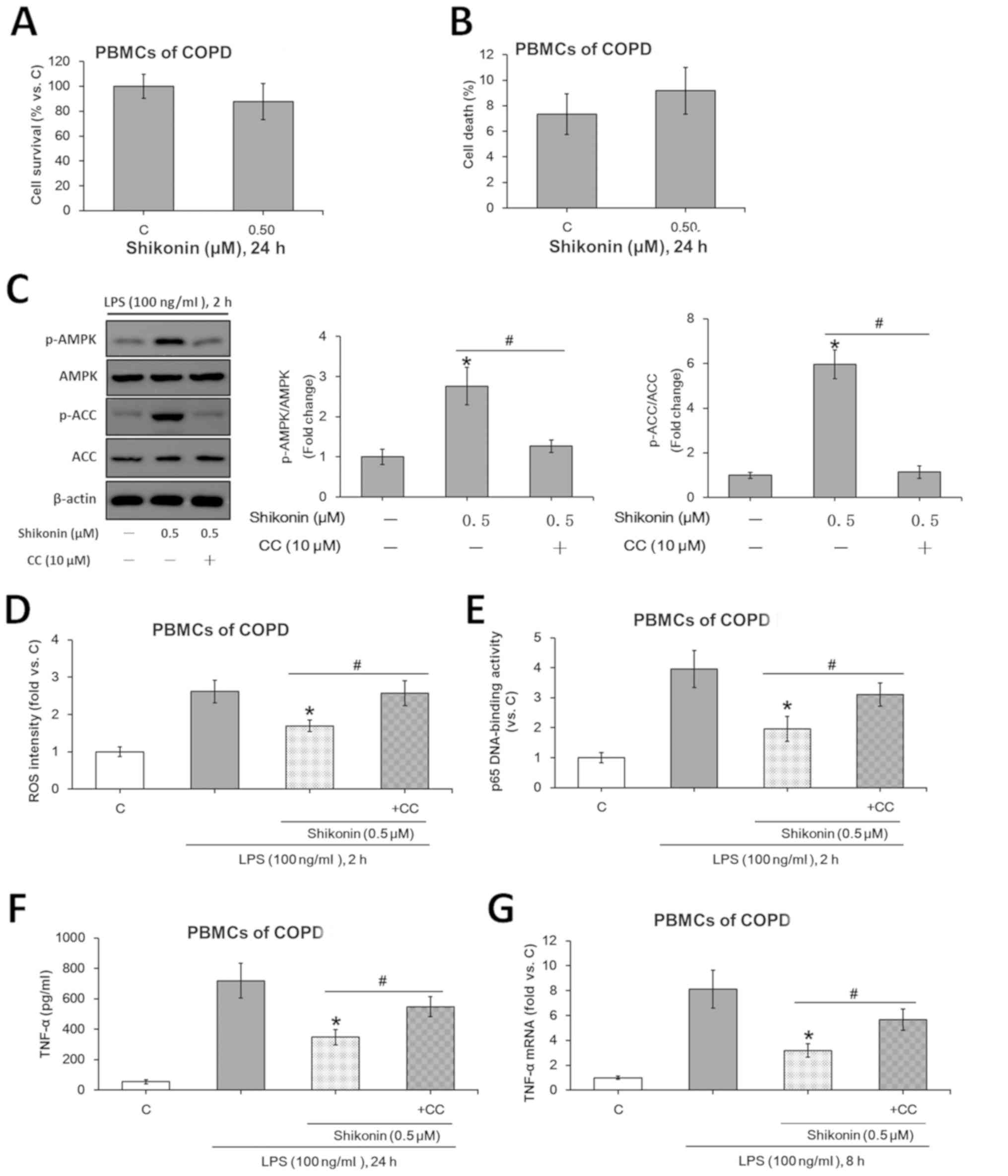 | Figure 5Non-cytotoxic dose of shikonin
suppresses LPS-induced TNF-α expression via AMPK activation in the
ex vivo cultured PBMCs from patients with COPD. After
treating with the indicated doses of shikonin for 24 h, the
survival and death rates of the ex vivo cultured PBMCs from
patients with COPD were evaluated by (A) MTT and (B) trypan blue
exclusion assays, respectively. (C) Protein expression levels of
ACC, p-ACC, AMPKα and p-AMPKα were detected by western blotting.
