Introduction
Kindler syndrome [KS; Online Mendelian Inheritance
in Man (OMIM) no. 173650] is a rare subtype of inherited
epidermolysis bullosa (1). Since KS
was first described by Theresa Kindler in 1954(2) and, to date, ~250 affected individuals
have been reported worldwide (1).
KS is characterized by skin fragility acral blister formation
beginning at birth or in early infancy, diffuse cutaneous atrophy,
poikiloderma, photosensitivity (which is severe during childhood
and usually weakens after adolescence), palmoplantar hyperkeratosis
and pseudo syndactyly (1). Mucosal
manifestations are also common and include hemorrhagic gingivitis
and mucositis, premature loss of teeth, periodontal disease and
labial leukokeratosis (1). Other
mucosal findings may include ectropion, anal stenosis, esophageal
strictures/stenosis, urethral stenosis/strictures, colitis and
severe phimosis (1). Severe
long-term complications of KS have been indicated to include
mucosal strictures, periodontitis and aggressive squamous cell
carcinomas (1). The phenotypic
spectrum ranges from mild to severe based on the severity of
symptoms, the organs involved and the age of onset (1). KS is inherited in an autosomal
recessive manner and FERMT1 has been identified to be a
pathogenic gene (1). To date,
>73 unique pathogenic variants in FERMT1 have been
reported in KS worldwide (1). The
majority of the pathogenic variants are nonsense mutations and
result in the loss of function of kindlin-1 gene (1). The diagnosis of KS is established in
patients with typical clinical manifestations and is based on the
identification of either biallelic FERMT1 pathogenic
variants on molecular genetic testing or suggestive histologic
findings and/or immunolabeling on skin biopsy (1). The current study reported a Chinese
patient with KS based on typical clinical manifestations and a
novel FERMT1 mutation, and presented a brief summary of all
pathogenic mutations in FERMT1 reported in KS between 1984
and May 2020.
Materials and methods
Ethics statement and clinical
participants
The current study was approved by the Ethics
Committee of Shanghai Skin Disease Hospital, and was performed
according to the principles of the Declaration of Helsinki
(3). Peripheral blood was collected
from the patient, his parents and 100 controls who were admitted to
the Dermatology Clinic of Shanghai Skin Disease Hospital, Shanghai,
China. The clinical information of all participants is presented in
Table I. Written informed consent
was obtained from all participants.
 | Table IClinical information of all
participants. |
Table I
Clinical information of all
participants.
| A, Patients |
|---|
| | Age, year | Sex | Location | Date of
recruitment |
|---|
| Proband | 33 | Male | Shanghai,
China | July, 2019 |
| Father | 60 | Male | Shanghai,
China | July, 2019 |
| Mother | 58 | Female | Shanghai,
China | July, 2019 |
| B,
Controlsa |
| Number, n | Age, years
(mean) | Sex,
Male:Female | Location | Date of
recruitment |
| 100 | 22-58(36) | 47:53 | Shanghai,
China | October 2019-July
2020 |
DNA extraction
DNA was extracted from peripheral blood using a
Blood Genomic DNA Miniprep kit (cat. no. AP-MN-BL-GDNA-50; Axygen;
Corning Life Sciences), according to the manufacturer's
protocol.
Multi-gene panel test
Genomic DNA was extracted from the patient as
aforementioned and a DNA library was constructed using a Kapa
Illumina HTP library kit (cat. no. 7138008001; Roche Diagnostics)
according to the manufacturer's protocol (Roche NimbleGen, Inc.). A
total of 2 µl of the DNA library was extracted and quantified using
a Qubit 3.0 fluorescence quantitative analyzer (DNA library yield:
38 ng/µl x50 µl). Based on the quantitative fluorescence results,
100 ng DNA library was used for electrophoresis detection with 2%
agarose gel, and a library fragment at ~300 bp was considered
qualified for subsequent hybridization. The DNA library was
hybridized using a probe labeled with biotin (47˚C for 16-20 h) and
the target area was captured with magnetic beads coated with
streptavidin following the protocol of Roche NimbleGen, Inc. 166 ng
DNA library was extracted, and a genetic skin disease gene
detection panel (designed by the current study) was used to capture
the target area according to the protocol of Roche NimbleGen, Inc.
The panel designed in the present study included 541 genes of
monogenic hereditary diseases, which comprised the following 21
bullous disease-associated genes: KRT5, KRT14, PLEC, DST,
KLHL24, TGM5, DSP, PKP1, JUP, EXPH5, COL7A1, LAMA3, LAMB3, LAMC2,
ITGA6, ITGB4, COL17A1, CD151, ITGA3, PLCG2 and
FERMT1.
