Introduction
Contrast-induced nephropathy (CIN) refers to acute
kidney injury (AKI) that occurs following intravascular application
of contrast medium (CM), in the absence of other causative factors.
CIN accounts for 12% of all cases of iatrogenic AKI (1), and is the third leading cause after
renal hypoperfusion and the use of nephrotoxic drugs. The morbidity
rate of CIN is ~7.1% (2), and it
may be higher among patients with acute coronary syndrome (3) or ST-segment elevation myocardial
infarction (4). Although the risk
of CIN has decreased due to technological advancements in recent
years, the total number of cases remains high due to an increase in
the number of patients requiring angiography. CM-induced decrease
in renal perfusion and the direct toxic effect of CM on renal
tubular cells are widely considered as the primary causes of CIN
(5). It is also accepted that renal
tubular obstruction, apoptosis, oxidative stress and immune
inflammatory reactions are implicated in contrast-induced AKI
(5-7).
However, the molecular mechanisms of CIN are yet to be fully
elucidated.
The toll-like receptors (TLRs) are important
components of the immune response to pathogens, of which TLR4 is a
vital regulator of the inflammatory response (8). TLR4-mediated downstream signaling
pathways include the myeloid differentiation primary response 88
(MyD88)-dependent and the MyD88-independent pathways; the former
primarily regulates the expression of a variety of
inflammation-associated genes, which transmit intracellular signals
through the TIR (Toll/IL-1 receptor) domain of MyD88. This
activates transcription factors, such as nuclear factor (NF)-κB,
thereby promoting the release of inflammatory factors, including
interleukin (IL)-1, IL-6 and tumor necrosis factor (TNF)-α
(9). Previous studies reported
that, in a model of AKI, blocking the TLR4/NF-κB signaling pathway
inhibited the expression of inflammatory factors and preserved
renal function (10,11); however, the expression levels of
signaling molecules upstream of NF-κB were not investigated.
Therefore, the aim of the present study was to
explore the role of TLR4/MyD88/NF-κB signaling in an in
vitro model of CIN by blocking the corresponding genetic loci
of the associated signaling proteins, and to provide a new
experimental basis for investigating the molecular mechanisms of
CIN.
Materials and methods
Cell culture
Human renal proximal tubular cells (HK-2) were
cultured in Dulbecco's modified Eagle's medium/Nutrient Mixture
F-12 (DMEM/F12; GE Healthcare Life Sciences) containing 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) in a
humidified atmosphere at 37˚C (5% CO2). The medium was
replaced every 2 days and the cells were passaged at ~80%
confluence using trypsin (GE Healthcare Life Sciences). Between
passages 4 and 6, cells in the logarithmic growth phase were
transferred into 6-, 12- or 96-well plates. The cells were
serum-starved by replacing the culture medium with serum-free
DMEM/F12, and cultured for 12 h prior to the addition of iohexol
(GE Healthcare Life Sciences).
Groupings
Serum-starved HK-2 cells were divided into the
following groups: i) The blank group, cultured in serum-free
DMEM/F12 only; ii) the iohexol group, where 100 mg/ml iohexol was
added at 12 h post-serum starvation; iii) the NF-κB RNAi group; and
iv) the TLR4 RNAi group, both of which were transfected with the
corresponding siRNAs and treated with 100 mg/ml iohexol; v) the
pyrrolidine dithiocarbamate (PDTC) group; and vi) the CLI-095
group, in which, 2 h prior to iohexol treatment, the cells were
treated at room temperature with 100 µmol/l PDTC (Sigma; Merck
KGaA) or 3 µmol/l CLI-095 (InvivoGen). The corresponding indicators
mentioned below were detected in all groups after 48 h of iohexol
treatment.