Relative ROS intensity and NF-κB activation were examined by (D)
flow cytometry and (E) p65 DNA-binding activity assay,
respectively. Extracellular contents and relative mRNA levels of
TNF-α were examined using (F) ELISA and (G) reverse
transcription-quantitative PCR, respectively. *P<0.05
vs. LPS alone group. #P<0.05 vs. corresponding
shikonin combined with LPS group. C, control; LPS,
lipopolysaccharide; TNF-α, tumor necrosis factor α; p-,
phosphorylated; ACC, acetyl-CoA carboxylase; AMPK, AMP-activated
protein kinase; IKKα/β, IκB kinase α/β; CC, Compound C; ROS,
reactive oxygen species; COPD, Chronic obstructive pulmonary
disease; PBMCs, peripheral blood mononuclear cells. |
Low-toxic dose of shikonin suppresses
LPS-induced endotoxin shock and TNF-α expression in mice
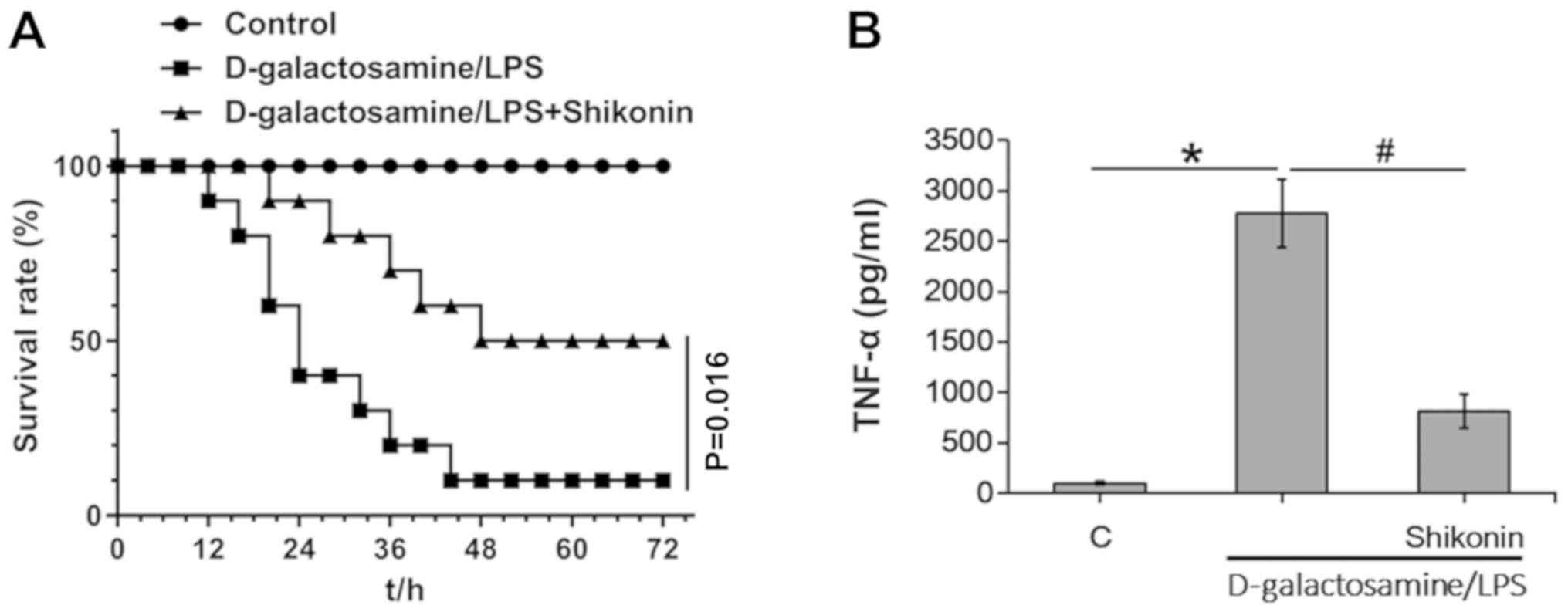
The present study investigated the protective role
of shikonin in LPS-induced inflammation in vivo in mice;
D-galactosamine and LPS were intraperitoneally injected into the
mice. D-galactosamine is a hepatotoxic transcriptional inhibitor
that strengthens the toxicity of TNF-α (50). Consistent with a previous study
(28), LPS and D-galactosamine
co-treatment triggered septic shock and led to mortality of mice
(Fig. 6A). In addition,
co-administration of shikonin (2.5 mg/kg BW) significantly
protected mice against endotoxin shock (Fig. 6A). The low-toxic dose of shikonin
(2.5 mg/kg BW) for in vivo application was determined based
on our previous work (data not published) and results from previous
study (51), wherein no obvious
toxicities were observed when administered to tested mice alone.