A total of 2 µl of the DNA library was extracted and
quantified using a Qubit 3.0 fluorescence quantifier (capture
library output, 9.2 ng/µl x20 µl). According to the quantitative
fluorescence results, 1 ng of the captured library was used to
detect the molecular size of the library using an Agilent 2100
Bioanalyzer (Agilent Technologies, Inc.), and a library fragment of
~300 bp was considered qualified for subsequent high-throughput
sequencing. An Illumina HiSeq X Ten sequencer (Illumina, Inc.) was
used for high-throughput sequencing of ~1 Gb/sample based on the
dilution capture library of the Qubit 3.0 fluorescence quantifier
and the Agilent 2100 Bioanalyzer. The sequencing reaction was
performed using the paired end direction and a 2x150 bp length. PCR
details are presented in Table
SI.
Data analysis and interpretation
The original data files were converted from bcl to
fastq format using Illumina CASAVA1.8 (Illumina, Inc.) and reads
were compared to the GRCh38/hg38 human genome reference using
SAMtools (v1.10.2; https://sourceforge.net/projects/samtools/),
Burrows-Wheeler Aligner (v0.7.17; https://sourceforge.net/projects/bio-bwa/), Genome
Analysis Toolkit (v4.1.4.1; https://github.com/broadinstitute/gatk/releases) and
Picard (v1.119; https://sourceforge.net/projects/picard/) to remove
repeated sequences and identify genetic variants. Nonsynonymous
genetic variants in the exon region were identified. Frequencies of
the identified genetic variants were required to be equal to 0 or
<1% in the Asian population in all used databases including
1,000 genomes databases (internationalgenome.org/data-portal/sample) and
ExAC (exac.broadinstitute.org/dbsnp). All identified
variants were evaluated by browsing through databases using gene
name or disease name as search terms, including Online Mendelian
Inheritance in Man (omim.org/), National Center for
Biotechnology Information (NCBS) Single Nucleotide Polymorphisms
database (ncbi.nlm.nih.gov/SNP/), NCBI ClinVar (ncbi.nlm.nih.gov/clinvar/) and Human Gene
Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php). The putative
effects on the proteins of the identified variants were explored
using various prediction algorithms, including PolyPhen2
(http://genetics.bwh.harvard.edu/pph2), MutationTaster
(mutationtaster.org/) and Sorting
Intolerant From Tolerant (sift.bii.a-star.edu.sg). The pathogenicity of the
identified variants was evaluated according to criteria established
by the American College of Medial Genetics and Genomics (ACMG)
(4).
Sanger sequencing
To verify the accuracy of the multi-gene panel test,
direct Sanger sequencing (5) was
performed to confirm whether the variants co-segregated with the
disease phenotype in the patient, his parents and the control group
using an ABI PRISM 3730XL automated sequencer (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The reverse and forward primers
were as follows: 5'-AGCGGTGAATGTATGTTGTC-3' and
5'-TCCTTGTCAGAGTTTAAGCTTCT-3'.
Literature review
The term ‘Kindler syndrome’ was used to search the
literature using PubMed between 1984 and May 2020 (https://pubmed.ncbi.nlm.nih.gov/) and the full
texts of retrieved literatures were read carefully. Pathogenic
mutations in FERMT1 identified in KS were summarized in
Table SII.
Results
Clinical features of the patient
The patient was a 33-year-old man born to healthy
non-consanguineous Chinese parents and had no siblings or children.
