Introduction
Brain edema is a common complication following
traumatic brain injury (TBI) around the world, which is associated
with high disability, mortality and morbidity rates. In 2014, the
Centers for Disease Control and Prevention documented 2.53 million
TBI-related emergency department visits and there were
approximately 288,000 TBI-related hospitalizations and 56,800
TBI-related deaths (1). As a
result, it severely affects the physical and mental wellbeing of
patients (2). In addition, brain
edema is an important secondary pathophysiological reaction
following severe cerebral trauma, which is characterized by the
pathological excessive water accumulation in spaces inside and
outside brain cells and an enlarged brain volume (3). This in turn induces and aggravates
intracranial hypertension. In some severe cases, it may induce
brain shift and cerebral hernia, which are major factors leading to
disability and mortality (4).
Although dehydration remains to be the major treatment option for
brain edema, the pathogenesis of traumatic brain edema is highly
complex and is generally induced by the combined action of multiple
factors, including disturbances in the microcirculation and the
blood-brain barrier (BBB), disorder of brain cell energy
metabolism, oxygen free radical damage, excitatory amino acid
poisoning, calcium ion overload, nitric oxide damage, lactic
acidosis and monoamine neurotransmitter toxicity (5). In recent years, aquaporin 4 (AQP4) and
the inflammatory response, which have been reported to participate
in the pathogenesis and therapeutic mechanism of brain edema, have
attracted increasing attention (6).
The AQP family consists of water channel proteins
that participate in water homeostasis regulation (7). It consists of 13 members (AQP1-13)
that are conserved across mammalian species, among which 7 (AQP1,
3, 4, 5, 8, 9 and 11) can be detected in the central nervous system
(CNS) (8). AQP4 is mostly expressed
in astrocytes and is the water channel with the highest abundance
in brain, which serves a vital role in water and ion homeostasis
(9). In addition, AQP4 has also
been widely suggested to exert a central and multifunctional role
in brain edema formation (6). In a
previous study, AQP4-deficient mice were not found to differ from
wild-type mice in terms of brain morphology and other physiological
functions in a previous study (10). However, during pathological
processes such as fluid infusion into the parenchyma, focal frozen
injury of the cortex and the implantation of melanoma cells,
AQP4-deficient mice exhibited increased severe brain swelling
compared with that in wild-type mice (10). However, a recent study by Yao et
al (11) found that, AQP4
knockout has been previously demonstrated to reduce the infarct
volume and brain edema, to provide a neuroprotective effect in mice
with permanent middle cerebral artery occlusion (11). AQP4 has also been reported to be
associated with inflammation, where AQP4 downregulation could
downregulate inflammatory cytokines, including interleukin (IL)-6,
IL-1β, tumor necrosis factor (TNF)-α and IL-10, which can protect
neonatal rats from brain edema induced by hypoxia-ischemia
(12). In addition, a previous
study revealed that 3% hypotonic saline (HS) can reduce brain edema
resulting from TBI by downregulating TNF-α and AQP4, reducing cell
apoptosis (13). However, the
effect of AQP4 on HS-mediated protection from traumatic brain
edema, the related mechanism and the regulatory mechanism of the
inflammatory response are not fully understood. Therefore, the aim
of the present study was to assess the effect of HS on TBI-induced
brain edema and the associated inflammatory response using a rat
model.
Materials and methods
Animals and TBI model
A total of 60 adult male Sprague-Dawley (SD) rats
(weight range, 220-270 g, 7 weeks old) were provided by Beijing
Vital River Laboratory (Beijing, China). Animal treatments and
experiments were carried out in accordance with the ‘Guide for the
Care and Use of Laboratory Animals' of the National Institutes of
Health (14). The present research
protocol was approved the Institutional Animal Care and Use
Committee of Jining No. 1 People's Hospital (Jining, China). Each
rat was maintained in an individual cage with free access to water
and food and allowed 1 week of adaptive feeding at 24˚C and
relative humidity of 55-65% on a normal light cycle (12/12-h,
lights on from 7:00 a.m. to 7:00 p.m.) prior to experimental
surgery to minimize animal suffering.
A controlled cortical impact model of TBI was
induced in vivo in rats as described previously (15). Animals were anesthetized by
isoflurane inhalation (4% for induction and 1.8% for maintenance)
mixed with pure oxygen at a flow rate of 500 ml/min (cat. no. R630;
Shenzhen Ruiwode Life Technology Co., Ltd.); animals were
maintained at 37±0.5˚C throughout the operation using thermal mats.
Rats were placed on a stereotaxic frame and secured with an incisor
bar and two ear bars. Subsequently, a craniotomy (diameter, 5 mm)
was performed on the right parietal cortex (3 mm lateral and
posterior from Bregma) with a dental drill. Disruption of the dura
and the associated vasculature was avoided as much as possible
during the process of craniotomy. A PCI 3000 PinPoint Precision
Cortical Impactor (Hatteras Instruments, Inc.), equipped with an
impactor tip (diameter, 4 mm), was used to deliver an impact at a
velocity of 4.5 m/sec. the dwell time was 75 msec and the
deformation was 1 mm. Following the induction of TBI, the piece of
resected skull was immediately replaced and secured with bone wax.
The incision was then closed using interrupted 4-0 silk stitches.
An identical procedure with the exception of impact delivery was
performed on sham rats.
Experimental procedure
Rats were randomized into three groups: i) Sham
group (n=18); ii) vehicle-treated TBI group (n=21); and iii)
HS-treated TBI group (n=21). For HS treatment, animals were given
an intravenous injection of 10% HS (10 g NaCl in 100 ml deionized
water) via the tail vein (0.08 ml/g; infusion rate, 0.3 ml/h) 3 h
after the induction of TBI (16,17).
For vehicle-treated rats, an equivalent amount of normal saline
(0.9 g NaCl in 100 ml deionized water) was infused intravenously.
The rats were sacrificed 2 days after treatment for further
analyses.
MRI
Sequential MRI was conducted in 21 rats (including
5, 8 and 8 from the sham, HS-treated and vehicle-treated groups,
respectively) at 0 and 48 h after TBI using a 7.0 T scanner
(BioSpec Products, Inc.) equipped with a four-channel phased array
rat head coil.
T2-weighted turbo spin-echo images (T2WI) were
obtained using the fast spin-echo sequence (under conditions of
field-of-view, 35x35 mm; matrix, 256x256; number of scans, 20;
slice thickness, 1 mm; echo time, 33 msec; repetition time, 2838.2
msec; number of averages, 1). The obtained images were analyzed
using the OsiriX software (version 4.2.2; http://www.osirix-viewer.com). To quantify the size of
the lesion on T2WI, both the brain and lesion areas were delineated
on a single slice by two researchers who reached a consensus, where
the region of interest defined by the researchers on each slice
containing the lesion was used. The brain and lesion volumes were
summed for all animals.
Over the course of TBI, the contralateral brain area
was determined according to the aforementioned method and was also
delineated in the obtained images in accordance with rescaled
drawings from the Paxinos and Watson Atlas (18). Hyperintense pixels in the
ipsilateral cortex on the T2WI were regarded as significantly
higher signals (P<0.05) compared with those in the contralateral
hemisphere. Additionally, lesion volume on the T2WI (Vu) was
calculated by multiplying the slice thickness by the hyperintense
area on each slice. To compensate for the effect of brain swelling,
the adjusted brain swelling (swelling %) and lesion volume (Ve %),
which were presented in the form of volume increases in the
affected hemisphere, were computed according to the equation below
(19):
where Vu and Ve represent the uncorrected and
corrected lesion volumes, respectively and Vc and Vi represent the
contralateral and ischemic hemisphere volumes, respectively.
Neurological deficit tests
The modified neurological severity score (mNSS) of
each group was evaluated at 48 h following TBI in a blinded manner
according to a previously described method (20). Neurological function scores ranged
from 0-18, with a higher score indicating a more severe injury
(20).