Furthermore, the present ELISA results of TNF-α in serum samples
indicated that shikonin (2.5 mg/kg BW) significantly suppressed
LPS/D-galactosamine-induced TNF-α expression in vivo
(Fig. 6B). Collectively, the
present results suggested that shikonin suppresses LPS-induced
TNF-α production and protects mice against endotoxin shock.
Discussion
This study showed that the non-cytotoxic doses of
shikonin remarkably suppressed LPS-induced TNF-α expression in
mouse macrophages (RAW 264.7 and primary cells). In addition, the
expression of TNF-α was downregulated by shikonin in LPS-stimulated
PBMCs isolated from patients with COPD. Furthermore, the underlying
molecular mechanisms of shikonin-mediated anti-TNF-α activities may
be regulated via activation of AMPK signaling pathway.
As a key sensor of intracellular energy status, AMPK
is able to regulate vital metabolic pathways in cells (14). It was been shown that AMPK plays an
important role in modulating inflammatory responses (24). A series of AMPK activators such as
AICAR, A769662, GSK621 and cordycepin have been reported to
suppress LPS-induced TNF-α production and NF-κB activation via AMPK
activation (31). As an extract of
traditional Chinese medicine, shikonin affects several important
transcription factors and signaling pathways, some of which are
closely related to the production of inflammatory cytokines
(32,36,52).
However, the present results suggested that shikonin at
non-cytotoxic doses suppressed LPS-induced TNF-α production in
macrophages, mainly via the activation of AMPK signaling pathway.
AMPK can be activated by the upstream AMPK kinases liver kinase B1
or Ca2+/calmodulin-dependent protein kinase kinase β in
cells (53). Several
anti-inflammatory compounds, such as metformin, AICAR and hydrogen
sulfide can activate AMPK via these two upstream kinases (54-56).
A previous study speculated that the mechanism of shikonin
activating AMPK is similar to that of metformin (35). However, the detailed underlying
mechanisms require further investigation.
Numerous studies have suggested a vital function of
AMPK for preventing oxidative stress (14,43).
AMPK activation can scavenge ROS production by maintaining NADPH
levels during energy stresses (14,43).
AMPK activation also inhibits H2O2-induced
oxidative stress by regulating the NADPH signaling pathway
(57). In addition, previous
studies have reported that AMPK attenuates LPS-induced ROS
generation and subsequently inhibits NF-κB activation (28,30,31).
Consistent with these studies, the present results indicated that
shikonin-activated AMPK could scavenge LPS-induced ROS generation.
Previous studies have also shown that shikonin can increase the
intracellular ROS level via various mechanisms, leading to
oxidative stress and cytotoxicity (34,58,59).
However, the present results are inconsistent with these existing
studies. These different between results may be due to the various
doses used; the high doses of shikonin promote intracellular ROS
production, while the non-cytotoxicity doses of shikonin scavenge
ROS via activating AMPK signaling. Additionally, the present
results may have valuable implications for investigating the
antioxidant effects of shikonin at non-cytotoxicity doses.
