Introduction
Cerebral ischemia-reperfusion (IR) injury is a
complex pathophysiological process that primarily occurs during
transient ischemic attack, cardiac arrest, shock and severe head
trauma (1). Due to the high
metabolic rates, neurons in the hippocampal CA1 region are
particularly vulnerable to the deleterious effects of ischemic
insult, resulting in further damage to learning and memory
(2). Although the exact mechanism
of which remains unclear, a number of pathological processes,
including disturbances in energy metabolism and oxidative stress
have been reported to be involved in neuronal damage, which are
mainly caused by excessive levels of reactive oxygen species (ROS),
excitotoxicity, necrotic and apoptotic cell death (3-6).
Autophagy is a process in which dysfunctional
organelles and proteins are degraded, in a manner that is dependent
on the type of lysosome involved. Under physiological conditions,
autophagy operating at basal levels ensures that damaged organelles
and abnormally aggregated proteins are removed (7). However, aberrant regulation of
autophagy has been previously associated with neurodegenerative
disorders, stroke, cancer and heart diseases, where excessive
activation of autophagy can trigger cell death (8). Cellular processes in neurons are
highly dynamic, such that cell growth, synaptic formation or
synaptic plasticity are highly dependent on the adequate regulation
of protein synthesis and degradation (9). However, excessive autophagy can
trigger extensive degradation of essential proteins and organelles,
leading to the collapse of cellular functions. Accumulating
evidence have suggested that excessive autophagy induced by
cerebral IR is detrimental to neurons that eventually leads to
neuronal cell death (10-12).
Additionally, as a consequence of prolonged, elevated cell stress,
it could be hypothesized that excessive activation of autophagy is
persistent. Therefore, prevention of excessive autophagy in
cerebral IR-induced hippocampal neuronal damage may alleviate
cognitive impairment.
Lithium, a commonly applied therapeutic agent for
depression (13), has been
demonstrated to exert protective effects on neuronal cells both
in vitro and in vivo (14). A previous study has reported that
lithium chloride can ameliorate deficits in spatial learning and
memory induced by repeated cerebral IR injury by increasing Akt and
GSK3β phosphorylation in addition to increasing BDNF expression in
hippocampal neurons (15,16). Therefore, the present study was
designed to investigate whether LiCl can alleviate cognitive
deficits as a result of repeated cerebral IR by targeting the
autophagy signaling pathway in mouse models, where the potential
mechanism was explored.
Materials and methods
Animals
Male C57BL/6 mice (n=120, weight, 22-26 g; age,
10-12 weeks) were purchased from Beijing Vital River Laboratory
Animal Technology Company (license no. SCXK 2013-011). All animal
usage, humane endpoints and procedures complied with the laboratory
animal management regulations of the Ministry of Science and
Technology in China [1988] no. 134 (17-19).
Ethics approval was obtained from the Ethics Committee of Hebei
General Hospital (approval no. 201909).
Animal health and behavior were monitored twice
daily, once in the morning and once in the evening. The animals
were kept in the Animal Center of Hebei People's Hospital
(Shijiazhuang, China) under 12-h light: Dark cycle at an ambient
temperature of 22-25˚C and humidity range of 30-70%. Anesthesia (50
mg/kg pentobarbital sodium, intraperitoneal) was applied prior to
model establishment and before the animals were sacrificed for
specimen collection.
Repeated cerebral IR model
Repeated cerebral IR was induced according to the
protocols described previously with some modifications (15), GCI was achieved by isolation of the
common carotid arteries through a ventral midline incision in the
neck, followed by bilateral occlusion of the arteries using cotton
thread for 5 min. Tension on the cotton thread was removed for 10
min and then initiated again for another 5 min. At the end of the
occlusion, the cotton thread was completely removed, the arteries
were visually inspected for reflow and the midline incision was
sutured. Sham-operated animals underwent a similar procedure, with
the exception of arterial occlusion. The mice were anesthetized by
an intraperitoneal injection with 50 mg/kg pentobarbital sodium,
following which they were subjected to cerebral ischemia for 20 min
by bilateral common carotid artery occlusion, with this treatment
repeated three more times at 10-min intervals. Sham-operated mice
underwent identical surgical procedures but their bilateral common
carotids were exposed and not occluded. Body temperature was
maintained at 37.0±0.5˚C throughout the operation. All moribund
animals were provided with pain relief and none were found dead
following surgery.
Experimental design
The present study was divided into two parts.
Time-course expression of mTOR, p-mTOR,
microtubule-associated protein light chain 3(LC3) II/I and Beclin1
in the hippocampus following repeated cerebral IR. In total, 72
mice were randomly assigned into six groups after repeated cerebral
IR: i) Sham-control; ii) day 1; iii) day 3; iv) day 7; v) day 14;
and vi) day 28 (n=8 in each group). The mice were euthanized 1, 3,
7, 14 and 28 days following model establishment, where the number
of mice that survived surgery in each group was 12, 11, 12, 10 and
11, respectively. In total, 6 mice were randomly selected from each
group for experimentation, the remaining mice were used for
subsequent analyses.
LiCl treatment and Morris water maze test.
LiCl (Sigma-Aldrich; Merck KGaA) was dissolved in normal saline
(NS) and was injected intraperitoneally. A total of 48 mice were
divided into 4 groups, with n=12 for each group that underwent the
following treatment: i) Sham group, where the mice received an
equal volume of NS for 14 days; ii) vehicle group, where the mice
received an equal volume of NS for 14 days following repeated
cerebral IR; iii) Pre-Li group, where the mice received 84 mg/kg
LiCl for 7 days before repeated cerebral IR and then were treated
with NS for 14 d after cerebral IR; and iv) Li group, where the
mice received 2 mmol/kg LiCl for 14 days after repeated cerebral
IR. The optimal concentration of LiCl was determined as previously
described (16). The number of mice
that survived surgery was 12 in the sham operation group, 11 in the
model group, 11 in the Pre-Li group and 10 in the Li group, where
intolerance to surgery was the cause of death. Mice in each group
were euthanized following the completion of the water maze test.