Hematoxylin and eosin (H&E) and
immunohistochemical (IHC) staining
Coronal sections of the cerebral cortical tissues
around the contusion site were selected for H&E and IHC
staining. Rats were sacrificed and perfused with PBS via the left
ventricle followed by 4% paraformaldehyde for fixation. Then, the
collected cerebral tissues were fixed at room temperature in
buffered paraformaldehyde (4%) for 24 h embedded in paraffin and
sectioned into 4-µm slices. H&E staining was performed after
deparaffinization by xylene followed by a descending ethanol
gradient at room temperature, where the stained tissues were
examined under a light microscope (magnification, x400).
For IHC staining, rats were sacrificed and perfused
with PBS via the left ventricle followed by 4% paraformaldehyde for
fixation. Cortical tissues around the contusion site were collected
and fixed at room temperature in 4% buffered paraformaldehyde for
24 h embedded in paraffin and sectioned into 4-µm slices, which
were then deparaffinized using xylene and rehydrated by descending
ethanol gradient. Hydrogen peroxide (3%) was diluted with deionized
water and used to block endogenous peroxidase activity for 10 min
at room temperature. Slices were incubated in citrate buffer (at
pH=6.0) for 10 min at 98˚C for antigen retrieval and incubated with
10% goat serum (cat. no. ZLI-9022; Origene Technologies, Inc.)
blocking solution for 20 min at 37˚C. Subsequently, the slices were
incubated with the following primary antibodies: Rabbit anti-rat
glial fibrillary acidic protein (GFAP; 1:2,000; cat. no. Z0334;
Dako; Agilent Technologies, Inc.), chicken anti-rat albumin
(1:1,000; cat. no. ab106582; Abcam) and rabbit anti-AQP4 (1:1,000;
cat. no. ab128906; Abcam), overnight at 4˚C. The slices were then
washed twice with PBS and then incubated at 37˚C with the relevant
polymeric horseradish peroxidase (HRP)-labeled anti-rabbit
immunoglobulin G secondary antibody (Super Vision IHC kit; cat. no.
SV0002; Boster Biological Technology) for 30 min at 37˚C. After
staining with 3,3'-diaminobenzidine for ~3 min at room temperature,
all slices were counterstained with hematoxylin for 2 min at 37˚C,
dehydrated and mounted on coverslips for light microscopic
examination. IHC staining for albumin, GFAP and AQP4 was examined
by ImageJ software 1.8.0 (National Institutes of Health). In total,
six visual fields from each slice (magnification, x400) were
examined and the average optical density (OD) was determined for
all images.
Determination of brain water content
(BWC)
Brain edema was evaluated by calculating the BWC of
all animals according to a previously described method (21). The dry and wet weights of the brain
were determined, where the water content was calculated for all
samples based on the following equation: 100% x [(wet weight - dry
weight)/wet weight].
Blood sodium concentration assay
Before the experiment ended, a 1-ml blood sample was
collected from the rat tail vein under anesthesia as described
previously (22). Blood sodium
concentration was analyzed immediately using an ABL80 type
automatic blood gas system (Radiometer Medical Ltd.) by another
technician blinded to the experimental groups.
ELISA
The plasma level of IL-1β was determined using
ELISA. Blood samples (1 ml) were collected from the rat abdominal
aorta under anesthesia as aforementioned and then subjected to 15
min centrifugation at 800 x g at 4˚C. The plasma IL-1β
concentration was measured using a commercial ELISA kit (cat. no.
F15810, Shanghai Xitang Biological Technology Co., Ltd.) in
accordance with the manufacturer's protocol. Then, the OD at 450 nm
was determined using a microplate reader.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from cells and brain tissue
from the area surrounding the edema using RNAiso plus (cat. no.
9109; Takara Biotechnology Co., Ltd.). RNA quantification was
carried out using a spectrophotometer (NanoDrop 2000; PEQLAB
Biotechnologie GmbH), and cDNA was synthesized from 1.0 µg RNA by
reverse transcriptase M-MLV (cat. no. 2641; Takara Biotechnology
Co., Ltd.) according to manufacturer's protocol. The temperature
and duration for reverse transcription was as follows: 42˚C for 10
min and 95˚C for 2 min. The primer sequences used for RT-PCR were
as follows: NF-κB forward, 5'-ACAGCCTGGTAGTGCGGTCGT-3' and reverse,
5'-TCAGCAAGTGGCTAGTCTGT-3'; IL-1β forward,
5'-AAAAGCTTGGTGATGTCTGG-3' and reverse, 5'-TTT CAACACGCAGGACAGG-3';
AQP4 forward, 5'-CTTTCT GGAAGGCAGTCTCAG-3' and reverse,
5'-CCACACCGA GCAAAACAAAGAT-3'; and GADPH (the reference gene),
forward, 5'-CGGATTTGGTCGTATTGGG-3' and reverse,
5'-CTGGAAGATGGTGATGGGATT-3'. qPCR was performed with
SYBR® Premix Ex™ Taq (Takara Biotechnology Co., Ltd)
using an ABI 7500 RT PCR System (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The thermocycling conditions were as follows:
95˚C for 10 sec, followed by 40 cycles of 95˚C for 5 sec and 60˚C
for 30 sec. A dissociation curve was used to examine the detected
signal specificity, which constituted a single peak. The
2-ΔΔCq method was used for
quantification of expression (23).
Western blotting
The perilesional brain tissue was isolated from each
treatment group 2 days following TBI, which was subjected to
homogenization with cytoplasmic buffer supplemented with KCl (10
mM), HEPES (10 mM, at pH=7.9), EGTA (0.1 mM), EDTA (0.1 mM),
Nonidet P-40 (0.15%), DTT (1 mM), NaF (10 mM), β-glycerophosphate
(50 mM), and Na3VO4 (5 mM) and phosphatase
inhibitors (Roche Diagnostics). The homogenates were subjected to
30 min centrifugation at 12,000 x g at 4˚C, following which the
supernatant was discarded and the pellet was not disturbed. To
extract nuclear protein, a nuclear buffer was supplemented with
NaCl (400 mM), HEPES (20 mM, at pH=7.9), EGTA (1 mM), EDTA (1 mM),
Nonidet P-40 (0.50%), DTT (1 mM), NaF (10 mM),
Na3VO4 (5 mM), β-glycerophosphate (50 mM), in
addition to the protease inhibitor cocktail and the buffer was
added to the aforementioned pellet. Subsequently, the homogenates
were subjected to a further 15 min incubation on ice after
vortexing at room temperature for 20 sec, which was repeated four
times. The homogenates were then subjected to 15 min of
centrifugation at 12,000 x g at 4˚C to assess the nuclear
NF-κB.
AQP4 expression was measured using total protein
extracts. Protein was collected from brain tissues around the
injury site using a total protein extraction kit (Bei Jing Pu Li
Lai Gene Technology Co., Ltd.) in accordance with the
manufacturer's protocol. Then, the resultant homogenates were
subjected to 30 min of centrifugation at 12,000 x g at 4˚C and the
supernatants were collected to analyze the amount of total protein
extracted using a bicinchoninic acid protein assay kit (cat. no.
P0012S; Beyotime Institute of Biotechnology). Protein lysates (20
µg) were subjected to 15 min of denaturation at 90˚C and 10%
SDS-PAGE was performed before the proteins were transferred onto
PVDF membranes (EMD Millipore). Subsequently, the membranes were
subjected to 2 h blocking using 5% non-fat milk at 37˚C and
incubated overnight at 4˚C with primary antibodies, including
anti-NF-κB (1:1,000; cat. no. 8242; Cell Signaling Technology,
Inc.), anti-AQP4 (1:800; cat. no. ab46182; Abcam), anti-zonula
occludens-1 (ZO-1; 1:400; cat. no. 61-7300; Invitrogen; Thermo
Fisher Scientific, Inc.), anti-claudin-5 (1:1,000; cat. no.