Moreover, AMPK and ROS production may be involve in mitochondrial
function. Gasparrini et al (60) found that strawberry extract (a
mixture containing 0.58 mg vitamin C per g fresh weight, 2.52 mg of
polyphenol gallic acid equivalent per g fresh weight, 0.66 mg
flavonoid catechin equivalent per g fresh weight and a variety of
other trace ingredients) can efficiently counteract LPS-induced
oxidative stress, reduce the amount of ROS and nitrite production,
stimulate endogenous antioxidant enzyme activities, enhance
protection against lipid, protein and DNA damage, and improve
mitochondria functionality by activating the AMPK pathway.
Moreover, Jung et al (61)
reported that liquiritigenin, an AMPK activator, can protect
hepatocytes against oxidative hepatic injury and mitochondrial
dysfunction induced by nutrition deprivation. Kajiwara et al
(62) demonstrated that the
commonly used AMPK activator, metformin, can suppress the growth of
L. pneumophila in macrophages by inducing mitochondrial ROS,
but not phagosomal NADPH oxidase-derived ROS, in a time- and
concentration-dependent manner. However, whether shikonin affects
mitochondrial function via activating AMPK pathway requires further
study.
COPD is a major health threat worldwide, affecting
over 10% of the adult population and contributing to 3.2 million
deaths annually (63). In patients
with COPD, the content of TNF-α is significantly elevated in the
bronchoalveolar lavage fluids, sputum, plasma and lung tissues,
which can serve as a main cause for lung damages (11-13,28).
Strategies to reduce the secretion of TNF-α can effectively prevent
the inflammatory damage in patients with COPD (11-13).
Shikonin has been reported to exert multiple pharmacological
effects, including anti-inflammation properties (35,36).
However, to the best of our knowledge, there no previous studies
have investigated the potential use of shikonin in treating
patients with COPD. The present results suggested that shikonin
inhibited LPS-induced TNF-α production via activation of the AMPK
pathway, suggesting that this herbal extract may facilitate the
treatment of COPD. Several other AMPK activators have been
demonstrated to exert potential therapeutic effects on COPD
(28,31). However, whether shikonin has
advantages over these compounds for the treatment of COPD requires
more investigation.
In conclusion, shikonin has been reported to have a
variety of medical functions, but the strong non-selective
cytotoxicity limits its use in clinic (36). The present results suggested that
the non-cytotoxic doses of shikonin inhibit LPS-induced TNF-α
expression in macrophages cell line, primary murine BMDMs and ex
vivo cultured PBMCs from patients with COPD. Moreover, the
low-toxic dose of shikonin suppressed LPS-induced endotoxin shock
and TNF-α production in mice. The present results may facilitate
the clinical application of shikonin as an anti-inflammatory agent
in treating COPD and other TNF-α-related inflammatory
disorders.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from The National
Natural Science Foundation of China (grant nos. 8187102538 and
81172222), The Natural Science Foundation of Shaanxi Province
(grant no. 2019JQ889), The Foundation of The State Key Laboratory
of Cancer Biology of China (grant nos. CBSKL201710 and
CBSKL2017Z09), and The Foundation of Xi'an Medical University
(grant no. 2018DOC02).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TP, XR and ZL conceived and designed the
experiments. TP and FZ performed the study. XW and XG assisted in
performing the experiments. FZ analyzed the data and created the
figures. TP and FZ wrote the manuscript. XR and ZL revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by The Institutional Review
Board of Xi Jing Hospital of Fourth Military Medical University.
The experiments involving human subjects were performed after
obtaining informed consent, and in accordance with the relevant
guidelines and regulations. All animal studies were approved by The
Animal Ethics Committee of Fourth Military Medical University.