The duration of the entire experiment was 20 days, consisting of 14
days after model establishment followed by 6 days of Morris water
maze testing.
Morris water maze test
The Morris water maze test is a widely applied
procedure for assessing cognitive impairment associated with the
hippocampal region (20). The maze
consisted of a large circular pool that was 120 cm in diameter, 60
cm in height and 45 cm in depth. The temperature of the water was
maintained at 23±1˚C. Within the maze, a platform was located at
the center of the north-east quadrant of the water tank, positioned
~1 cm below the water surface. To start each experiment, the mouse
was gently placed in the water at the edge of a randomly selected
quadrant but not the north-east quadrant, with its nose pointing
toward the wall. The time taken for the mouse to navigate to the
platform was automatically recorded using the DigBehv Animal
Behavior Video-tracking System (JLBehv-MWMG-1; Shanghai Jiliang
Software Technology Co., Ltd.). Each experiment was considered as
complete when the mouse mounts the platform or at 60 sec
regardless. If the mice failed to reach the platform within 60 sec,
they were guided manually to the platform and allowed to stay on it
for 30 sec. Its escape latency, the time taken to reach the
platform, was accepted as 60 sec. The experiments were conducted
six times daily for five consecutive days.
On the sixth day of the test, a probe trial was
conducted without the platform. At the start of the experiment, the
mice were placed in a quadrant which did not contain the platform.
The mice were allowed to swim freely for 60 sec in the pool. The
time spent in the target quadrant where the platform had been
located was recorded.
Histopathology
Immediately after the behavioral tests, mice were
anesthetized with pentobarbital sodium followed by transcardial
perfusion with 4% paraformaldehyde. Following perfusion, the brain
tissues were immediately removed and immersed in 4%
paraformaldehyde at 4˚C, for 48 h before being embedded in
paraffin. Coronal brain sections were subsequently cut into 5-µm
thick sections and underwent Nissl staining with 1% toluidine blue
at 37˚C for 10 min, according to manufacturer's protocols (cat. no.
C0117; Beyotime Institute of Biotechnology). Two slides that were
selected from the same site of each mouse were observed under light
microscopy (Nikon 50i; Nikon).
Western blot analysis
Anesthetized mice (n=6 at each time point in each
group) were first sacrificed by cervical dislocation. The
hippocampal tissues were then removed, frozen in liquid nitrogen
and stored at -80˚C until protein extraction for western blot
analysis. Tissues were first homogenized in ice-cold RIPA buffer
(Beijing Solarbio Science & Technology Co., Ltd.) and incubated
on ice for 30 min, which were then centrifuged at 4˚C at a speed of
12,000 x g for 10 min. The dialyzed supernatants were obtained
after centrifugation. Protein concentration was determined using
the Bicinchoninic acid method (Pierce; Thermo Fisher Scientific,
Inc.). A total of 40 µg of protein were electrophoresed on a 7-12%
SDS-polyacrylamide gel at 80 V for 2 h and then transferred 2 h
onto PVDF membranes (EMD Millipore). Membranes were blocked with 5%
skimmed milk for 2-4 h at room temperature and then incubated
overnight with the following primary antibodies at 4˚C: mTOR
(1:2,000 dilution; Signalway Antibody LLC; cat. no. 41187), p-mTOR
(1:1,000 dilution; Anbo Biotechnology Co., Ltd.; cat. no.
ab109268), LC3B (1:200 dilution; Abgent, Inc.; cat. no. AP1806a)
and Beclin1 (1:200 dilution; Abgent, Inc.; cat. no. AP52755). The
next day, following three rounds of washing with TBS supplemented
with 10% Tween-20 (TBST), the membranes were incubated with the
anti-rabbit horseradish peroxidase-conjugated secondary antibody
(1:10,000 dilution; Proteintech Group Inc., cat. no. HRP-60008) for
2 h at room temperature. Following a further three rounds of
washing with TBST and one round of TBS wash, the protein on the
membranes were detected using the ECL method (High sensitive ECL
luminescence reagent; Sangon Biotech Co., Ltd.; cat. no. C500044),
where β-actin (Proteintech Group, Inc.; cat. no. HRP-60008) was
served as a loading control. The bands were scanned and analyzed
using the ImageJ analysis software (version 1.30v; National
Institutes of Health).
Statistical analysis
Data were expressed as the mean ± standard error of
the mean and processed using the GraphPad Prism 8.0 (GraphPad
Software, Inc.) software. For the Morris water maze test, latencies
to find the platform were analyzed using repeated-measures ANOVA
followed by Tukey's test for multiple comparisons among different
groups. All the other data were analyzed using one-way analysis of
variance (ANOVA) followed by Tukey's test for intergroup
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
LiCl treatment attenuates learning and
memory impairment in repeated cerebral ischemia-reperfusion mouse
models
Following the final LiCl injection, all mice were
trained in the Morris water maze for 5 days consecutively to assess
their learning capabilities, followed by memory examination on the
final day prior to sacrifice. Mice in the vehicle group required
significantly more time to find the platform (P<0.01) compared
with those in the sham-operated group. Mice in the Pre-Li and Li
groups demonstrated shorter mean latencies compared with those in
the vehicle group (P<0.05, Pre-Li vs. Li groups; P<0.01,
Pre-Li or Li group vs. the vehicle group) on day 3. Over the next
two days, mice in the Pre-Li and Li groups exhibited significantly
shorter escape latencies compared with those in the vehicle group
(both P<0.01).
During the probe trial without the platform, the
vehicle group spent significantly less time in the target quadrant
compared with those in the sham-operated group (P<0.01).
Compared with the vehicle group, mice in the Pre-Li group spent
significantly more time in the target quadrant (P<0.01).