35-2500; Invitrogen; Thermo Fisher Scientific, Inc.), anti-occludin
(1:400; cat. no. 33-1500; Invitrogen; Thermo Fisher Scientific,
Inc.), anti-GAPDH (1:1,000; cat. no. AF1186; Beyotime Institute of
Biotechnology) and anti-Histone H3 (1:1,000; cat. no. AH433;
Beyotime Institute of Biotechnology). Then, the membranes were
rinsed with PBST containing 0.1% Tween-20 for 10 min and repeated
five times. Subsequently, the PVDF membranes were incubated with a
HRP-labeled goat anti-rabbit IgG (1:5,000; cat. no. abs20002; Absin
Biotechnology Co., Ltd.) or HRP-labeled goat anti-mouse IgG
secondary antibody (1:5,000; cat. no. abs20001; Absin) at 25˚C for
1 h. The bands were visualized using the ECL (cat. no. P0018;
Beyotime Institute of Biotechnology) method and analyzed with the
ImageJ software 1.8.0 (National Institutes of Health). GAPDH was
used to normalize the expression of the proteins, whilst histone H3
was used to normalize the expression of NF-κB in the nuclear
extracts.
Statistical analysis
Each value was measured three times and data are
presented as the mean ± SD. SPSS 18.0 (IBM Corp.) was used for
statistical analyses. No repeated-measure (matched) values were
available in the current study, and all of the values were examined
by one-way ANOVA and Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
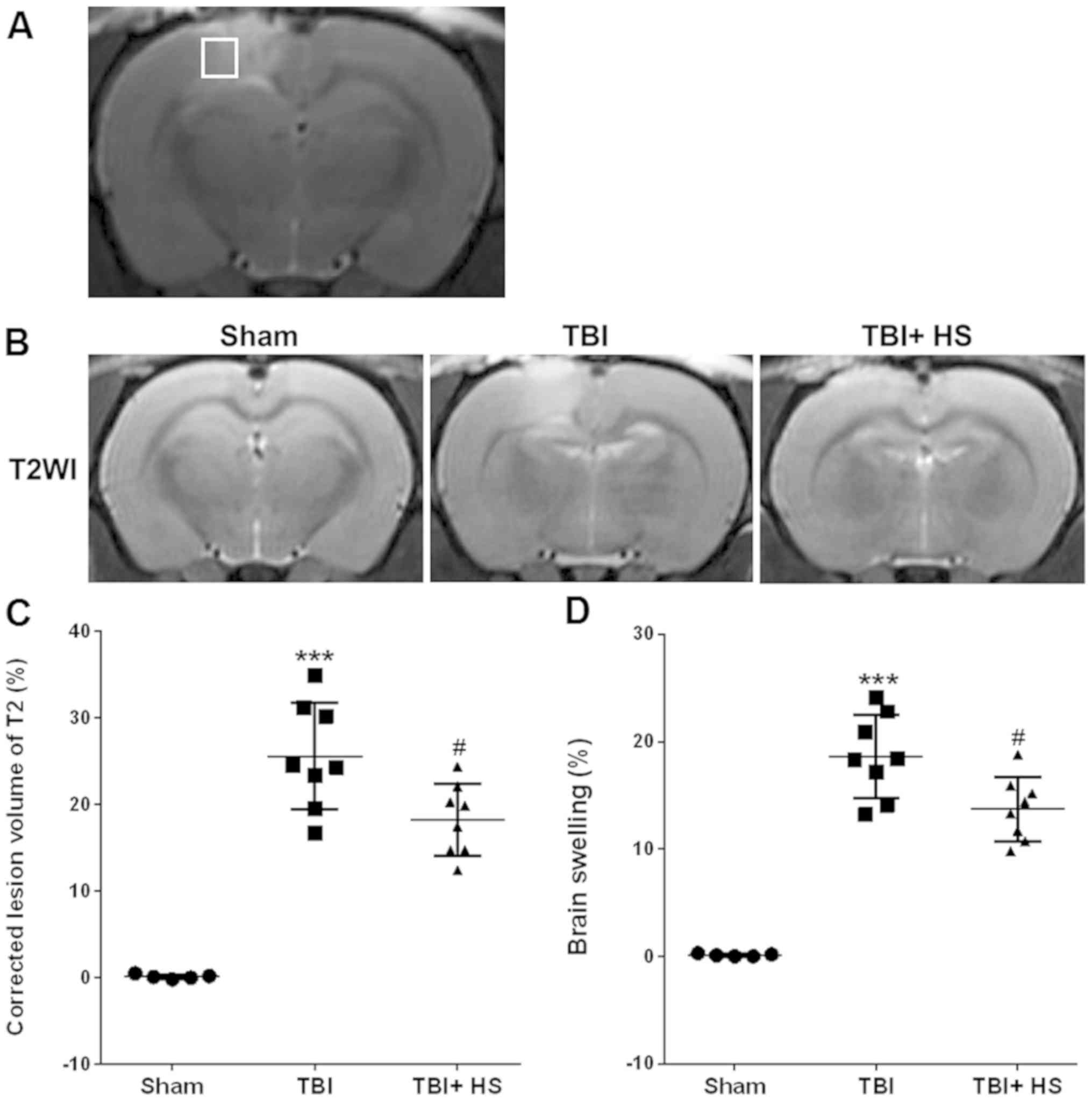
HS treatment reduces the volume of
TBI-induced lesions on T2WI
MRI was first performed to determine the efficacy of
HS treatment on lesions induced by TBI, as observed on the T2WI
(Fig. 1A). On day 3 following TBI,
the corrected T2WI Ve were found to be 18.25±4.13 and 25.59±6.13%
in the HS and vehicle groups, respectively (P<0.05; Fig. 1B and C). In addition, HS treatment significantly
alleviated brain swelling compared with that in the vehicle group,
where the HS and vehicle groups exhibited values of 13.73±2.98 and
18.63±3.87%, respectively (P<0.05; Fig. 1D).
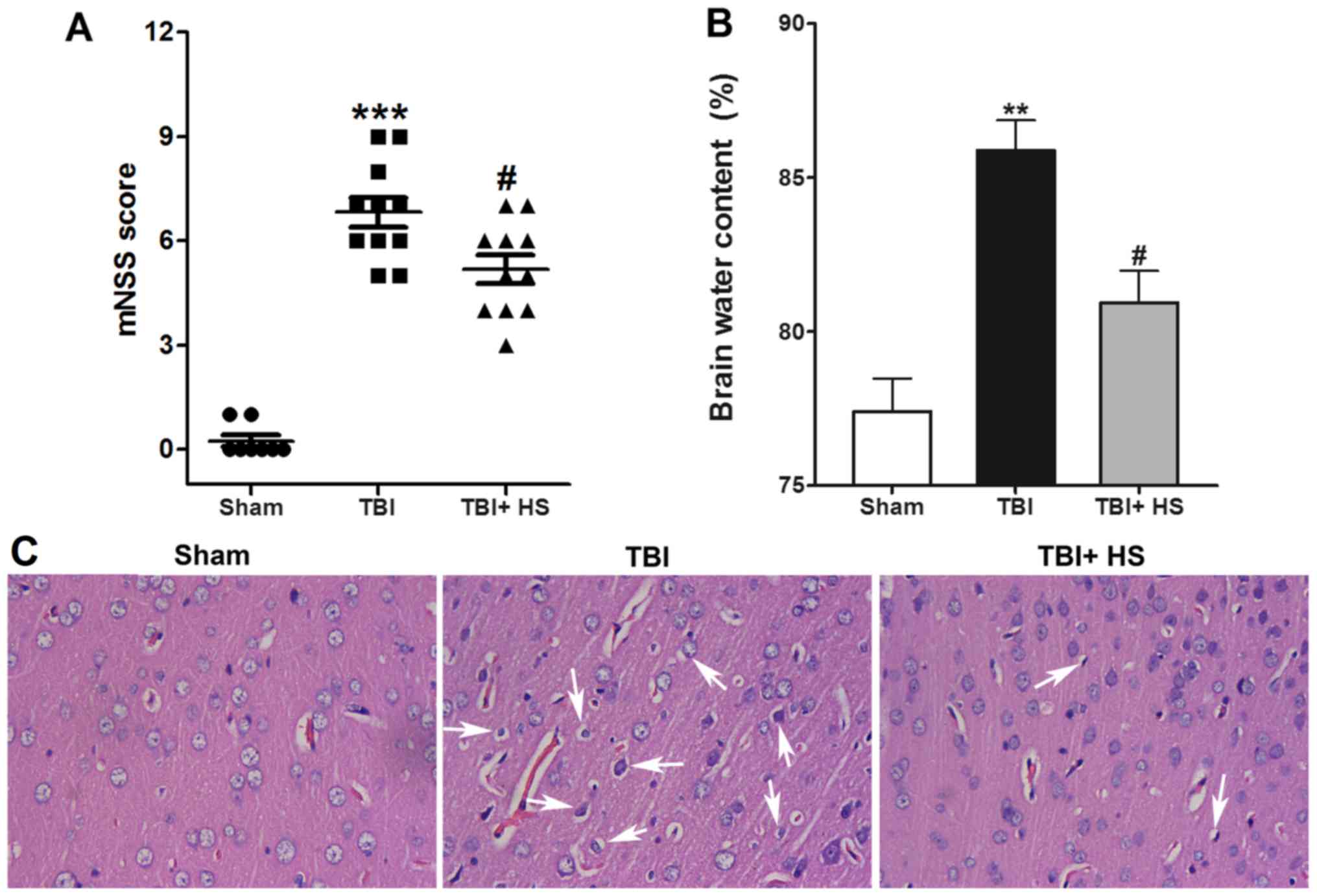
HS improves TBI-induced neurological
functional deficits and brain edema
Compared with the sham group, the mNSS scores of the
TBI group were found to be significantly increased at 48 h
post-TBI. HS treatment significantly reduced the mNSS scores
compared with those of TBI alone, suggesting that HS significantly
reduced neurological deficits (P<0.05; Fig. 2A). Subsequently, TBI-induced brain
edema was evaluated by measuring the BWC, where it was demonstrated
that the BWC was significantly increased in the ipsilateral cortex
in the TBI group compare with that in the sham group, which was
significantly reversed by HS treatment (Fig. 2B).