The patient's skin was fragile and blistered easily following
injury or friction at birth mainly in the knee, elbow, heel and hip
that undergo high friction. Furthermore, the patient's skin
gradually developed a parchment-like appearance, telangiectasia was
present on the face and neck, and diffuse cutaneous atrophy and
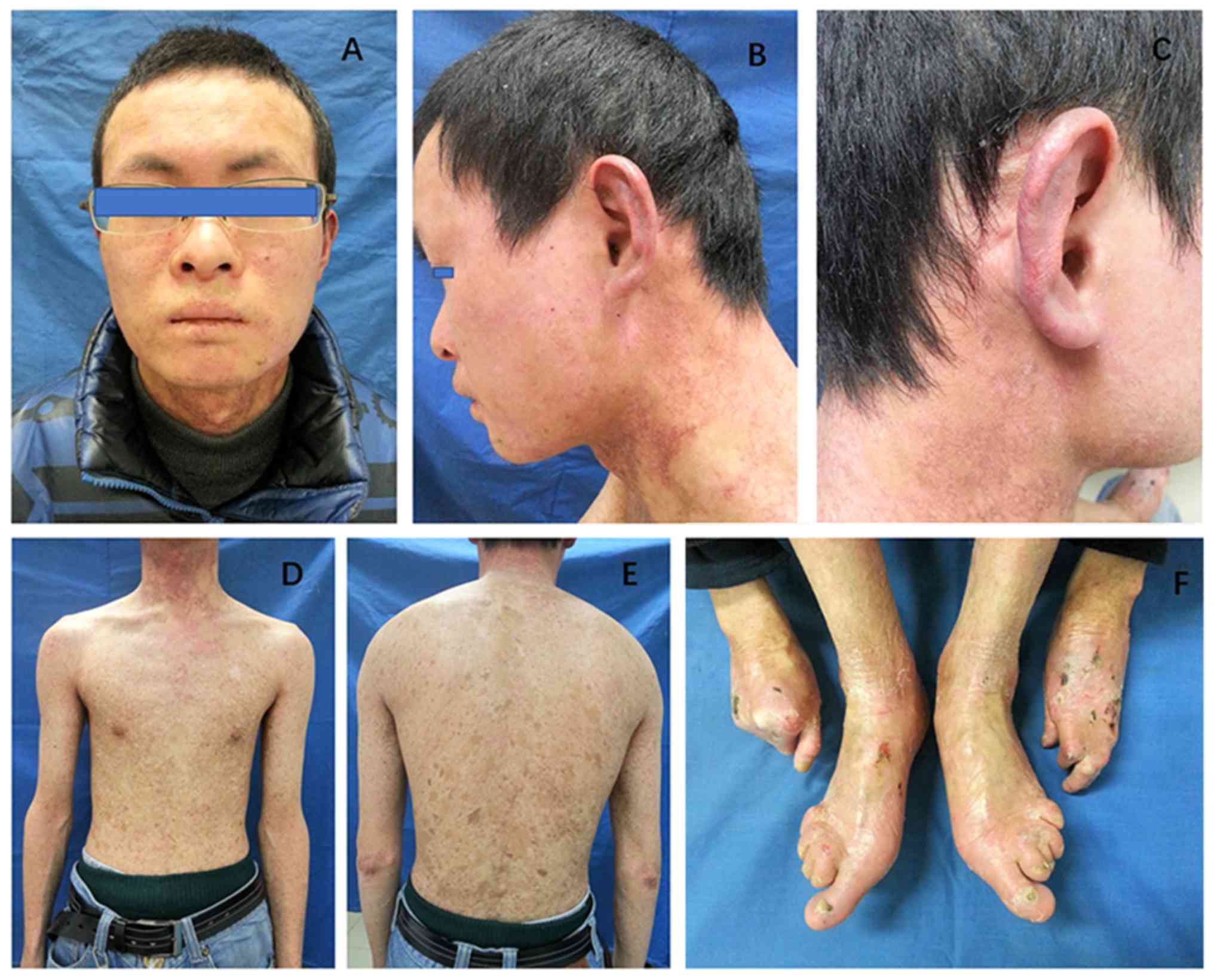
poikiloderma was present across the whole body (Fig. 1). The patient exhibited
photosensitivity in childhood, which eased followed puberty. The
patient's fingernails and toenails began to exhibit damage when at
~7 years old and gradually developed contracture, nail dystrophy,
muscular dystrophy and muscle weakness (Fig. 1). The patient underwent pneumothorax
at 22 years old and recovered following treatment. The patient also
exhibited significant eye damage including nearsightedness (left
eye, 500 degrees; right eye, 600 degrees), astigmatism, proneness
to eye fatigue and frequent keratitis and corneal ulcers.
Furthermore, the patient exhibited oral damage, including oral
ulcers, gingivitis, periodontal disease, persistent gingival
bleeding, prominent cavities in the right teeth and significantly
decreased chewing function. The patient was diagnosed with
tympanitis 2 years ago, which improved following treatment.
Additionally, the patient exhibited dysphagia and a narrow
esophagus, and could not swallow pills. Intestinal obstruction
occurred when the patient was young; but following treatment,
normal function was restored. The patient's foreskin had been
removed when young. Additionally, keratinizing plaques occurred
repeatedly on his palmar metatarsals. These plaques would fall off
by themselves and caused the patient apparent pain following
shedding. At the end of the study period, the condition of the
patient was stable and the patient was able to nurse himself.