Although mice in the Li group also spent more time in the target
quadrant compared with mice in vehicle group, no significant
difference was observed between the two groups (Fig. 1B).
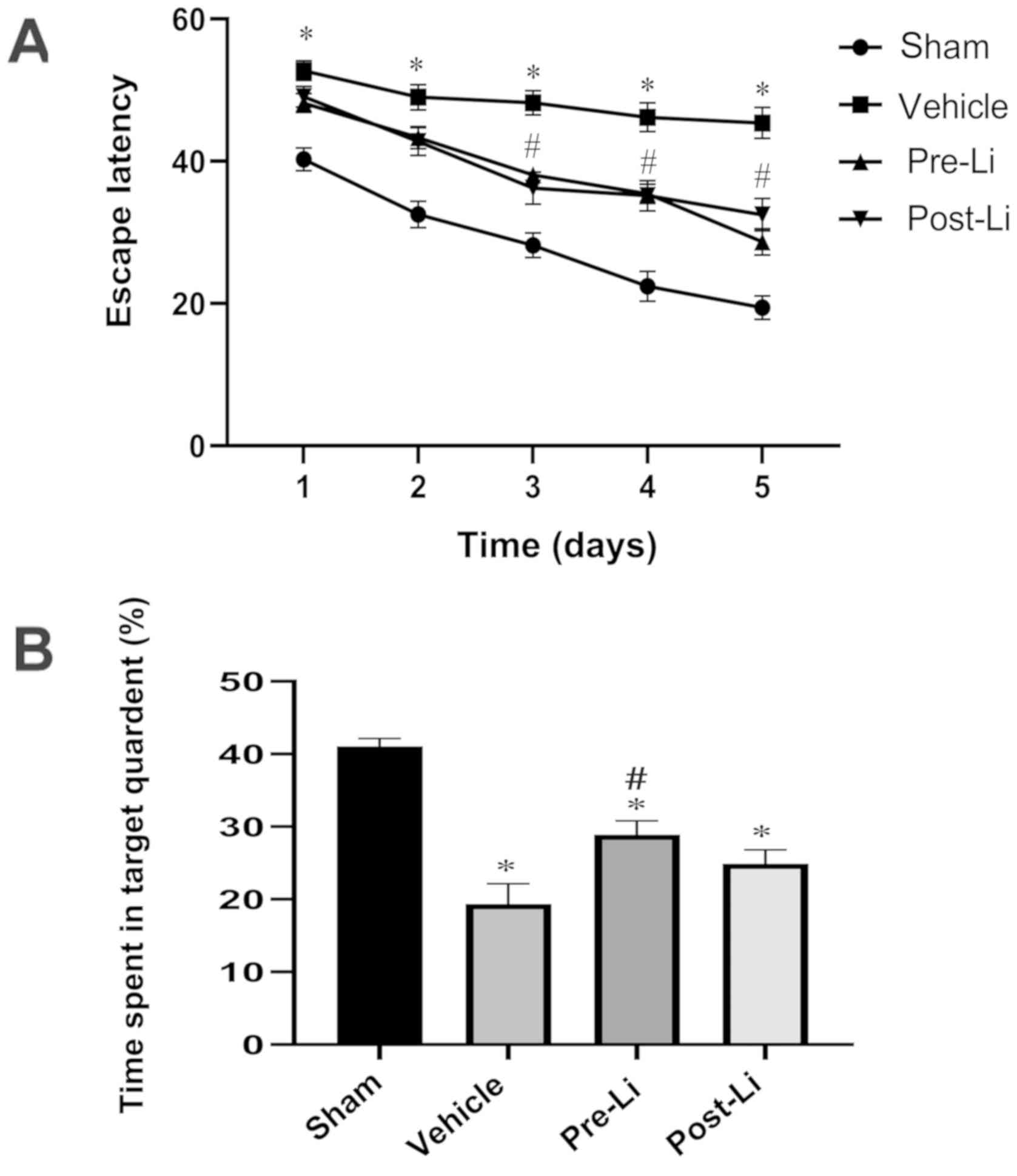 | Figure 1Morris water maze experiment data,
showing the effect of LiCl treatment on spatial cognitive
impairment in mice following repeated cerebral ischemia. (A) Escape
latency, showing the time taken for the mice to find the hidden
platform. For repeated-measures ANOVA, all mice exhibited
progressive declines in the escape latency, where the main factors
of day [F(4,160)=72.736; P<0.01] and group [F(3,40)=46.731; P<0.01] were found to be
statistically significant. (B) Time spent in the target quadrant in
the probe trail. n=12 for sham group, n=11 for vehicle and Pre-Li
groups, and n=10 for Li group, F(3,40)=22.636. *P<0.01 vs. Sham
and #P<0.01 vs. Vehicle. Li, LiCl. |
LiCl treatment reverses morphologic
alterations in the repeated cerebral IR mice
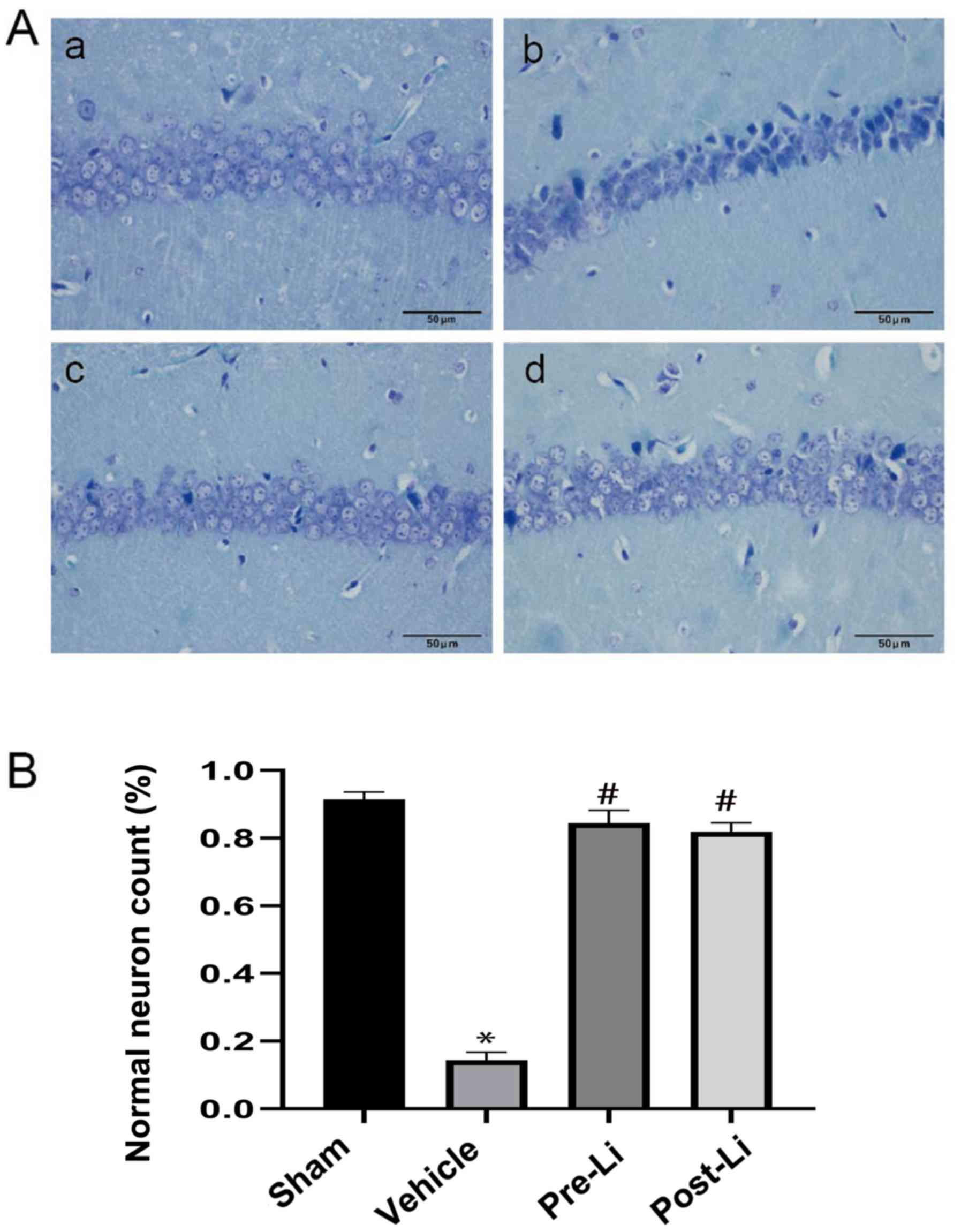
Nissl staining results demonstrated that the
pyramidal neurons in the hippocampus CA1 region in the sham group
were tightly ranked in order, where the neurons were clear and
moderate in size, synonymous with normal microstructure (Fig. 2A-a). Compared with the sham group,
hippocampal samples from the vehicle group exhibited fewer
pyramidal neurons that are loosely arranged with neuronal shrinkage
(Fig. 2A-b). Administration of LiCl reversed the morphologic