H&E staining results identified a regular
arrangement of neurons in the sham group, along with capillary
morphogenesis and normal glial cells. However, 2 days following
TBI, the majority of cells were disorderly arranged where the
nuclei became pyknotic or were severely shrunken. In addition,
cellular swelling and hypervacuolization were observed in certain
areas. HS treatment significantly improved TBI induced nuclei
shrinking and cell swelling (Fig.
2C).
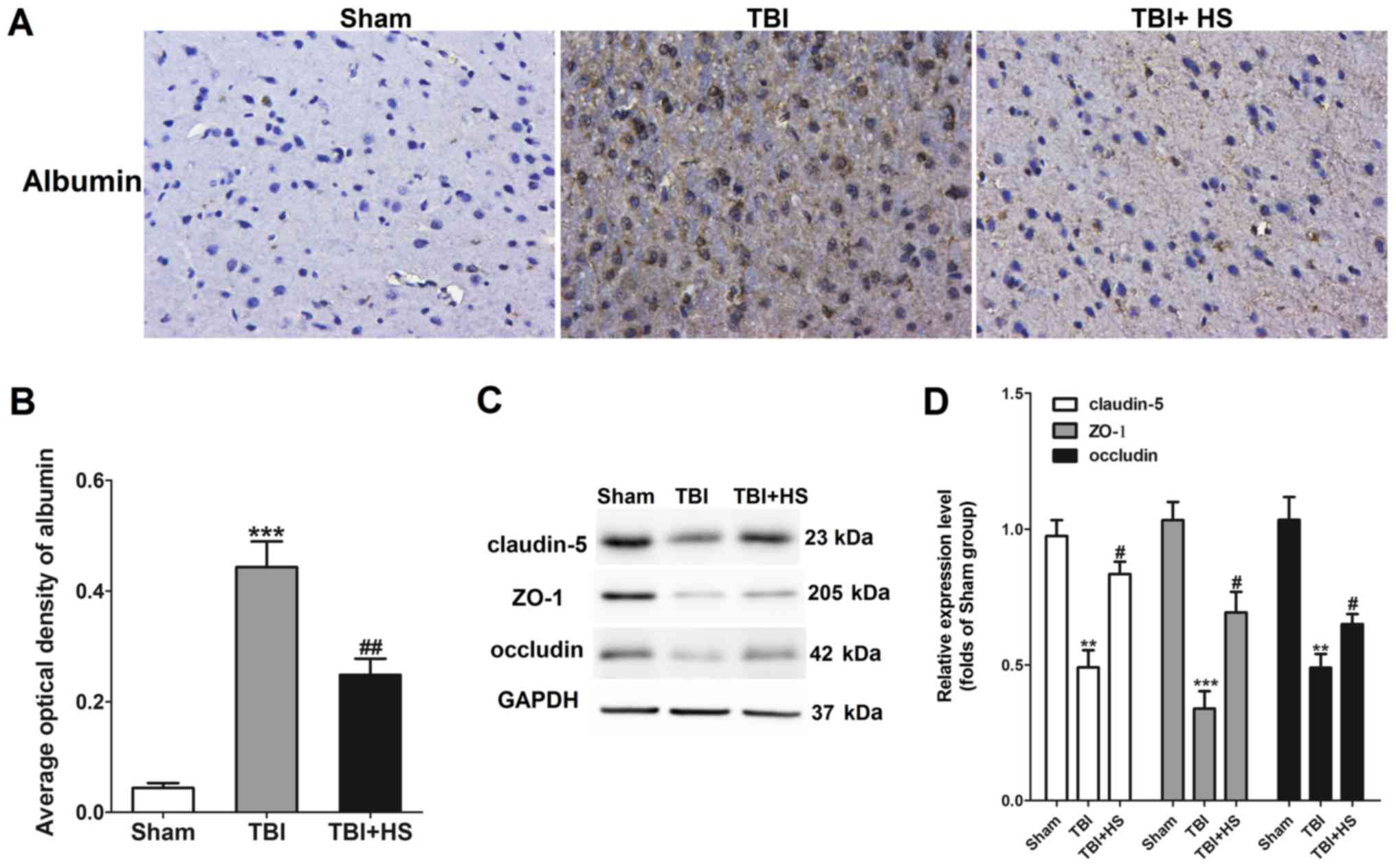
HS improves TBI-induced BBB
injury
BBB leakage was next assessed by IHC staining for
albumin and western blotting for claudin-5, ZO-1 and occludin. It
was found that albumin staining was largely negative in the sham
group, whilst a significant increase in albumin expression was
observed in tissues from the TBI group (Fig. 3A and B), indicating BBB injury. Albumin
expression was found to be significantly downregulated in tissues
in the HS group compared with that in the TBI group alone.
In addition, tight junction (TJ)-associated
proteins, including occludin, ZO-1 and claudin-5, were also
revealed to be significantly downregulated by TBI, which was
significantly reversed by HS treatment (Fig. 3D). Therefore, these results
suggested that HS treatment alleviated BBB destruction following
TBI.