Patient consent for publication
Written informed consent was obtained from all
patients prior to publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lu Y, Chang R, Yao J, Xu X, Teng Y and
Cheng N: Effectiveness of long-term using statins in COPD-a network
meta-analysis. Respir Res. 20(17)2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Park HY, Kang D, Lee H, Shin SH, Kang M,
Kong S, Rhee CK, Cho J and Yoo KH: Impact of chronic obstructive
pulmonary disease on mortality: A large national cohort study.
Respirology. 25:726–734. 2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lopez AD, Shibuya K, Rao C, Mathers CD,
Hansell AL, Held LS, Schmid V and Buist S: Chronic obstructive
pulmonary disease: Current burden and future projections. Eur
Respir J. 27:397–412. 2006.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Li P, Wu Y, Li M, Qiu X, Bai X and Zhao X:
AS-703026 inhibits LPS-induced TNFα production through MEK/ERK
dependent and independent mechanisms. PLoS One.
10(e0137107)2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Barnes PJ: New anti-inflammatory targets
for chronic obstructive pulmonary disease. Nat Rev Drug Discov.
12:543–559. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Brusasco V and Martinez F: Chronic
obstructive pulmonary disease. Compr Physiol. 4:1–31.
2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Roversi S, Roversi P, Spadafora G, Rossi R
and Fabbri LM: Coronary artery disease concomitant with chronic
obstructive pulmonary disease. Eur J Clin Invest. 44:93–102.
2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Cosio MG, Saetta M and Agusti A:
Immunologic aspects of chronic obstructive pulmonary disease. N
Engl J Med. 360:2445–2454. 2009.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Lamela J and Vega F: Immunologic aspects
of chronic obstructive pulmonary disease. N Engl J Med.
361(1024)2009.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Singh D, Smyth L, Borrill Z, Sweeney L and
Tal-Singer R: A randomized, placebo-controlled study of the effects
of the p38 MAPK inhibitor SB-681323 on blood biomarkers of
inflammation in COPD patients. J Clin Pharmacol. 50:94–100.
2010.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ouagued M, Martin-Chouly CA, Brinchault G,
Leportier-Comoy C, Depincé A, Bertrand C, Lagente V, Belleguic C
and Pruniaux MP: The novel phosphodiesterase 4 inhibitor, CI-1044,
inhibits LPS-induced TNF-alpha production in whole blood from COPD
patients. Pulm Pharmacol Ther. 18:49–54. 2005.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Rabinovich RA, Figueras M, Ardite E, Carbó
N, Troosters T, Filella X, Barberà JA, Fernandez-Checa JC, Argilés
JM and Roca J: Increased tumour necrosis factor-alpha plasma levels
during moderate-intensity exercise in COPD patients. Eur Respir J.
21:789–794. 2003.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Profita M, Chiappara G, Mirabella F, Di
Giorgi R, Chimenti L, Costanzo G, Riccobono L, Bellia V, Bousquet J
and Vignola AM: Effect of cilomilast (Ariflo) on TNF-alpha, IL-8,
and GM-CSF release by airway cells of patients with COPD. Thorax.
58:573–579. 2003.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Jeon S-M, Chandel NS and Hay N: AMPK
regulates NADPH homeostasis to promote tumour cell survival during
energy stress. Nature. 485:661–665. 2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Jansen T, Kvandová M, Daiber A, Stamm P,
Frenis K, Schulz E, Münzel T and Kröller-Schön S: The AMP-activated
protein kinase plays a role in antioxidant defense and regulation
of vascular inflammation. Antioxidants (Basel).
9(525)2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Saisho Y: Metformin and inflammation: Its
potential beyond glucose-lowering effect. Endocr Metab Immune
Disord Drug Targets. 15:196–205. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Salt IP and Palmer TM: Exploiting the
anti-inflammatory effects of AMP-activated protein kinase
activation. Expert Opin Investig Drugs. 21:1155–1167.
2012.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Dandapani M and Hardie DG: AMPK: Opposing
the metabolic changes in both tumour cells and inflammatory cells?