changes and increased the number of normal neurons (Fig. 2).
Expression of LC3II/I, Beclin1 and
mTOR phosphorylation after repeated cerebral IR in mice
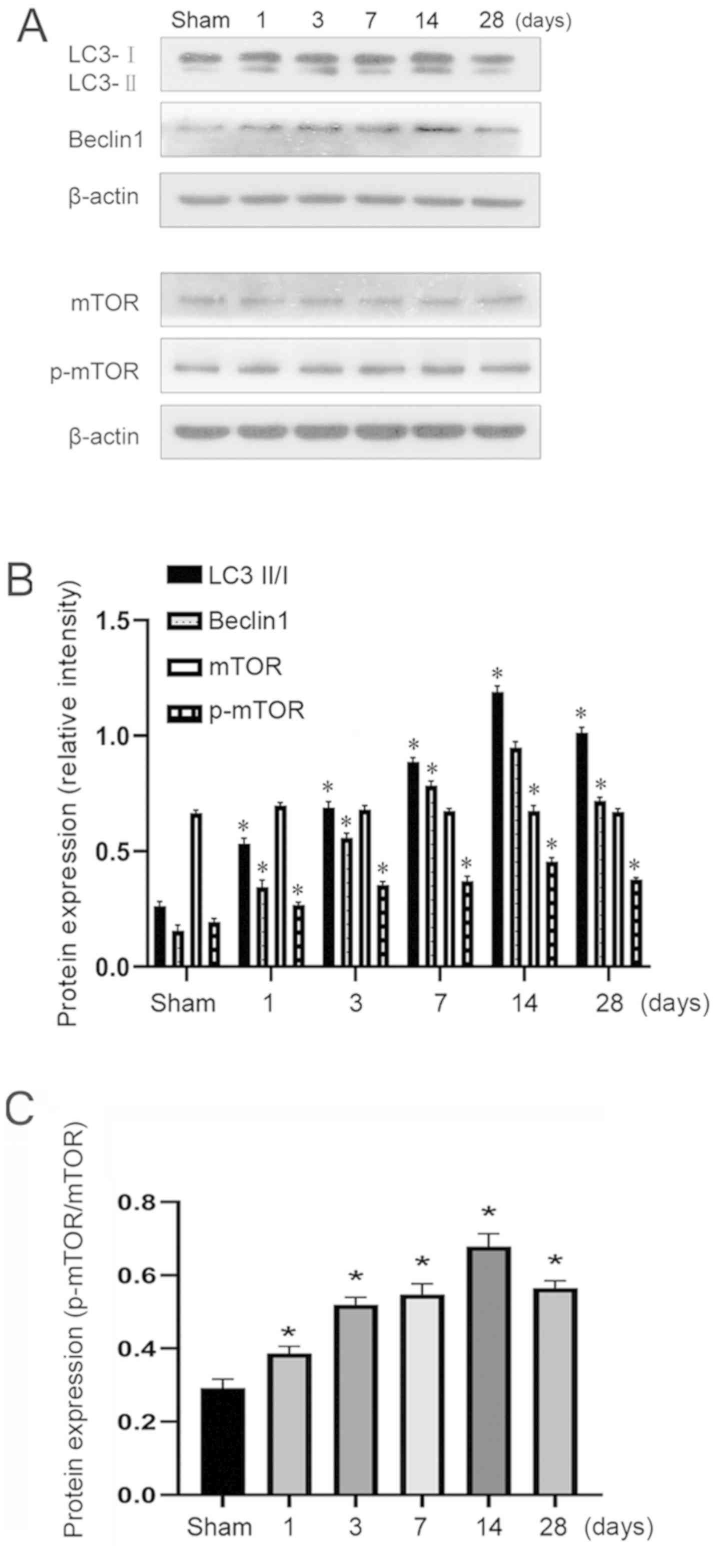
Western blotting was used to measure the expression
levels of LC3II/I and Beclin1 in addition to mTOR phosphorylation
after repeated cerebral ischemia. There was no marked difference in
total mTOR expression between the six groups (Fig. 3). mTOR phosphorylation was
significantly increased in both pre-Li and Li treatment groups
compared with that in the sham group (P<0.01). The gradual
increase in LC3II/I and Beclin1 expression suggested that autophagy
was in progress, which rose to the highest level on day 14 but was
reduced on day 28 (Fig. 3). The
reductions in LC3II/I and Beclin1 expression 28 days after model
establishment compared with those on day 14 suggested the
occurrence of hippocampal repair, but the level of autophagy
remained higher compared with that in the sham group (Fig. 3).
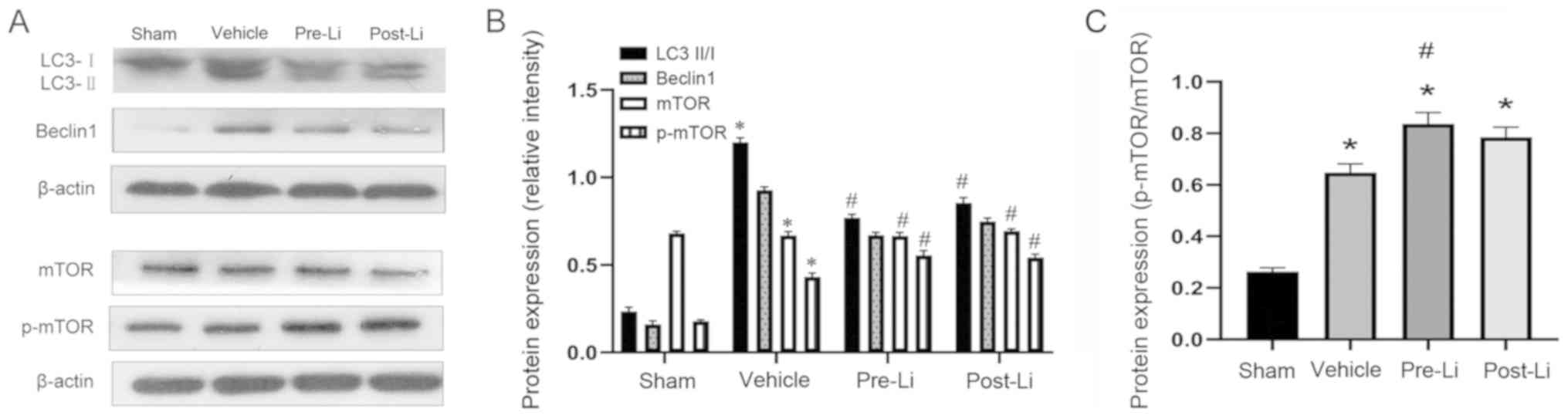
LiCl regulates p-mTOR phosphorylation,
Beclin1 expression and the LC3II/I ratio in the mouse
hippocampus
The LC3II/I ratio, Beclin1 expression and p-mTOR
phosphorylation were examined using western blot analysis 14 days
after repeated cerebral IR. p-mTOR phosphorylation was
significantly increased in the Pre-Li and Li groups compared with
that in the vehicle group (both P<0.01; Fig. 4). The LC3II/I ratio and Beclin1