Small interfering (si)RNA
transfection
The siRNAs were designed and synthesized by Shanghai
GenePharma Co., Ltd., and subsequent experiments were conducted
following confirmation of a gene-silencing efficiency of ≥80%
(Fig. 1). Negative control was used
for RNA interference and the sequences were as follows: Forward,
5'-UUCUCCGAACGUGUCACGUTT-3' and reverse,
5'-ACGUGACACGUUCGGAGAATT-3'. The siRNA sequences were as follows:
NF-κB forward, 5'-GCACCAUCAACUAUGAUGATT-3' and reverse,
5'-UCAUCAUAGUUGAUGGUGCTT-3'; and TLR4 forward,
5'-GGAAUGAGCUAGUAAAGAATT-3' and reverse,
5'-UUCUUUACUAGCUCAUUCCTT-3'. Lipofectamine® 3000 (Thermo
Fisher Scientific, Inc.) was incubated for 5 min at room
temperature with moderate serum-free DMEM/F12, and was added to the
diluted siRNA solution, which was diluted with an equal volume of
serum-free medium (20 pmol siRNA per 2 µl Lipofectamine solution).
The mixture was then co-cultured with cells seeded into 6-well
plates (1x105 cells/well) overnight, to a final
concentration of 40 nmol/l for both siRNAs.
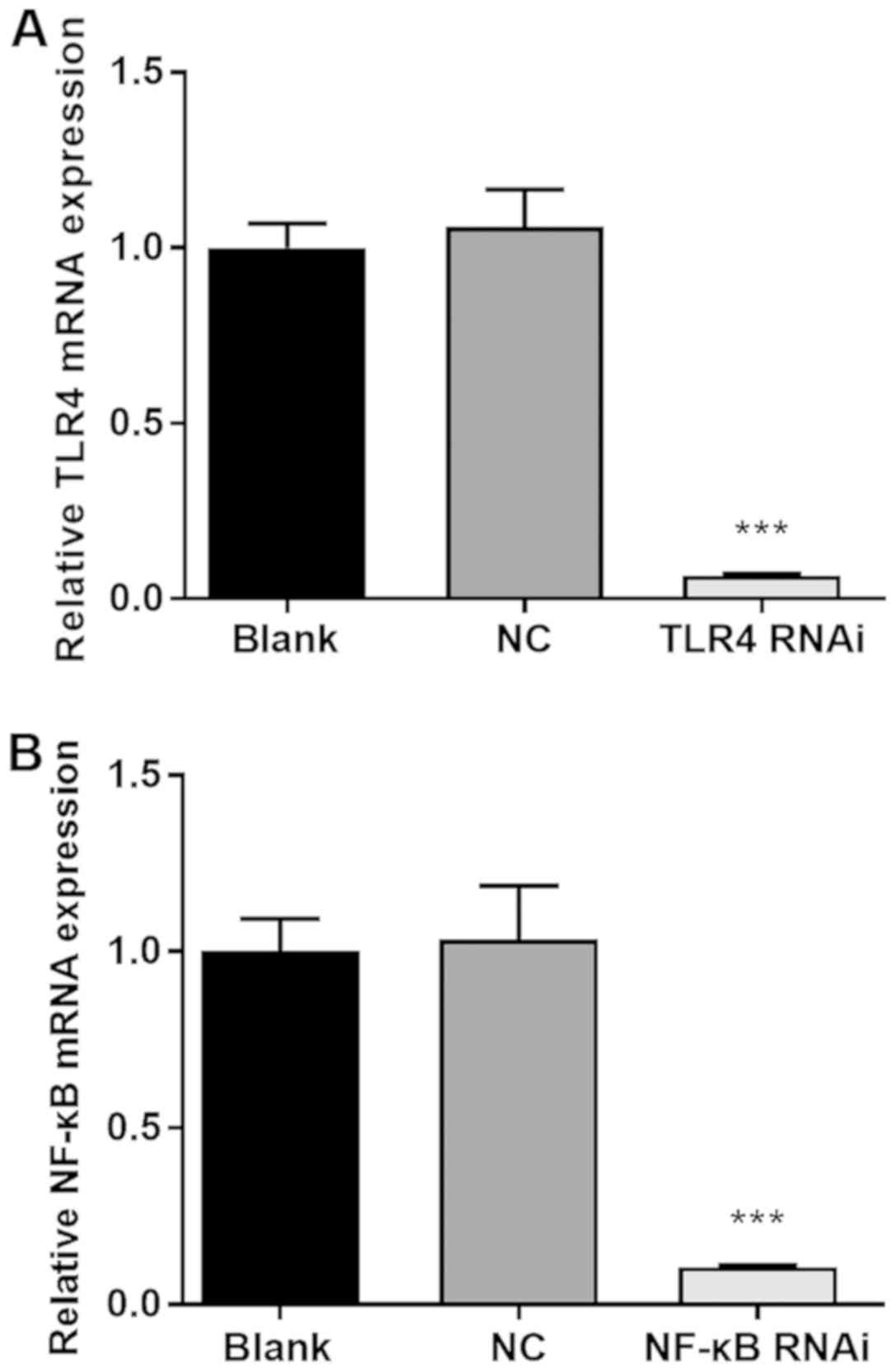 | Figure 1siRNA transfection achieves a high
level of gene silencing efficiency. (A) NC group or TLR4 RNAi
group: Using Lipofectamine® 3000, HK-2 cells were
transfected with NC or TLR4 siRNA, respectively, to a final
concentration of 40 nmol/l; blank group: Lipofectamine®
3000 alone. (B) NC group or NF-κB RNAi group: Using
Lipofectamine® 3000, HK-2 cells were transfected with NC
or NF-κB siRNA, respectively, to a final concentration of 40
nmol/l; blank group: Lipofectamine® 3000 alone. After 48