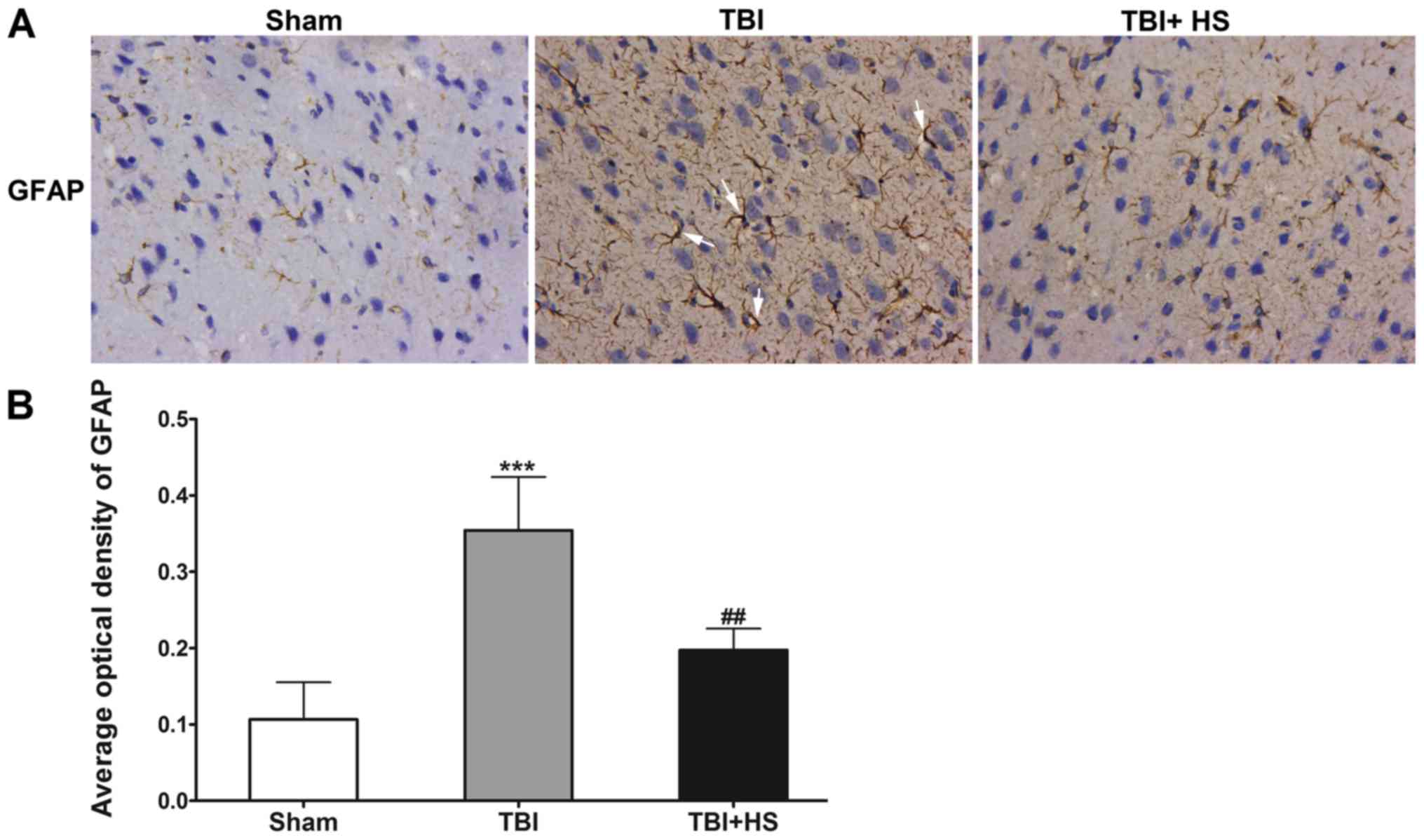
HS reduces TBI-induced astrocyte
activation
Astrocyte activation was evaluated by IHC staining
for GFAP (Fig. 4). GFAP expression
was detected in the sham group, which identified few astrocytes
with finely branched protrusions. In the TBI group, GFAP expression
was demonstrated to be elevated with astrocyte hyperplasia, cell
body enlargement, protrusions, thickening and extravascular
collagen fibers all observed, suggesting the activation of
astrocytes. However, after HS treatment, GFAP expression remained
to be elevated compared with that in the sham group, but was
significantly lower compared with that in the TBI group, suggested
that the astrocyte protrusions did not completely recover their
fine morphology. Therefore, it was speculated that HS mitigated
astrocyte activation around the site of TBI.
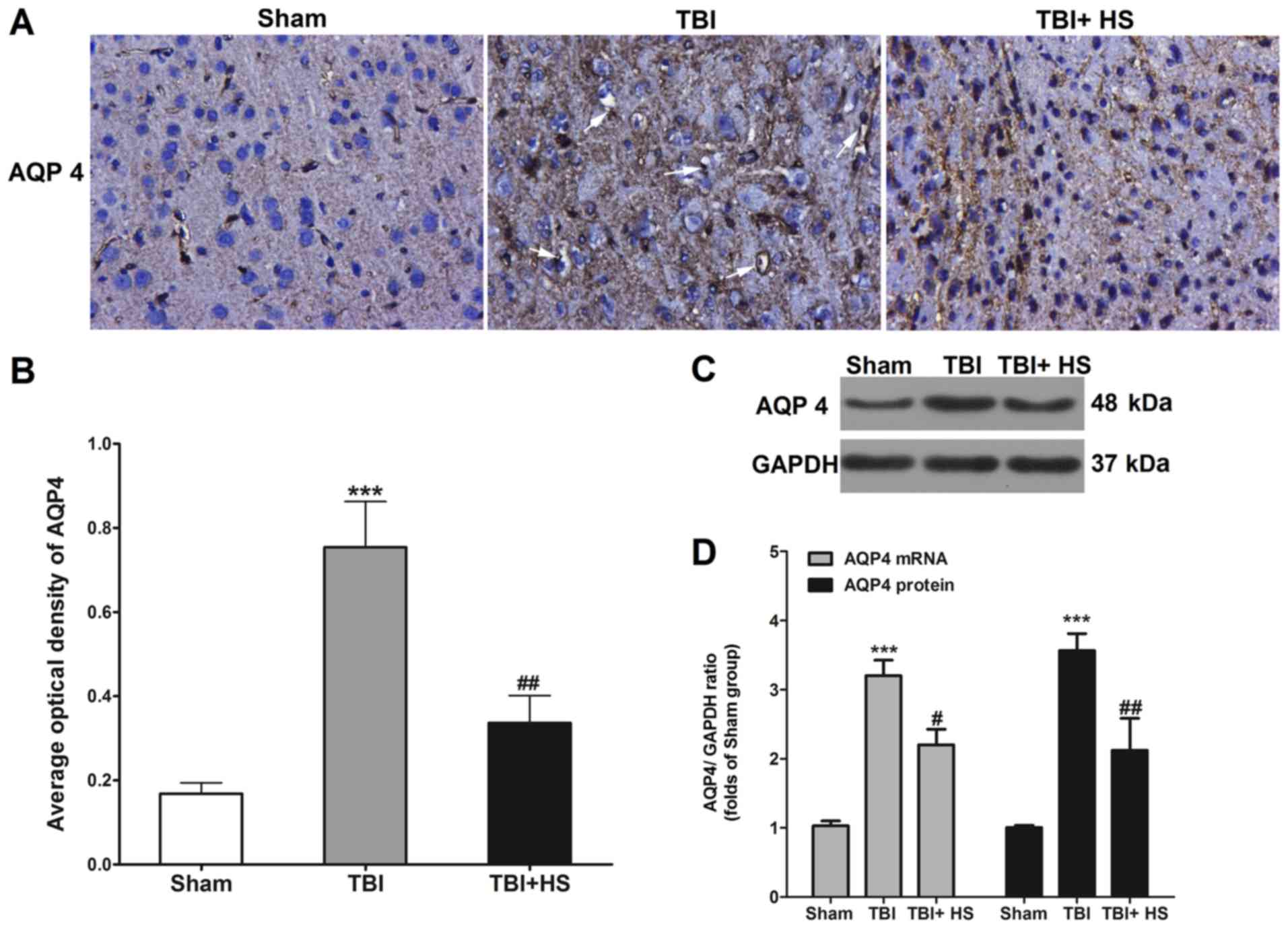
AQP4 expression is reduced by HS
treatment
AQP4 expression tissues around the contusion site
was evaluated using IHC (Fig. 5). A
small amount of AQP4 expression was observed in the sham group,
which was significantly upregulated in the TBI group, where AQP4
staining was clearly observed around the blood vessels. After HS
treatment, AQP4 expression was significantly downregulated compared
with that in the TBI group, where the positively stained particles
were substantially smaller.
AQP 4 expression in peri-TBI tissues was also
measured using western blotting and RT-qPCR. Compared with the sham
group, the protein and mRNA expression levels of AQP4 in the TBI
group were found to be markedly increased, which was reversed by HS
treatment.