expression were significantly reduced in the Pre-Li and Li groups
compared with that in the vehicle group (both P<0.01; Fig. 4).
Discussion
Although a number of studies have previously
demonstrated that cerebral IR may induce excessive autophagy in the
early stages (21-23),
whether mechanisms associated with autophagy serve a persistent
role in cognitive impairment as a result of repeated cerebral IR
remains unknown. Therefore, in the present study, a repeated
cerebral IR mouse model was used to monitor the long-term dynamic
changes in the expression of autophagy markers. In a previous study
(15), 2 mmol/kg LiCl has been
demonstrated to exert protective effects against spatial cognitive
impairment in mouse repeated cerebral IR injury models by
increasing Akt and GSK3β phosphorylation in the hippocampus. A
subsequent study demonstrated that pre- and post-treatment with
LiCl can alleviate spatial learning and memory impairment (16). In the present study,
pre-administration with LiCl at a dose of 2 mmol/kg significantly
protected against impaired learning and memory impairment as
indicated by markedly shorter escape latencies and longer time
spent in the target quadrant. However, post-treatment with 2
mmol/kg LiCl partially alleviated spatial cognitive impairment with
slightly shorter escape latencies, whilst both LiCl pre- and
post-treatment effectively increased mTOR phosphorylation and
inhibited autophagy.