Mutation analyses
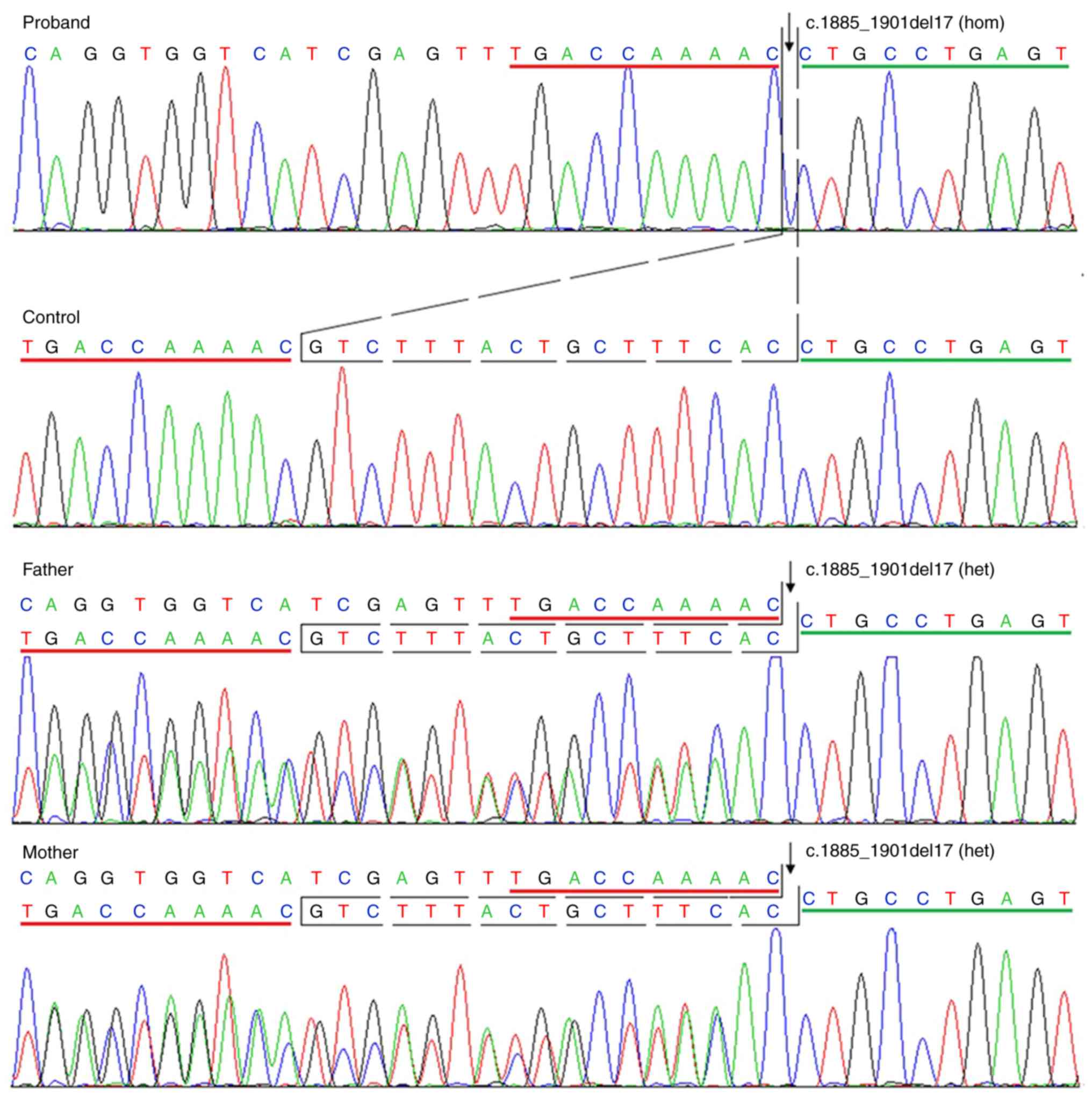
The multi-gene panel test revealed a homogeneous
mutation in FERMT1, c.1885_1901del (p.Val629fs), on exon 15
in the patient. An identical, heterogeneous mutation was reported
in the patient's parents, which was further confirmed by Sanger
sequencing. Furthermore, this mutation was absent in the 100
controls analyzed by Sanger sequencing (Fig. 2). Combined with the clinical
features and genetic analysis, the patient was diagnosed with KS.
Furthermore, a heterogeneous mutation c.745C>T (p.R249X) on exon
7 in the serpin family B member 7 (SERPINB7) gene was
identified in the patient using the multi-gene panel test.
Review of 83 FERMT1 mutations reported
in KS
Literature review from 1984 up to May 2020, and
inclusive of the data in the current study, revealed ~83 different
mutations in FERMT1 in different populations and included
frameshift (insertions and deletions), nonsense, missense and
splice mutations. Mutations were located in exons 1-15, introns 1,
7-11, 13-14 and regulatory regions. Only 23 were identified to be
recurrent in >1 report (Table
SII). The results demonstrated that >50% of the 83 mutations
produced a premature termination codon and may lead to absent
kindlin-1 protein or production of nonfunctional proteins.