h, cells from each group were harvested and the relative expression
levels of NF-κB or TLR4, respectively, were determined by reverse
transcription-quantitative PCR. ***P<0.001 vs. the NC
group. siRNA, small interfering RNA; NC, negative control; NF-κB,
nuclear factor κB; TLR4, toll-like receptor 4. |
Cell Counting Kit-8 (CCK-8) assay
Cells in the logarithmic growth phase were seeded
into 96-well plates at a density of 5x103 cells/well;
each group contained six wells, including the A0 group, which was
incubated with DMEM/F12 only. Following grouping, CCK-8 reagent (10
µl; Dojindo Molecular Technologies, Inc.) was added to each well,
and the plates were incubated for 2 h at 37˚C in the dark. The
absorbance at 450 nm was then determined by Synergy Mx (Bio Tek,
Inc.).
Flow cytometry
The Annexin V-FITC/propidium iodide (PI) Apoptosis
Detection Kit (Nanjing KeyGen Biotech Co., Ltd.) was used to detect
apoptosis by flow cytometry. Cells were seeded into 12-well plates
(5x104 cells/well), and the treated cells were
collected, centrifuged for 5 min at 4˚C (350 x g) and washed three
times with cold PBS. The cells were resuspended in 500 µl binding
buffer and 5 µl Annexin V-FITC was added. After 5 min of incubation
in the dark at room temperature, 5 µl PI was added and incubated
under the same conditions for 10 min. Finally, apoptosis was
detected by CytoFLEX (Beckman, Inc.) and analyzed using CytExpert
2.3 (Beckman, Inc.).
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted from treated HK-2 cells
using TRIzol® reagent (Thermo Fisher Scientific, Inc.).