Although repeated cerebral ischemia attack followed
by reperfusion injury does not result in limb movement disorder,
damage to the neurons in the hippocampus can occur (24). The hippocampus is known as the
predominant location for the regulation of learning and memory,
where neuronal damage is closely associated with cognitive
impairment (25). The C57Bl/6 mouse
model used for the present study are particularly vulnerable to
brain ischemia due to poorly-developed posterior communicating
arteries (26-28).
In previous studies (15,16,29),
20-min cerebral ischemia for three times at 10-min intervals
induced neuronal injury in the hippocampus, resulting in impaired
learning and memory. For the present study, identical models were
established, in which mice in the repeated cerebral IR group
exhibited longer escape latencies and spent significantly less time
in the target quadrant in the Morris water maze test compared with
those in the sham group, suggesting that the successful model can
be applied to evaluate the effects of LiCl on spatial cognition
following repeated cerebral IR.
Although the mood stabilizing effects of lithium is
well documented, a number of studies have suggested that the
benefits of lithium can also be extended to increased protection
against cognitive impairment induced by acute cerebrovascular
disease (30-34).
Additional studies have also demonstrated that treatment or
pre-treatment with lithium improved impairments in spatial learning
and memory as a result of global cerebral IR (35,36).
In the present study, pre-Li treatment significantly improved the
impaired learning and memory deficits which were consistent with
the aforementioned studies, but Li treatment only partially
alleviated the cognitive deficits. This difference could be due to
the treatment time (16). In a
previous study (15), it was
demonstrated that repeated cerebral IR mice treated with 2 mmol/kg
LiCl for 28 days significantly reduced escape latency and increased
time spent in the target quadrant in Morris water maze experiments.
Coupling this previous observation with data from the present
study, LiCl treatment may be an effective protective agent against
cognitive impairment in a time-dependent manner.
In spite of controversies regarding the role of
autophagy in neuronal cell death, accumulating evidence have
reported that autophagy is activated in focal or global cerebral
ischemia or hypoxia-ischemia models in rats and mice (10,37,38).
Beclin1 knockdown has been demonstrated to reduce infarct volume,
histological injury and neurological deficits induced by focal
cerebral ischemia in adult rats (39). These findings support the notion
that autophagy serves a detrimental role in acute cerebral ischemia
(40). Yang et al (41) found that inhibition of autophagy by
3-methyladenine (3-MA) could markedly reduce infarct size, edema
formation and neurological deficits after permanent middle cerebral
artery occlusion. However, the extent to which autophagy is
involved in repeated cerebral IR-induced cognitive impairment has
not been elucidated in those previous studies. In the present
study, a time course experiment was performed by repeating cerebral
ischemia for 20 min, with 10 min reperfusions three times. The
animals were then euthanized as various time points (1, 3, 7, 14
and 28 days) after the treatment and hippocampal tissue samples
were collected to measure the expression of proteins associated
with autophagy. The expression of Beclin1 and LC3-II in the
hippocampus increased significantly from day 1 to day 28 after
cerebral IR, with maximal induction observed on day 14. This
observation suggest that increased autophagy is linked with neuron
damage in the hippocampus and subsequent impairments in spatial
cognitive function following cerebral IR. Following LiCl
administration, autophagy was reduced to a certain extent along
with a reduction in the population of dying neurons; however, it
remains unclear whether the reduction of autophagy was the sole
cause of this effect.
The mTOR signaling pathway is a central regulator of
protein synthesis and is considered to be a key controller of cell
growth, cell survival and autophagy (42-44).
In mammalian cells, mTOR assembles into two functionally distinct
protein complexes-mTORC1 and mTORC2. mTORC1 can serve as a
regulator of autophagy and is highly sensitive to rapamycin
(45). Although the role of mTOR in
ischemic injury is unclear, mTOR has been previous demonstrated to
exhibit neuroprotective properties (46,47).
In focal cerebral IR models of neonatal rats, 3-MA exerted
neuroprotective effects even after 4 h ischemia (48). The present study revealed that mTOR
phosphorylation in the hippocampus increased from days 1 to 28
following cerebral IR. Additionally, LiCl treatment significantly
increased mTOR phosphorylation whilst reducing the ratio of LC3II/I
and Beclin1 expression, suggesting that mTOR may serve as a
therapeutic target in cases of repeated cerebral IR injury.
However, by contrast, induction of autophagy by upregulating mTOR
phosphorylation has been previously reported to promote neuronal
survival (49). It could be
hypothesized that the discrepancy between these findings may be
associated with distinct cell types and models across different
studies. In animal experiments, the mice were treated with LiCl
prior to the establishment of IR model with the aim of minimizing
the off-target effects of model establishment whilst maximizing the
potential effects of LiCl (16).
However, application of this methodology in clinical practice
remains to be explored and requires further validation.
For future studies, supplementing a group of
positive controls treated with autophagic inhibitors, to detect the
mechanism of autophagy in repeated IR-induced injury, could be used
to further assist in elucidating the potential mechanism of lithium
chloride on autophagy.
In summary, the present study suggests that
excessive autophagy can contribute to neuronal injury in a repeated
cerebral IR mouse model. Additionally, LiCl inhibits excessive
autophagy by activating mTOR signaling and ameliorating repeated
IR-induced the neuronal injury in the hippocampus. LiCl may serve
as a promising therapeutic agent for the treatment of cognitive
impairment caused by cerebral IR injury.
Acknowledgements
Not applicable.
Funding
This project was supported by the National Natural
Science Foundation of China (grant no. 81241037), the Natural
Science Foundation of Hebei (grant no. H2013307046) and the Major
Medical Scientific Research Subject of Hebei (grant no.
zd2013005).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YX designed the study, performed the experiments
mainly including the surgery, behavioral test and the western blot,
carried out the statistical analysis, and prepared the manuscript.