Discussion
KS is caused by mutations in FERMT1 on
chromosomal locus 20p12.3(1).
FERMT1 contains 15 exons and spans 48.5 kb of genomic DNA
and encodes the 677-amino acid protein kindlin, a member of the
protein family of kindlin, which is an essential integrin activator
and component of keratinocyte focal adhesions, and serves a
regulatory role in keratinocyte migration, adhesion and
proliferation (6). These features
of KS may depend on the functions of kindlin-1. Numerous unique
pathogenic variants in FERMT1 have been reported in KS
(1). The current study reported a
case of KS and identified a novel mutation in FERMT1.
Additionally, a brief summary of pathogenic mutations in
FERMT1 was presented.
The patient exhibited a wide range of clinical
feature including fragile skin, blister formation,
photosensitivity, diffuse cutaneous atrophy and poikiloderma,
telangiectasia of the face and neck, contracture of the ends of
limbs, nail dystrophy, muscle, eye and oral damage, tympanitis,
dysphagia, a narrow esophagus, pneumothorax and palmoplantar
keratoderma. Furthermore, apart from pneumothorax and tympanites,
other aforementioned patient symptoms have been reported by
previous studies. Additionally, numerous symptoms that have been
previously reported in patients with KS were absent in the patient
outlined in the current study, including web formation, pseudo
syndactyly, genital malformation, osteoporosis (7), brachydactyly, bladder cancer (8), squamous carcinoma hyperhidrosis
(9), loss of teeth, ichthyosis,
microstomia cheilitis, halitosis (10), ectropion, urethral stenosis,
constipation, anal atresia (11),
xerosis, hypertelorism (12),
ulcerative colitis, cholangitis, cirrhosis (13), alopecia areata (14), depression and thyroid adenoma
(15). However, it remains unclear
whether some of these symptoms were a result of KS or were
coincidental. According to the literature search of the present
study, hundreds of cases of KS have been reported. These cases have
been demonstrated to exhibit similar and different symptoms, and
different disease-causing mutations without an obvious
genotype-phenotype correlation.
Genetic analyses revealed a homogeneous mutation in
the patient: c.1885_1901del (p.Val629fs) on exon 15. Furthermore, a
heterogeneous identical mutation was reported in the patient's
parents. To the best of our knowledge, this mutation has not been
previously reported. This mutation was determined to be pathogenic
according to the ACMG guidelines (4). Based on the clinical manifestation and
genetic test results, the patient was diagnosed with KS. The
mutation c.1885_1901del, which is a 17 bp deletion, generated a
frameshift change of the nucleotide sequence and a premature
termination codon 28 codons downstream. The resulting reading frame
coded for a mutant polypeptide lacking 16 amino acids at the C
terminus. This protein region homologous to the talin band
4.1/ezrin/radixin/moesin domain and its elimination is predicted to
lead to the loss of kindlin-1 linkage function between actin
cytoskeleton and integrin-associated signal platforms (10). Loss of kindlin-1 functions leads to
the destruction of epithelial barriers and the inflammatory
intestinal phenotype characteristic of KS (16). The current study did not perform a
mutant protein expression analysis as no further samples of the
patient's skin were available.
To date, c.1885_1901del (p.Val629fs) is the 5th
reported mutation in Chinese patients with KS, with the other four
mutations including three deletion mutations: c.994_995delCA
(17), a 17252-bp deletion mutation
(18) and g.63601_66617del
(14), and one missense mutation,
c.1343T>A (p.Met448Lys) (19).
c.994_995delCA was the only mutation that was recurrent and was
also reported in the Iranian population (20). Additionally, one heterogeneous
mutation c.745C>T (p.R249X) on exon 7 in the SERPINB7
gene was identified in the patient in the current study by the
multi-gene panel test. Mutations in SERPINB7 have been
reported to cause Nagashima-type palmoplantar keratosis (NPPK; OMIM
no. 615598), which is an autosomal recessive palmoplantar
keratoderma (21). However, the
c.745C>T (p.R249X) mutation was heterogeneous in this patient.
Therefore, the mutation did not cause NPPK. Therefore, although the
patient exhibited palmoplantar keratoderma, this symptom was caused
by KS, not NPKK.