Biochem Soc Trans. 41:687–693. 2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Bai A, Ma AG, Yong M, Weiss CR, Ma Y, Guan
Q, Bernstein CN and Peng Z: AMPK agonist downregulates innate and
adaptive immune responses in TNBS-induced murine acute and
relapsing colitis. Biochem Pharmacol. 80:1708–1717. 2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Yang Z, Kahn BB, Shi H and Xue BZ:
Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK)
antagonizes fatty acid-induced inflammation through SIRT1. J Biol
Chem. 285:19051–19059. 2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wang S, Zhang M, Liang B, Xu J, Xie Z, Liu
C, Viollet B, Yan D and Zou MH: AMPKalpha2 deletion causes aberrant
expression and activation of NAD(P)H oxidase and consequent
endothelial dysfunction in vivo: Role of 26S proteasomes. Circ Res.
106:1117–1128. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Salminen A, Hyttinen JM and Kaarniranta K:
AMP-activated protein kinase inhibits NF-κB signaling and
inflammation: Impact on healthspan and lifespan. J Mol Med (Berl).
89:667–676. 2011.PubMed/NCBI View Article : Google Scholar
|
|
23
|
O'Neill LA and Hardie DG: Metabolism of
inflammation limited by AMPK and pseudo-starvation. Nature.
493:346–355. 2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Jeon SM: Regulation and function of AMPK
in physiology and diseases. Exp Mol Med. 48(e245)2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Galic S, Fullerton MD, Schertzer JD,
Sikkema S, Marcinko K, Walkley CR, Izon D, Honeyman J, Chen ZP, van
Denderen BJ, et al: Hematopoietic AMPK β1 reduces mouse adipose
tissue macrophage inflammation and insulin resistance in obesity. J
Clin Invest. 121:4903–4915. 2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kauppinen A, Suuronen T, Ojala J,
Kaarniranta K and Salminen A: Antagonistic crosstalk between NF-κB
and SIRT1 in the regulation of inflammation and metabolic
disorders. Cell Signal. 25:1939–1948. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Shen J, Liang L and Wang C: Perifosine
inhibits lipopolysaccharide (LPS)-induced tumor necrosis factor
(TNF)-α production via regulation multiple signaling pathways: New
implication for Kawasaki disease (KD) treatment. Biochem Biophys
Res Commun. 437:250–255. 2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wu YH, Li Q, Li P and Liu B: GSK621
activates AMPK signaling to inhibit LPS-induced TNFα production.
Biochem Biophys Res Commun. 480:289–295. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ducommun S, Ford RJ, Bultot L, Deak M,
Bertrand L, Kemp BE, Steinberg GR and Sakamoto K: Enhanced
activation of cellular AMPK by dual-small molecule treatment: AICAR
and A769662. Am J Physiol Endocrinol Metab. 306:E688–E696.
2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhang JL, Xu Y and Shen J: Cordycepin
inhibits lipopolysaccharide (LPS)-induced tumor necrosis factor
(TNF)-α production via activating amp-activated protein kinase
(AMPK) signaling. Int J Mol Sci. 15:12119–12134. 2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Li P, Li X, Wu Y, Li M and Wang X: A novel
AMPK activator hernandezine inhibits LPS-induced TNFα production.
Oncotarget. 8:67218–67226. 2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Guo C, He J, Song X, Tan L, Wang M, Jiang
P, Li Y, Cao Z and Peng C: Pharmacological properties and
derivatives of shikonin-A review in recent years. Pharmacol Res.
149(104463)2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Chen X, Yang L, Oppenheim JJ and Howard
MZ: Cellular pharmacology studies of shikonin derivatives.
Phytother Res. 16:199–209. 2002.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zhou G, Yang Z, Wang X, Tao R and Zhou Y:
TRAIL enhances shikonin induced apoptosis through ROS/JNK signaling
in cholangiocarcinoma cells. Cell Physiol Biochem. 42:1073–1086.
2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Velliquette RA, Rajgopal A, Rebhun J and
Glynn K: Lithospermum erythrorhizon Root and its
naphthoquinones repress SREBP1c and activate PGC1α through AMPKα.