MF, WJ, WAL and YJ were involved in the experiments such as the
surgeries and behavioral tests. YD, XJ, JX and NM performed data
collection. PL was responsible for the supervision of the entire
project. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Ethics approval was obtained from the Ethics
Committee of Hebei General Hospital (Shijiazhuang, China; approval
no. 201909).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chatauret N, Badet L, Barrou B and Hauet
T: Ischemia-reperfusion: From cell biology to acute kidney injury.
Prog Urol. 24 (Suppl 1):S4–S12. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Erfani S, Khaksari M, Oryan S, Shamsaei N,
Aboutaleb N, Nikbakht F, Jamali-Raeufy N and Gorjipour F: Visfatin
reduces hippocampal CA1 cells death and improves learning and
memory deficits after transient global ischemia/reperfusion.
Neuropeptides. 49:63–68. 2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Huang XP, Tan H, Chen BY and Deng CQ:
Combination of total astragalus extract and total Panax notoginseng
saponins strengthened the protective effects on brain damage
through improving energy metabolism and inhibiting apoptosis after
cerebral ischemia-reperfusion in mice. Chin J Integr Med.
23:445–452. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Guo C, Wang S, Duan J, Jia N, Zhu Y, Ding
Y, Guan Y, Wei G, Yin Y, Xi M and Wen A: Protocatechualdehyde
protects against cerebral ischemia-reperfusion-induced oxidative
injury via protein kinase Cepsilon/Nrf2/HO-1 Pathway. Mol
Neurobiol. 54:833–845. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zhang S, Zhang Y, Li H, Xu W, Chu K, Chen
L and Chen X: Antioxidant and anti-excitotoxicity effect of Gualou
Guizhi decoction on cerebral ischemia/reperfusion injury in rats.
Exp Ther Med. 9:2121–2126. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Yaidikar L and Thakur S: Punicalagin
attenuated cerebral ischemia-reperfusion insult via inhibition of
proinflammatory cytokines, up-regulation of Bcl-2, down-regulation
of Bax, and caspase-3. Mol Cell Biochem. 402:141–148.
2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: A double-edged sword. Science. 306:990–995.
2004.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kourtis N and Tavernarakis N: Autophagy
and cell death in model organisms. Cell Death Differ. 16:21–30.
2009.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Lee JA: Neuronal autophagy: A housekeeper
or a fighter in neuronal cell survival? Exp Neurobiol Mar. 21:1–8.
2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Shi R, Weng J, Zhao L, Li XM, Gao TM and
Kong J: Excessive autophagy contributes to neuron death in cerebral
ischemia. CNS Neurosci Ther. 18:250–260. 2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Rami A, Langhagen A and Steiger S: Focal
cerebral ischemia induces upregulation of Beclin 1 and
autophagy-like cell death. Neurobiol Dis. 29:132–141.
2008.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wen YD, Sheng R, Zhang LS, Han R, Zhang X,
Zhang XD, Han F, Fukunaga K and Qin ZH: Neuronal injury in rat
model of permanent focal cerebral ischemia is associated with
activation of autophagic and lysosomal pathways. Autophagy.
4:762–769. 2008.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Machado-Vieira R, Zanetti MV, DE Sousa RT,
Soeiro-DE-Souza MG, Moreno RA, Busatto GF and Gattaz WF: Lithium
efficacy in bipolar depression with flexible dosing: A six-week,
open-label, proof-of-concept study. Exp Ther Med. 8:1205–1208.
2014.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Inoki K, Ouyang H, Zhu T, Lindvall C, Wang
Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, et al: TSC2
integrates Wnt and energy signals via a coordinated phosphorylation
by MAPK and GSK3 to regulate cell growth. Cell. 126:955–968.
2006.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Fan M, Song C, Wang T, Li L, Dong Y, Jin W
and Lu P: Protective effects of lithium chloride treatment on
repeated cerebral ischemia-reperfusion injury in mice. Neurol Sci.
36:315–321. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Fan M, Jin W, Zhao H, Xiao Y, Jia Y, Yin
Y, Jiang X, Xu J, Meng N and Lv P: Lithium chloride administration
prevents spatial learning and memory impairment in repeated
cerebral ischemia-reperfusion mice by depressing apoptosis and
increasing BDNF expression in hippocampus. Behav Brain Res.
291:399–406. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ogden BE, Pang William W, Agui T and Lee
BH: Laboratory animal laws, regulations, guidelines and standards
in China mainland, Japan, and Korea. ILAR J. 57:301–311.
2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kilkenny C, Browne WJ, Cuthill IC, Emerson
M and Altman DG: Improving bioscience research reporting: The
ARRIVE guidelines for reporting animal research. Osteoarthritis
Cartilage. 20:256–260. 2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Science and Technology in China. Nature
162: 17, 1948.
|
|
20
|
Vorhees CV and Williams MT: Morris water
maze: Procedures for assessing spatial and related forms of
learning and memory. Nat Protoc. 1:848–858. 2006.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wang JY, Xia Q, Chu KT, Pan J, Sun LN,
Zeng B, Zhu YJ, Wang Q, Wang K and Luo BY: Severe global cerebral
ischemia-induced programmed necrosis of hippocampal CA1 neurons in
rat is prevented by 3-methyladenine: A widely used inhibitor of
autophagy. J Neuropathol Exp Neurol. 70:314–322. 2011.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zheng Y, Hou J, Liu J, Yao M, Li L, Zhang
B, Zhu H and Wang Z: Inhibition of autophagy contributes to
melatonin-mediated neuroprotection against transient focal cerebral
ischemia in rats. J Pharmacol Sci. 12:354–364. 2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Li L, Tian J, Long MK, Chen Y, Lu J, Zhou
C and Wang T: Protection against experimental stroke by Ganglioside
GM1 is associated with the inhibition of autophagy. PLoS One.