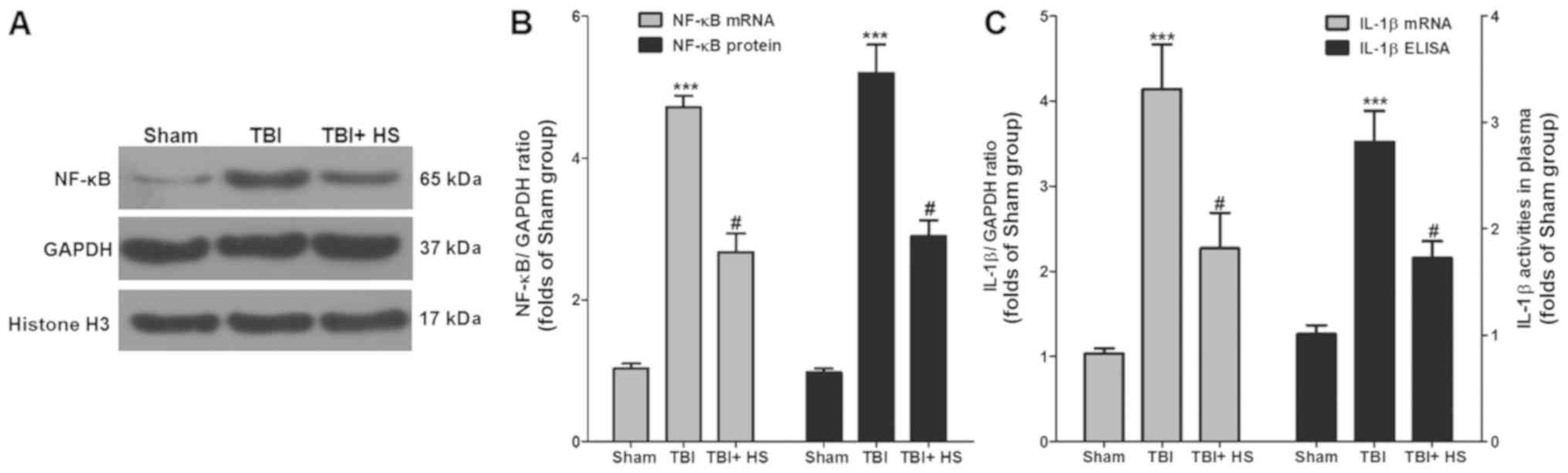
Detection of inflammatory factors
The blood sodium concentration was analyzed using an
automatic blood gas system, where the results showed that HS
treatment remarkably increased the blood sodium ion concentration
from 144.50±0.94 mmol/l (sham group) to 158.11±2.2 mmol/l
(P<0.05; Table I).
 | Table IBlood sodium concentrations in each
treatment group. |
Table I
Blood sodium concentrations in each
treatment group.
| Treatment
group | Sodium
concentration, mmol/l |
|---|
| Sham | 144.50±0.94 |
| TBI | 149.75±1.33 |
| TBI + HS |
158.11±2.2a |
The expression levels of inflammatory factors in
each group were next measured (Fig.
6). Western blotting and RT-qPCR results suggested that,
compared with the sham group, the protein and mRNA expression
levels of NF-κB in the TBI group were significantly higher whilst
HS treatment significantly reduced NF-κB expression compared with
those in the TBI group.
Serum IL-1β levels were next measured using ELISA.
Compared with the sham group, the serum IL-1β level in the TBI
group was found to be significantly higher, which was significantly
reversed by HS treatment. RT-qPCR was also performed in the tissue
samples to detect the changes in IL-1β mRNA expression. Compared
with the sham group, IL-1β mRNA expression in the TBI group was
significantly higher, but HS treatment significantly downregulated
IL-1β expression compared with that in the TBI group.
Discussion
HS has previously demonstrated efficacy in treating
brain edema in the clinic, but the precise underlying mechanism
remains unknown. In the present study, MRI was performed to
investigate the effects of HS on brain edema induced by TBI in
rats. It was demonstrated that HS treatment significantly reduced
the degree of brain edema in rats, restored the integrity of the
BBB, reduced the activation of astrocytes and downregulated AQP4
expression. In addition, detection of inflammatory factors
indicated that HS significantly reduced IL-1β and NF-κB
expression.
AQP4 is the most important isoform of aquaporin in
mammalian brain tissues, which regulate brain water homeostasis and
has been previously demonstrated to be closely correlated with the
formation of traumatic brain edema (6). Brain edema is related to AQP4 in
astrocytes peripheral to the BBB (24). Previous studies on brain edema have
focused on the balance between brain tissues and serum osmotic
pressure, where a reduction in serum osmotic pressure results in a
concentration gradient that induces brain edema (25,26).
However, it has been reported that edemas mainly form in the
astrocyte foot processes in capillaries instead of neurons
(27). In addition, AQP4 is mainly
expressed in only astrocytes and not neurons (28). Therefore, AQP4 represents an
important target for treating brain edema. A previous study
reported that AQP4 expression in astrocytes is increased under
numerous pathological conditions in which cytotoxic edema serves a
dominant role, including TBI, stroke, meningitis and hyponatremia
(29). Appelboom et al
(30) showed that the severity of
brain edema formed due to brain tissue hemorrhage is associated
with the AQP4 gene. Additionally, Suzuki et al (31) found that both protein and mRNA
expression levels of AQP 4 are upregulated in swollen glial cells
in the cerebrum and the contusion area. Sun et al (32) also showed that brain edema in rats
reaches a peak 24 h following TBI modeling, where AQP4 expression
in astrocytes in the injury area was significantly enhanced, whilst
that in the peripheral area is weakened compared with the injury
area and AQP4 expression in other areas remained unchanged. Yao
et al (11) also reported
that AQP4 knockout can evidently reduce brain edema and injury in
cerebral ischemia mice. In AQP4 knockdown mice, improved outcomes
could be achieved after TBI by reducing cytotoxic brain water
accumulation (33). Furthermore,
AER-271, an AQP4 inhibitor, has been previously demonstrated to
block acute brain edema, whilst improving the short-term outcomes
of a rat pediatric asphyxia cardiac arrest model (34). Previous studies have also indicated
that AQP4 deletion in mice can reduce neuronal death and brain
edema following acute ischemic stroke and water intoxication
(35,36). Therefore, suppressing AQP4 at an
appropriate time can reduce the accumulation of edema fluid in the
brain, thus lowering morbidity and mortality. The present results
also indicated that HS can be applied to treat TBI-induced brain
edema, with this effect is closely associated with the suppression
of AQP4.
TBI-induced brain edema involves a highly complex
mechanism, where the destruction of BBB integrity is an important
link between vasogenic brain edema and secondary injuries (37). TJ proteins, consisting of the
transmembrane complex (occludins and claudins) and cytoplasmic (ZO
family) proteins, are anchored to the actin cytoskeleton and are
markers of BBB integrity, which contribute to its structural
strength (38). It has been
reported in both animal and human TBI studies that increased BBB
permeability contributes to the pathophysiology of brain edema,
which may be caused by increased BBB permeability via the
degradation of TJ proteins, including claudin-5, occludin and ZO-1
(39,40). In addition, AQP4 expression is
abundant in structures associated with the BBB, including the foot
processes of astrocytes and the basolateral plasma membrane of the
ependymal epithelium (9). A
previous study has also revealed that AQP 4 content is closely
associated with the integrity of the BBB (41). The present study also suggested that
albumin expression in post-TBI tissues was significantly increased,
suggesting that the BBB was compromised, whilst HS treatment
restored BBB integrity in rats.
The activation of astrocytes is mainly characterized
by cell body hypertrophy of astrocytes with the notable thickening
of protrusions (42). Additionally,
it has been previously reported that astrocyte activation is a
common stress response in the CNS under numerous pathological
conditions (43). In the present
study, IHC staining suggested that the expression of GFAP was
enhanced. A previous study also revealed that 6 h after brain
trauma, astrocytes were found to be activated in the cortex around
the injury site, where additional reactive astrocytes were found in
the entire injured hemisphere at 24 h and were more pronounced than
at 72 h (44). In another study,
activated astrocytes were previously observed in the hippocampus 24
h, 3 and 7 days after brain injury, but the number of activated
astrocytes was not significantly changed (45). The present study indicated that 2
days after TBI, GFAP expression was significantly elevated,
suggesting that astrocytes were activated, which was reversed by HS
treatment. However, the dynamic changes that occur in astrocytes
during HS treatment require further investigation.
Elevated inflammation is characteristic of brain
edema, such that brain edema formation and posttraumatic
inflammation are two key pathological processes that are associated
with secondary brain injury (46).