Inclusive to the data of the current study, ~83
different pathogenic mutations in FERMT1 have been reported
from 1984 up to May 2020. These mutations were scattered throughout
the FERMT1 gene, including exons 1-15, introns 1, 7-11,
13-14 and regulatory regions exhibiting no obvious hotspots or
clustering. Numerous mutations were nonsense mutations or
frameshift variants (insertion and deletion variants). The majority
of these mutations were reported to be homogenous in patients and
few were compound heterogeneous mutations. Most of these mutations
result in loss of function and may lead to absent kindlin-1 protein
or the production of dysfunctional proteins (1). Only 23 mutations were revealed to be
recurrent in >1 report. Notably, c.676insC was the most common
mutation reported in 11 different reports (7,22-31)
in multiple populations, including German, Albanian, Kosovian,
Turkish, Serbian-Greek, Pakistani, Serbian, Australian, Indian,
Brazilian and Greek Caucasian, indicating that c.676insC may be a
mutation hotspot and serve as a founder effector in the genetic
pathogenesis of KS. Furthermore, c.910G>T occurred in 6 reports
(20,23,25,27,32,33),
c.328C>T in 5 reports (9,20,23,34,35),
c.1718+2T>C in 4 reports (20,22,36,37) in
multiple different populations and appeared to be relatively common
in FERMT1. Certain mutations occurred in the same population
in different reports, indicating population-specific pathogenic
mutations. For instance, c.1089delG (38-40)
was reported three times in only Japanese patients and
c.1848_1851dupGGAA (9,10,41)
was reported three times in only Turkish patients. Further
genotype-phenotype correlation studies are required to clarify
these associations.
In conclusion, the current study reported a case of
KS in a Chinese patient, identified one novel mutation
c.1885_1901del (p.Val629fs) on exon 15 in FERMT1, and
presented a summary of pathogenic FERMT1 mutations in KS.
These results expand the spectrum of known FERMT1 mutations
and provided a detailed mutation repertoire of FERMT1 in KS.
Early and accurate diagnosis of KS is pivotal for the evaluation of
potential complications and genetic counselling.
Supplementary Material
PCR details of a multi.gene panel
test.
Summary of fermitin family member
1mutations in Kindler syndrome.
Acknowledgements
Not applicable.
Funding
The current work was supported by Key Projects of
Shanghai Municipal Commission of Health and Family Planning (grant
no. 201640016).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the sequence read archive (SRA),
https://submit.ncbi.nlm.nih.gov/subs/bioproject/,
SRA access no.: PRJNA641496.
Authors' contributions
LM has been involved in drafting the manuscript and
revising it critically for important intellectual content, and the
acquisition of data, analysis and interpretation of data. XY, YW,
ZZ and LY have been involved in the acquisition of data, analysis
and interpretation of data. GZ, ML and XW have made substantial
contributions in the conception and design of the current study.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The current study was approved by the Ethics
Committee of the Shanghai Skin Disease Hospital, Shanghai, China
and the patient, his parents and all 100 controls provided written
informed consent.
Patient consent for publication
The patient provided written informed consent for
the publication of any associated data and accompanying images.
Competing interests
The authors declare that they have no conflicting
interests.
References
|
1
|
Youssefian L, Vahidnezhad H and Uitto J:
Kindler Syndrome In: GeneReviews((R)). Adam MP and Pagon RA (eds),
Seattle, WA, pp1-19, 2016.
|
|
2
|
Kindler T: Congenital poikiloderma with
traumatic bulla formation and progressive cutaneous atrophy. Br J
Dermatol. 66:104–111. 1954.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Rickham PP: Human experimentation Code of
ethics of the world medical association. Declaration of helsinki.
Br Med J. 2(177)1964.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Estrada-Rivadeneyra D: Sanger sequencing.
FEBS J. 284(4174)2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Jobard F, Bouadjar B, Caux F, Hadj-Rabia
S, Has C, Matsuda F, Weissenbach J, Lathrop M, Prud'homme JF and
Fischer J: Identification of mutations in a new gene encoding a
FERM family protein with a pleckstrin homology domain in kindler
syndrome. Hum Mol Genet. 12:925–935. 2003.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Mansur AT, Elcioglu NH, Aydingöz IE,
Akkaya AD, Serdar ZA, Herz C, Bruckner-Tuderman L and Has C: Novel
and recurrent KIND1 mutations in two patients with kindler syndrome
and severe mucosal involvement. Acta Derm Venereol. 87:563–565.
2007.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Fuchs-Telem D, Nousbeck J, Singer A,
McGrath JA, Sarig O and Sprecher E: New intragenic and promoter
region deletion mutations in FERMT1 underscore genetic homogeneity
in kindler syndrome. Clin Exp Dermatol. 39:361–367. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Lai-Cheong JE, Tanaka A, Hawche G, Emanuel
P, Maari C, Taskesen M, Akdeniz S, Liu L and McGrath JA: Kindler
syndrome: A focal adhesion genodermatosis. Br J Dermatol.