Obesity (Silver Spring). 26:126–134. 2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wang R, Yin R, Zhou W, Xu D and Li S:
Shikonin and its derivatives: A patent review. Expert Opin Ther
Pat. 22:977–997. 2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Zhang C, Zhang Y, Zhang C, Liu Y, Liu Y
and Xu G: Pioglitazone increases VEGFR3 expression and promotes
activation of M2 macrophages via the peroxisome
proliferator-activated receptor γ. Mol Med Rep. 19:2740–2748.
2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Shi-Lin D, Yuan X, Zhan S, Luo-Jia T and
Chao-Yang T: Trametinib, a novel MEK kinase inhibitor, suppresses
lipopolysaccharide-induced tumor necrosis factor (TNF)-α production
and endotoxin shock. Biochem Biophys Res Commun. 458:667–673.
2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Wright CJ, Agboke F, Muthu M, Michaelis
KA, Mundy MA, La P, Yang G and Dennery PA: Nuclear factor-κB
(NF-κB) inhibitory protein IκBβ determines apoptotic cell death
following exposure to oxidative stress. J Biol Chem. 287:6230–6239.
2012.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Si H, Zhang Y, Song Y and Li L:
Overexpression of adrenomedullin protects mesenchymal stem cells
against hypoxia and serum deprivation-induced apoptosis via the
Akt/GSK3β and Bcl-2 signaling pathways. Int J Mol Med.
41:3342–3352. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhu X, Jiang Y, Shan PF, Shen J, Liang QH,
Cui RR, Liu Y, Liu GY, Wu SS, Lu Q, et al: Vaspin attenuates the
apoptosis of human osteoblasts through ERK signaling pathway. Amino
Acids. 44:961–968. 2013.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Hu YB, Wu X, Qin XF, Wang L and Pan PH:
Role of endoplasmic reticulum stress in silica-induced apoptosis in
RAW264.7 cells. Biomed Environ Sci. 30:591–600. 2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Pan T, Zhang M, Zhang F, Yan G, Ru Y, Wang
Q, Zhang Y, Wei X, Xu X, Shen L, et al: NDRG2 overexpression
suppresses hepatoma cells survival during metabolic stress through
disturbing the activation of fatty acid oxidation. Biochem Biophys
Res Commun. 483:860–866. 2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Pan T, Zhang F, Li F, Gao X, Li Z, Li X
and Ren X: Shikonin blocks human lung adenocarcinoma cell migration
and invasion in the inflammatory microenvironment via the
IL-6/STAT3 signaling pathway. Oncol Rep. 44:1049–1063.
2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Cortizo AM, Bruzzone L, Molinuevo S and
Etcheverry SB: A possible role of oxidative stress in the
vanadium-induced cytotoxicity in the MC3T3E1 osteoblast and UMR106
osteosarcoma cell lines. Toxicology. 147:89–99. 2000.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Qian J, Jiang F, Wang B, Yu Y, Zhang X,
Yin Z and Liu C: Ophiopogonin D prevents H2O2-induced injury in
primary human umbilical vein endothelial cells. J Ethnopharmacol.