According to a previous study, 10% HS could alleviate brain edema
by inhibiting the Na-K-2Cl co-transporter isoform 1 (NKCC1), where
this process was found to be mechanistically due to the attenuation
of NKCC1 stimulation by IL-1β and TNF-α (47). Previous studies have also reported
the relationship between inflammation and brain edema. Holmin and
Mathiesen (48) revealed that
intraventricular injection of TNF-α and IL-1β, or the injection of
TNF-α into swine through the internal carotid artery can induce an
inflammatory response in the brain and increase BBB permeability in
the brain cortex, thus inducing vasogenic brain edema. In addition,
Ito et al (49) showed that
the lateral intraventricular injection of IL-1β can induce AQP4
expression in brain tissue in rats and that an inhibitor can
suppress the induction of astrocyte expression in vitro,
suggesting the possible participation of IL-1β in brain edema
formation via the activation of astrocytes through the induction
pathway. In addition, a previous study revealed that suppressing
IL-1β can mitigate TBI and brain edema and improve behaviors
associated with depression in rats after brain injury (50). In the present study, it was
demonstrated that the effect of HS on brain edema resulting from
TBI was closely associated with reductions in the levels of
TBI-induced inflammatory factors.
NF-κB serves as a key player in the
neuroinflammatory response in astrocytes during various
neurological disorders by promoting the transcription of
inflammatory mediators, including proinflammatory cytokines and
chemokines (51). IL-1β can be
triggered by binding to the receptor IL-1R, which is an upstream
signaling factor of NF-κB (52). It
has also been demonstrated that IL-1β can increase BBB permeability
and disruption by downregulating the expression of TJ proteins
(53). A recent study also revealed
that inhibiting NF-κB binding activity and translocation can reduce
inflammatory cell infiltration, reduce matrix metalloproteinase-9
expression and ameliorating BBB disruption (54). The present results suggested that HS
treatment inhibited NF-κB, IL-1β and BBB permeability, but no
direct evidence regarding the relationship between NF-κB/IL-1β and
BBB function as a result of HS treatment of brain edema was
identified, which require further study.
However, some limitations associated with the
current study should be noted. Firstly, the present study only
evaluated brain edema at the end point of the experiments using
T2WI. Therefore, monitoring of the progression of brain edema
through different MRI sequences should be addressed in future
studies. Additionally, although previous studies have reported cell
death after TBI (55,56), the present study did not evaluate
parameters of cell death, including that of neurons and various
types of glial cells, since it was mainly focused on brain edema
during TBI and HS treatment. Therefore, future studies should focus
on the mechanism of cell apoptosis in TBI. Another limitation of
the present study was that the treatment effect of only one dose
(10%) of HS on brain edema was evaluated. Increased attention
should be provided to the effects of different concentrations of HS
to identify the optimal treatment conditions for different types of
brain edema in future studies.
In conclusion, the present study identified the
protective effect of tail vein injections of HS in a rat model of
TBI-induced brain edema via MRI, BWC detection and histology. It
was demonstrated that HS significantly reduced the upregulated
expression of AQP4 induced by TBI. Further analysis of inflammatory
factors suggested that the efficacy of HS in brain edema resulting
from TBI was closely related to the downregulation of AQP4, the
restoration of BBB integrity and the suppression of inflammatory
factors IL-1β and NF-κB. However, the precise regulatory mechanism
remains to be further elucidated.
Acknowledgements
Not applicable.
Funding
This work was supported by Projects of medical and
health technology development program in Shandong province (grant
no. 2017WS037).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HZ performed animal experiments and prepared this
manuscript. JL performed the MRI experiment. YL performed the
H&E and IHC experiments. CS measured the BWC. GF performed
molecular biology experiments. WL performed statistical analysis.
LF designed this study and was a major contributor in writing the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All of the animal procedures were conducted in
accordance with the Guidelines for Care and Use of Laboratory
Animals, and were approved by the Animal Care and Use Committee at
Jining No. 1 People's Hospital (approval no. 2017037).
Patient consent for publication
Not Applicable
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Capizzi A, Woo J and Verduzco-Gutierrez M:
Traumatic brain injury: An overview of epidemiology,
pathophysiology, and medical management. Med Clin North Am.
104:213–238. 2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Honeybul S: Reconsidering the role of
hypothermia in management of severe traumatic brain injury. J Clin
Neurosci. 28:12–15. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Donkin JJ and Vink R: Mechanisms of
cerebral edema in traumatic brain injury: Therapeutic developments.
Curr Opin Neurol. 23:293–299. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Adeva MM, Souto G, Donapetry C, Portals M,
Rodriguez A and Lamas D: Brain edema in diseases of different
etiology. Neurochem Int. 61:166–174. 2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kochanek PM, Jackson TC, Ferguson NM,
Carlson SW, Simon DW, Brockman EC, Ji J, Bayır H, Poloyac SM,
Wagner AK, et al: Emerging therapies in traumatic brain injury.
Semin Neurol. 35:83–100. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Mamtilahun M, Tang G, Zhang Z, Wang Y,
Tang Y and Yang GY: Targeting Water in the Bra in: Role of
Aquaporin-4 in Ischemic Brain Edema. Curr Drug Targets. 20:748–755.
2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kitchen P, Day RE, Salman MM, Conner MT,
Bill RM and Conner AC: Beyond water homeostasis: Diverse functional
roles of mammalian aquaporins. Biochim Biophys Acta.
1850:2410–2421. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Verkman AS, Anderson MO and Papadopoulos
MC: Aquaporins: Important but elusive drug targets. Nat Rev Drug
Discov. 13:259–277. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Papadopoulos MC and Verkman AS: Aquaporin
water channels in the nervous system. Nat Rev Neurosci. 14:265–277.
2013.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Papadopoulos MC, Manley GT, Krishna S and
Verkman AS: Aquaporin-4 facilitates reabsorption of excess fluid in
vasogenic brain edema. FASEB J. 18:1291–1293. 2004.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Yao X, Derugin N, Manley GT and Verkman
AS: Reduced brain edema and infarct volume in aquaporin-4 deficient
mice after transient focal cerebral ischemia. Neurosci Lett.
584:368–372. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Liu S, Mao J, Wang T and Fu X:
Downregulation of Aquaporin-4 protects brain against hypoxia
ischemia via anti-inflammatory mechanism. Mol Neurobiol.
54:6426–6435. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yin J, Zhang H, Chen H, Lv Q and Jin X:
Hypertonic Saline Alleviates Brain Edema After Traumatic Brain
Injury via Downregulation of Aquaporin 4 in Rats. Med Sci Monit.
24:1863–1870. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press (US), Washington (DC), 2011.
|
|
15
|
Huang L, Cao W, Deng Y, Zhu G, Han Y and
Zeng H: Hypertonic saline alleviates experimentally induced
cerebral oedema through suppression of vascular endothelial growth
factor and its receptor VEGFR2 expression in astrocytes. BMC
Neurosci. 17(64)2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wen M, Ye J, Han Y, Huang L, Yang H, Jiang
W, Chen S, Zhong W, Zeng H and Li DY: Hypertonic saline regulates
microglial M2 polarization via miR-200b/KLF4 in cerebral edema
treatment. Biochem Biophys Res Commun. 499:345–353. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Niu F, Dong J, Xu X, Zhang B and Liu B:
Mitochondrial division inhibitor 1 prevents early-stage induction
of mitophagy and accelerated cell death in a rat model of moderate
controlled cortical impact brain injury. World Neurosurg.
122:e1090–e1101. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Paxinos G and Watson C: The rat brain in
stereotaxic coordinates. 6th Edition. Elsevier, 2007.
|
|
19
|
Gerriets T, Stolz E, Walberer M, Müller C,
Kluge A, Bachmann A, Fisher M, Kaps M and Bachmann G: Noninvasive
quantification of brain edema and the space-occupying effect in rat
stroke models using magnetic resonance imaging. Stroke. 35:566–571.
2004.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhao M, Liang F, Xu H, Yan W and Zhang J:
Methylene blue exerts a neuroprotective effect against traumatic
brain injury by promoting autophagy and inhibiting microglial
activation. Mol Med Rep. 13:13–20. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Khaksari M, Soltani Z, Shahrokhi N,
Moshtaghi G and Asadikaram G: The role of estrogen and
progesterone, administered alone and in combination, in modulating
cytokine concentration following traumatic brain injury. Can J
Physiol Pharmacol. 89:31–40. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Li W, Wang R, Xie H, Zhang J and Jia Z:
Changes of pathological and physiological indicators affecting drug
metabolism in rats after acute exposure to high altitude. Exp Ther
Med. 9:98–104. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Stokum JA, Kurland DB, Gerzanich V and
Simard JM: Mechanisms of astrocyte-mediated cerebral edema.