160:233–242. 2009.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kacar N, Semerci N, Ergin S, Pascucci M,
Zambruno G and Castiglia D: A novel frameshift mutation in the
KIND1 gene in Turkish siblings with kindler syndrome. Br J
Dermatol. 158:1375–1377. 2008.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Has C, Yordanova I, Balabanova M,
Kazandjieva J, Herz C, Kohlhase J and Bruckner-Tuderman L: A novel
large FERMT1 (KIND1) gene deletion in kindler syndrome. J Dermatol
Sci. 52:209–212. 2008.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kartal D, Borlu M, Has C and Fölster-Holst
R: A novel mutation in the fermt1 gene in Turkish siblings with
kindler syndrome. J Eur Acad Dermatol Venereol. 30:1233–1235.
2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Roda Â, Travassos AR, Soares-de-Almeida L
and Has C: Kindler syndrome in a patient with colitis and primary
sclerosing cholangitis: Coincidence or association? Dermatol Online
J 24: 2018.
|
|
14
|
Zhou C, Song S and Zhang J: A novel
3017-bp deletion mutation in the FERMT1 (KIND1) gene in a Chinese
family with kindler syndrome. Br J Dermatol. 160:1119–1122.
2009.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Fukushi R, Tsuboi R, Maeda T, Kanda Y,
Sakai N, Suzuki S and Harada K: A case of kindler syndrome in a
young Indian female with exon deletion. Int J Dermatol. 58:e19–e21.
2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Herz C, Aumailley M, Schulte C,
Schlotzer-Schrehardt U, Bruckner-Tuderman L and Has C: Kindlin-1 is
a phosphoprotein involved in regulation of polarity, proliferation,
and motility of epidermal keratinocytes. J Biol Chem.
281:36082–36090. 2006.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Oh SJ, Kim SE, Lee SE and Kim SC:
Homozygous deletion mutation of the fermt1 gene in a Chinese
patient with kindler syndrome. Ann Dermatol. 28:503–505.
2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Gao Y, Bai JL, Liu XY, Qu YJ, Cao YY, Wang
JC, Jin YW, Wang H and Song F: A novel large deletion mutation of
FERMT1 gene in a Chinese patient with Kindler syndrome. J Zhejiang
Univ Sci B. 16:957–962. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zheng BW, Zhu XZ, Lan Y, Ma JC and Li XQ:
Unique variants in the FLG gene and FERMT1 gene in a chinese
patient with ichthyosis and kindler syndrome. JAAD Case Rep.
5:1061–1064. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Youssefian L, Vahidnezhad H, Barzegar M,
Li Q, Sotoudeh S, Yazdanfar A, Ehsani AH, Kajbafzadeh AM, Mozafari
N, Daryani NE, et al: The kindler syndrome: A spectrum of FERMT1
mutations in Iranian families. J Invest Dermatol. 135:1447–1450.
2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kubo A: Nagashima-type palmoplantar
keratosis: A common Asian type caused by SERPINB7 protease
inhibitor deficiency. J Invest Dermatol. 134:2076–2079.
2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Techanukul T, Sethuraman G, Zlotogorski A,
Horev L, Macarov M, Trainer A, Fong K, Lens M, Medenica L, Ramesh
V, et al: Novel and recurrent FERMT1 gene mutations in kindler
syndrome. Acta Derm-Venereol. 91:267–270. 2011.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Has C, Herz C, Zimina E, Qu HY, He Y,
Zhang ZG, Wen TT, Gache Y, Aumailley M and Bruckner-Tuderman L:
Kindlin-1 is required for RhoGTPase-mediated lamellipodia formation
in keratinocytes. Am J Pathol. 175:1442–1452. 2009.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kiritsi D, He Y, Pasmooij AM, Onder M,
Happle R, Jonkman MF, Bruckner-Tuderman L and Has C: Revertant
mosaicism in a human skin fragility disorder results from slipped
mispairing and mitotic recombination. J Clin Invest. 122:1742–1746.
2012.PubMed/NCBI View
Article : Google Scholar
|
|
25
|
Kern JS, Herz C, Haan E, Moore D,
Nottelmann S, von Lilien T, Greiner P, Schmitt-Graeff A, Opitz OG,
Bruckner-Tuderman L and Has C: Chronic colitis due to an epithelial
barrier defect: The role of kindlin-1 isoforms. J Pathol.