11(e0144219)2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Chuang EH, Iwasaki K, Mishima K, Egashira
N and Fujiwara M: Repeated cerebral ischemia induced hippocampal
cell death and impairments of spatial cognition in the rat. Life
Sci. 72:609–619. 2002.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Voss JL, Bridge DJ, Cohen NJ and Walker
JA: A Closer look at the hippocampus and memory. Trends Cogn Sci.
21:577–588. 2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wellons JC III, Sheng H, Laskowita DT,
Mackensen GB, Pearlstein RD and Warner DS: A comparison of
strain-related susceptibility in two murine recovery models of
global cerebral ischemia. Brain Res. 868:14–21. 2000.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Fujii M, Hara H, Meng W, Vonsattel JP,
Huang Z and Moskowitz MA: Strain-related differences in
susceptibility to transient forebrain ischemia in SV-129 and
C57black/6 mice. Stroke. 28:1805–1810. 1997.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Maeda K, Hata R and Hossmann KA: Regional
metabolic disturbances and cerebrovascular anatomy after permanent
middle cerebral artery occlusion in C57black/6 and SA129 mice.
Neurobiol Dis. 6:101–108. 1999.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wang T, Lv P, Jin W, Zhang H, Lang J and
Fan M: Protective effect of donepezil hydrochloride on cerebral
ischemia/reperfusion injury in mice. Mol Med Rep. 9:509–514.
2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Fiorentini A, Rosi MC, Grossi C, Luccarini
I and Casamenti F: Lithium improves hippocampal neurogenesis,
neuropathology and cognitive functions in APP mutant mice. PLoS
One. 5(e14382)2010.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Contestabile A, Greco B, Ghezzi D, Tucci
V, Benfenati F and Gasparini L: Lithium rescues synaptic plasticity
and memory in Down syndrome mice. J Clin Invest. 123:348–361.
2013.PubMed/NCBI View
Article : Google Scholar
|
|
32
|
Li N, Zhang X, Dong H, Zhang S, Sun J and
Qian Y: Lithium ameliorates LPS-induced astrocytes activation
partly via inhibition of toll-like receptor 4 expression. Cell
Physiol Biochem. 38:714–725. 2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhu ZF, Wang QG, Han BJ and William CP:
Neuroprotective effect and cognitive outcome of chronic lithium on
traumatic brain injury in mice. Brain Res Bull. 83:272–277.
2010.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Gold AB, Herrmann N and Lanctot KL:
Lithium and its neuroprotective and neurotrophic effects: Potential
treatment for post-ischemic stroke sequelae. Curr Drug Targets.
12:243–255. 2011.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Yan XB, Hou HL, Wu LM, Liu J and Zhou JN:
Lithium regulates hippocampal neurogenesis by ERK pathway and
facilitates recovery of spatial learning and memory in rats after
transient global cerebral ischemia. Neuropharmacology. 53:487–495.
2007.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Bian Q, Shi T, Chuang DM and Qian Y:
Lithium reduces ischemia-induced hippocampal CA1 damage and
behavioral deficits in gerbils. Brain Res. 1184:270–276.
2007.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Adhami F, Liao G, Morozov YM, Schloemer A,
Schmithorst VJ, Lorenz JN, Dunn RS, Vorhees CV, Wills-Karp M, Degen
JL, et al: Cerebral ischemia-hypoxia induces intravascular
coagulation and autophagy. Am J Pathol. 169:566–583.
2006.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Li WL, Yu SP, Chen D, Yu SS, Jiang YJ,
Genetta T and Wei L: The regulatory role of NF-κB in autophagy-like
cell death after focal cerebral ischemia in mice. Neuroscience.
244:16–30. 2013.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Yang Y, Gao K, Hu Z, Li W, Davies H, Ling
S, Rudd JA and Fang M: Autophagy upregulation and apoptosis
downregulation in DAHP and triptolide treated cerebral ischemia.
Mediators Inflamm. 2015(120198)2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Zheng YQ, Liu JX, Li XZ, Xu L and Xu YG:
RNA interference- mediated downregulation of Beclin1 attenuates
cerebral ischemic injury in rats. Acta Pharmacol Sin. 30:919–927.
2009.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Yang Z, Zhong L, Zhong S, Xian R and Yuan
B: Hypoxia induces microglia autophagy and neural inflammation
injury in focal cerebral ischemia model. Exp Mol Pathol.
98:219–224. 2015.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Laplante M and Asbatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Zhou J, Tan SH Nicolas V, Bauvy C, Yang
ND, Zhang J, Xue Y, Codogno P and Shen HM: Activation of lysosomal
function in the course of autophagy via mTORC1 suppression and
autophagosome-lysosome fusion. Cell Res. 23:508–523.
2013.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Wullschleger S, Loewith R and Hall MN:
mTOR signaling in growth control and disease. Cell. 124:471–484.
2006.
|
|
45
|
Jung CH, Ro SH, Cao J, Otto NM and Kim DH:
mTOR regulation of autophagy. FEBS Lett. 584:1287–1295.
2010.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Chi OZ, Mellender SJ, Barsoum S, Liu X,
Damito S and Weiss HR: Effects of rapamycin pretreatment on
blood-brain barrier disruption in cerebral ischemia-reperfusion.
Neurosci Lett. 620:132–136. 2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Fu L, Huang L, Cao C, Yin Q and Liu J:
Inhibition of AMP-activated protein kinase alleviates focal
cerebral ischemia injury in mice: Interference with mTOR and
autophagy. Brain Res. 1650:103–111. 2016.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Puyal J, Vaslin A, Mottier V and Clarke
PG: Postischemic treatment of neonatal cerebral ischemia should
target autophagy. Ann Neurol. 66:378–389. 2009.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Wang PR, Wang JS, Zhang C, Song XF, Tian N
and Kong LY: Huang-Lian-Jie-Du-Decotion induced protective
autophagy against the injury of cerebral ischemia/reperfusion via
MAPK-mTOR signaling pathway. J Ethnopharmacol. 149:270–280.
2013.PubMed/NCBI View Article : Google Scholar
|