Neurochem Res. 40:317–328. 2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kimelberg HK: Current concepts of brain
edema. Review of laboratory investigations. J Neurosurg.
83:1051–1059. 1995.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Hatashita S, Hoff JT and Salamat SM:
Ischemic brain edema and the osmotic gradient between blood and
brain. J Cereb Blood Flow Metab. 8:552–559. 1988.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wasterlain CG and Torack RM: Cerebral
edema in water intoxication. II. An ultrastructural study. Arch
Neurol. 19:79–87. 1968.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Hara-Chikuma M and Verkman AS:
Physiological roles of glycerol-transporting aquaporins: The
aquaglyceroporins. Cell Mol Life Sci. 63:1386–1392. 2006.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Papadopoulos MC and Verkman AS:
Aquaporin-4 gene disruption in mice reduces brain swelling and
mortality in pneumococcal meningitis. J Biol Chem. 280:13906–13912.
2005.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Appelboom G, Bruce S, Duren A, Piazza M,
Monahan A, Christophe B, Zoller S, LoPresti M and Connolly ES:
Aquaporin-4 gene variant independently associated with oedema after
intracerebral haemorrhage. Neurol Res. 37:657–661. 2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Suzuki R, Okuda M, Asai J, Nagashima G,
Itokawa H, Matsunaga A, Fujimoto T and Suzuki T: Astrocytes
co-express aquaporin-1, -4, and vascular endothelial growth factor
in brain edema tissue associated with brain contusion. Acta
Neurochir Suppl (Wien). 96:398–401. 2006.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Sun MC, Honey CR, Berk C, Wong NL and Tsui
JK: Regulation of aquaporin-4 in a traumatic brain injury model in
rats. J Neurosurg. 98:565–569. 2003.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Yao X, Uchida K, Papadopoulos MC, Zador Z,
Manley GT and Verkman AS: Mildly reduced brain swelling and
improved neurological outcome in aquaporin-4 knockout mice
following controlled cortical impact brain injury. J Neurotrauma.
32:1458–1464. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Wallisch JS, Janesko-Feldman K, Alexander
H, Jha RM, Farr GW, McGuirk PR, Kline AE, Jackson TC, Pelletier MF,
Clark RSB, et al: The aquaporin-4 inhibitor AER-271 blocks acute
cerebral edema and improves early outcome in a pediatric model of
asphyxial cardiac arrest. Pediatr Res. 85:511–517. 2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Manley GT, Fujimura M, Ma T, Noshita N,
Filiz F, Bollen AW, Chan P and Verkman AS: Aquaporin-4 deletion in
mice reduces brain edema after acute water intoxication and
ischemic stroke. Nat Med. 6:159–163. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
36
|
Katada R, Akdemir G, Asavapanumas N,
Ratelade J, Zhang H and Verkman A: Greatly improved survival and
neuroprotection in aquaporin-4-knockout mice following global
cerebral ischemia. FASEB J. 28:705–714. 2014.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Jha RM, Kochanek PM and Simard JM:
Pathophysiology and treatment of cerebral edema in traumatic brain
injury. Neuropharmacology. 145 (Pt B):230–246. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Alves JL: Blood-brain barrier and
traumatic brain injury. J Neurosci Res. 92:141–147. 2014.
|
|
39
|
Lu L, Wang M, Yuan F, Wei X and Li W:
Roles of elevated 20 HETE in the breakdown of blood brain barrier
and the severity of brain edema in experimental traumatic brain
injury. Mol Med Rep. 17:7339–7345. 2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Kim JY, Ko AR, Hyun HW and Kang TC: ETB
receptor-mediated MMP-9 activation induces vasogenic edema via ZO-1
protein degradation following status epilepticus. Neuroscience.
304:355–367. 2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Nico B, Frigeri A, Nicchia GP,
Quondamatteo F, Herken R, Errede M, Ribatti D, Svelto M and Roncali
L: Role of aquaporin-4 water channel in the development and
integrity of the blood-brain barrier. J Cell Sci. 114:1297–1307.
2001.PubMed/NCBI
|
|
42
|
Escartin C and Bonvento G: Targeted
activation of astrocytes: A potential neuroprotective strategy. Mol
Neurobiol. 38:231–241. 2008.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Hol EM and Pekny M: Glial fibrillary
acidic protein (GFAP) and the astrocyte intermediate filament
system in diseases of the central nervous system. Curr Opin Cell
Biol. 32:121–130. 2015.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Okimura Y, Tanno H, Fukuda K, Ohga M,
Nakamura M, Aihara N and Yamaura A: Reactive astrocytes in acute
stage after experimental brain injury: Relationship to extravasated
plasma protein and expression of heat shock protein. J Neurotrauma.
13:385–393. 1996.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Burda JE, Bernstein AM and Sofroniew MV:
Astrocyte roles in traumatic brain injury. Exp Neurol. 275:305–315.
2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Hopp S, Nolte MW, Stetter C, Kleinschnitz
C, Sirén AL and Albert-Weissenberger C: Alleviation of secondary
brain injury, posttraumatic inflammation, and brain edema formation
by inhibition of factor XIIa. J Neuroinflammation.
14(39)2017.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Huang LQ, Zhu GF, Deng YY, Jiang WQ, Fang
M, Chen CB, Cao W, Wen MY, Han YL and Zeng HK: Hypertonic saline
alleviates cerebral edema by inhibiting microglia-derived TNF-α and
IL-1β-induced Na-K-Cl Cotransporter up-regulation. J
Neuroinflammation. 11(102)2014.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Holmin S and Mathiesen T: Intracerebral
administration of interleukin-1beta and induction of inflammation,
apoptosis, and vasogenic edema. J Neurosurg. 92:108–120.
2000.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Ito H, Yamamoto N, Arima H, Hirate H,
Morishima T, Umenishi F, Tada T, Asai K, Katsuya H and Sobue K:
Interleukin-1beta induces the expression of aquaporin-4 through a
nuclear factor-kappaB pathway in rat astrocytes. J Neurochem.
99:107–118. 2006.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Fenn AM, Skendelas JP, Moussa DN,
Muccigrosso MM, Popovich PG, Lifshitz J, Eiferman DS and Godbout
JP: Methylene blue attenuates traumatic brain injury-associated
neuroinflammation and acute depressive-like behavior in mice. J
Neurotrauma. 32:127–138. 2015.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Skaper SD, Facci L, Zusso M and Giusti P:
An inflammation-centric view of neurological disease: Beyond the
neuron. Front Cell Neurosci. 12(72)2018.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Liu R, Pan MX, Tang JC, Zhang Y, Liao HB,
Zhuang Y, Zhao D and Wan Q: Role of neuroinflammation in ischemic
stroke. Neuroimmunol Neuroinflamm. 4:158–166. 2017.
|
|
53
|
Blamire AM, Anthony DC, Rajagopalan B,
Sibson NR, Perry VH and Styles P: Interleukin-1β -induced changes
in blood-brain barrier permeability, apparent diffusion
coefficient, and cerebral blood volume in the rat brain: A magnetic
resonance study. J Neurosci. 20:8153–8159. 2000.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Lee WT, Tai SH, Lin YW, Wu TS and Lee EJ:
YC 1 reduces inflammatory responses by inhibiting nuclear factor κB
translocation in mice subjected to transient focal cerebral
ischemia. Mol Med Rep. 18:2043–2051. 2018.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Sater AP, Rael LT, Tanner AH, Lieser MJ,
Acuna DL, Mains CW and Bar-Or D: Cell death after traumatic brain
injury: Detrimental role of anoikis in healing. Clin Chim Acta.
482:149–154. 2018.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Raghupathi R: Cell death mechanisms
following traumatic brain injury. Brain Pathol. 14:215–222.
2004.PubMed/NCBI View Article : Google Scholar
|