213:462–470. 2007.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Lai-Cheong JE, Moss C, Parsons M, Almaani
N and McGrath JA: Revertant mosaicism in kindler syndrome. J Invest
Dermatol. 132:730–732. 2012.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Ashton GH, McLean WH, South AP, Oyama N,
Smith FJ, Al-Suwaid R, Al-Ismaily A, Atherton DJ, Harwood CA, Leigh
IM, et al: Recurrent mutations in kindlin-1, a novel keratinocyte
focal contact protein, in the autosomal recessive skin fragility
and photosensitivity disorder, kindler syndrome. J Invest Dermatol.
122:78–83. 2004.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Martignago BC, Lai-Cheong JE, Liu L,
McGrath JA and Cestari TF: Recurrent KIND1 (C20orf42) gene
mutation, c.676insC, in a Brazilian pedigree with kindler syndrome.
Br J Dermatol. 157:1281–1284. 2007.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Shaiq PA, Klausegger A, Muzaffar F, Bauer
JW, Khan MI, Khanum A, Qamar R and Raja GK: Founder mutation
c.676insC in three unrelated kindler syndrome families belonging to
a particular clan from Pakistan. J Dermatol. 39:640–641.
2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Thomson MA, Ashton GH, McGrath JA, Eady RA
and Moss C: Retrospective diagnosis of kindler syndrome in a
37-year-old man. Clin Exp Dermatol. 31:45–47. 2006.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Gkaitatzi M, Kalloniati E, Has C, Kiritsi
D, Spiliopoulos T and Georgiou S: Kindler syndrome: A rare case
report from Greece. Oxf Med Case Rep. 2019(omz003)2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Has C, Wessagowit V, Pascucci M, Baer C,
Didona B, Wilhelm C, Pedicelli C, Locatelli A, Kohlhase J, Ashton
GH, et al: Molecular basis of kindler syndrome in Italy: novel and
recurrent Alu/Alu recombination, splice site, nonsense, and
frameshift mutations in the KIND1 gene. J Invest Dermatol.
126:1776–1783. 2006.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Has C and Bruckner-Tuderman L: A novel
nonsense mutation in kindler syndrome. J Invest Dermatol.
122:84–86. 2004.PubMed/NCBI View Article : Google Scholar
|
|
34
|
de Almeida Jr HL, Heckler GT, Fong K,
Lai-Cheong J and McGrath J: Sporadic Kindler syndrome with a novel
mutation. An Bras Dermatol. 88 (Suppl 1):S212–S215. 2013.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Has C, Burger B, Volz A, Kohlhase J,
Bruckner-Tuderman L and Itin P: Mild clinical phenotype of kindler
syndrome associated with late diagnosis and skin cancer.
Dermatology. 221:309–312. 2010.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Fassihi H, Wessagowit V, Jones C,
Dopping-Hepenstal P, Denyer J, Mellerio JE, Clark S and McGrath JA:
Neonatal diagnosis of Kindler syndrome. J Dermatol Sci. 39:183–185.
2005.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Diociaiuti A, Zambruno G, Giancristoforo
S, Proto V, Boldrini R, Castiglia D and El Hachem M: Acral skin
atrophy in an infant: An early clue to Kindler syndrome diagnosis.
J Eur Acad Dermatol Venereol. 30:1046–1049. 2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Siegel DH, Ashton GH, Penagos HG, Lee JV,
Feiler HS, Wilhelmsen KC, South AP, Smith FJ, Prescott AR,
Wessagowit V, et al: Loss of kindlin-1, a human homolog of the
Caenorhabditis elegans actin-extracellular-matrix linker protein
UNC-112, causes kindler syndrome. Am J Human Genet. 73:174–187.
2003.PubMed/NCBI View
Article : Google Scholar
|
|
39
|
Wada M, Masuda K, Tsuruta D, Tamai K,
Lai-Cheong JE, McGrath JA and Katoh N: Case of kindler syndrome
resulting from mutation in the FERMT1 gene. J Dermatol.
39:1057–1058. 2012.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Natsuga K, Nishie W, Shinkuma S, Nakamura
H, Matsushima Y, Tatsuta A, Komine M and Shimizu H: Expression of
exon-8-skipped kindlin-1 does not compensate for defects of kindler
syndrome. J Dermatol. 61:38–44. 2011.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Yildirim TT, Kaya FA, Taskesen M, Dündar
S, Bozoglan A, Tekin GG and Akdeniz S: Aggressive periodontitis
associated with kindler syndrome in a large kindler syndrome
pedigree. Turk J Pediatr. 59:56–61. 2017.PubMed/NCBI View Article : Google Scholar
|