128:438–445. 2010.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Watcharanurak K, Zang L, Nishikawa M,
Yoshinaga K, Yamamoto Y, Takahashi Y, Ando M, Saito K, Watanabe Y
and Takakura Y: Effects of upregulated indoleamine 2, 3-dioxygenase
1 by interferon γ gene transfer on interferon γ-mediated antitumor
activity. Gene Ther. 21:794–801. 2014.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Ji G, Zhang Y, Yang Q, Cheng S, Hao J,
Zhao X and Jiang Z: Genistein suppresses LPS-induced inflammatory
response through inhibiting NF-κB following AMP kinase activation
in RAW 264.7 macrophages. PLoS One. 7(e53101)2012.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Dumitru CD, Ceci JD, Tsatsanis C,
Kontoyiannis D, Stamatakis K, Lin JH, Patriotis C, Jenkins NA,
Copeland NG, Kollias G and Tsichlis PN: TNF-alpha induction by LPS
is regulated posttranscriptionally via a Tpl2/ERK-dependent
pathway. Cell. 103:1071–1083. 2000.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Thakur R, Trivedi R, Rastogi N, Singh M
and Mishra DP: Inhibition of STAT3, FAK and Src mediated signaling
reduces cancer stem cell load, tumorigenic potential and metastasis
in breast cancer. Sci Rep. 5(10194)2015.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Andújar I, Rios JL, Giner RM and Recio MC:
Pharmacological properties of shikonin-a review of literature since
2002. Planta Med. 79:1685–1697. 2013.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Neumann D: Is TAK1 a direct upstream
kinase of AMPK? Int J Mol Sci. 19(2412)2018.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Jiang T, Yu JT, Zhu XC, Wang HF, Tan MS,
Cao L, Zhang QQ, Gao L, Shi JQ, Zhang YD and Tan L: Acute metformin
preconditioning confers neuroprotection against focal cerebral
ischaemia by pre-activation of AMPK-dependent autophagy. Br J
Pharmacol. 171:3146–3157. 2014.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Kurumbail RG and Calabrese MF: Structure
and regulation of AMPK. Exp Suppl. 107:3–22. 2016.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Zhou X, Cao Y, Ao G, Hu L, Liu H, Wu J,
Wang X, Jin M, Zheng S, Zhen X, et al: CaMKKβ-dependent activation
of AMP-activated protein kinase is critical to suppressive effects
of hydrogen sulfide on neuroinflammation. Antioxid Redox Signal.
21:1741–1758. 2014.PubMed/NCBI View Article : Google Scholar
|
|
57
|
She C, Zhu LQ, Zhen YF, Wang XD and Dong
QR: Activation of AMPK protects against hydrogen peroxide-induced
osteoblast apoptosis through autophagy induction and NADPH
maintenance: New implications for osteonecrosis treatment? Cell
Signal. 26:1–8. 2014.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Lu B, Gong X, Wang ZQ, Ding Y, Wang C, Luo
TF, Piao MH, Meng FK, Chi GF, Luo YN and Ge PF: Shikonin induces
glioma cell necroptosis in vitro by ROS overproduction and
promoting RIP1/RIP3 necrosome formation. Acta Pharmacol Sin.
38:1543–1553. 2017.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Zhang X, Cui JH, Meng QQ, Li SS, Zhou W
and Xiao S: Advance in anti-tumor mechanisms of shikonin, alkannin
and their derivatives. Mini Rev Med Chem. 18:164–172.
2018.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Gasparrini M, Forbes-Hernandez TY,
Giampieri F, Afrin S, Alvarez-Suarez JM, Mazzoni L, Mezzetti B,
Quiles JL and Battino M: Anti-inflammatory effect of strawberry
extract against LPS-induced stress in RAW 264.7 macrophages. Food
Chem Toxicol. 102:1–10. 2017.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Jung EH, Lee JH, Kim SC and Kim YW: AMPK
activation by liquiritigenin inhibited oxidative hepatic injury and
mitochondrial dysfunction induced by nutrition deprivation as
mediated with induction of farnesoid X receptor. Eur J Nutr.
56:635–647. 2017.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Kajiwara C, Kusaka Y, Kimura S, Yamaguchi
T, Nanjo Y, Ishii Y, Udono H, Standiford TJ and Tateda K: Metformin
mediates protection against legionella pneumonia through activation
of AMPK and mitochondrial reactive oxygen species. J Immunol.
200:623–631. 2018.PubMed/NCBI View Article : Google Scholar
|
|
63
|
GBD 2015 Chronic Respiratory Disease
Collaborators. Global, regional, and national deaths, prevalence,
disability-adjusted life years, and years lived with disability for
chronic obstructive pulmonary disease and asthma, 1990-2015: A
systematic analysis for the global burden of disease study 2015.
Lancet Respir Med. 5:691–706. 2017.PubMed/NCBI View Article : Google Scholar
|