Reverse transcription was performed with 1 µg total RNA using the
PrimeScript™ RT reagent Kit with gDNA Eraser (Takara
Bio, Inc.) according to the manufacturer's protocol. The cDNA was
then amplified using the Custom gene RT-qPCR Quantitation Kit
(Shanghai GenePharma Co., Ltd.) as per the manufacturer's
instructions; the primer sequences are listed in Table I. The RT-qPCR thermocycling
conditions were as follows: Reverse transcription, one cycle at
95˚C for 3 min; qPCR, 40 cycles at 95˚C for 12 sec and 62˚C for 40
sec. The mRNA expression levels were quantified with the Bio-Rad
CFX Manager (Bio-Rad Laboratories, Inc.) using the
2-∆∆Cq method (12),
with GAPDH as the reference gene.
 | Table IPrimers designed for reverse
transcription-quantitative PCR analysis. |
Table I
Primers designed for reverse
transcription-quantitative PCR analysis.
| mRNA | Primer pairs
(5'-3') |
|---|
| GAPDH | Forward:
AAAATCAAGTGGGGCGATGC |
| | Reverse:
GATGACCCTTTTGGCTCCCC |
| TLR4 | Forward:
GTCTCCTCCACATCCTCCCT |
| | Reverse:
CTCCCAGAACCAAACGATG |
| MyD88 | Forward:
GTCTCCTCCACATCCTCCCT |
| | Reverse:
CAGTTGCCGGATCTCCAAGT |
| NF-κB | Forward:
TTGGGAATGGTGAGGTCACTCTAAC |
| | Reverse:
TCTCCTGTCACCGCGTAGTCG |
| TNF-α | Forward:
TTCTGCCTGCTGCACTTTGGAG |
| | Reverse:
AGGGCTGATTAGAGAGAGGTCCCTG |
| IL-1β | Forward:
AGCACCTTCTTTCCCTTCATCTTTG |
| | Reverse:
CATAAGCCTCGTTATCCCATGTGTC |
| IL-6 | Forward:
GCCAGAGCTGTGCAGATGAGT |
| | Reverse:
TGGCATTTGTGGTTGGGTCAG |
Western blotting
Treated HK-2 cells were washed twice with cold PBS,
and lysed on ice for 1 h with RIPA lysis buffer containing 1 mM
PMSF (both from Beyotime Institute of Biotechnology). Total protein
was quantified using the Enhanced BCA Protein Assay Kit (Beyotime
Institute of Biotechnology). Protein samples (40 µg/lane) were
separated by SDS-PAGE using a 10% gel, and transferred onto PVDF
membranes (Thermo Fisher Scientific, Inc.), which were subsequently
blocked with 5% skimmed milk for 2 h at room temperature. The
membranes were probed with the following primary antibodies at 4˚C
overnight: Anti-caspase-3 (1:1,000; cat. no. 19677-1-AP;
ProteinTech Group, Inc.), anti-caspase-9 and cleaved (c)-caspase-9
(1:1,000; cat. no. 10380-1-AP; ProteinTech Group, Inc.), anti-NF-κB
p65 (1:1,000; cat. no. 10745-1-AP; ProteinTech Group, Inc.),
anti-MyD88 (1:1,000; cat. no. 23230-1-AP; ProteinTech Group, Inc.),
anti-TLR4 (1:800; cat. no. 19811-1-AP; ProteinTech Group, Inc.) and
anti-β-actin (1:5,000; cat. no. ab8227; Abcam). After washing
thrice with PBS-T at room temperature, the membranes were incubated
with horseradish peroxidase-conjugated goat anti-rabbit IgG
secondary antibodies (1:8,000; cat. no. ab 97051; Abcam) at room
temperature for 1 h. Finally, the treated membranes were visualized
using BeyoECL Star reagent (Beyotime Institute of Biotechnology)
according to the manufacturer's instructions, and then quantified
using Quantity One 1-D Analysis Software Version 4.6.8 (Bio-Rad
Laboratories, Inc.) with β-actin as the loading control.
Statistical analysis
All statistical analyses were conducted using SPSS
version 22.0 (IBM Corp.) and all data are presented as the mean ±
standard deviation. Variations between groups were statistically
assessed using a Student's t-test or one-way analysis of variance
followed by Tukey's multiple comparisons test, as appropriate.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Iohexol stimulates the expression of
TLR4, MyD88 and NF-κB
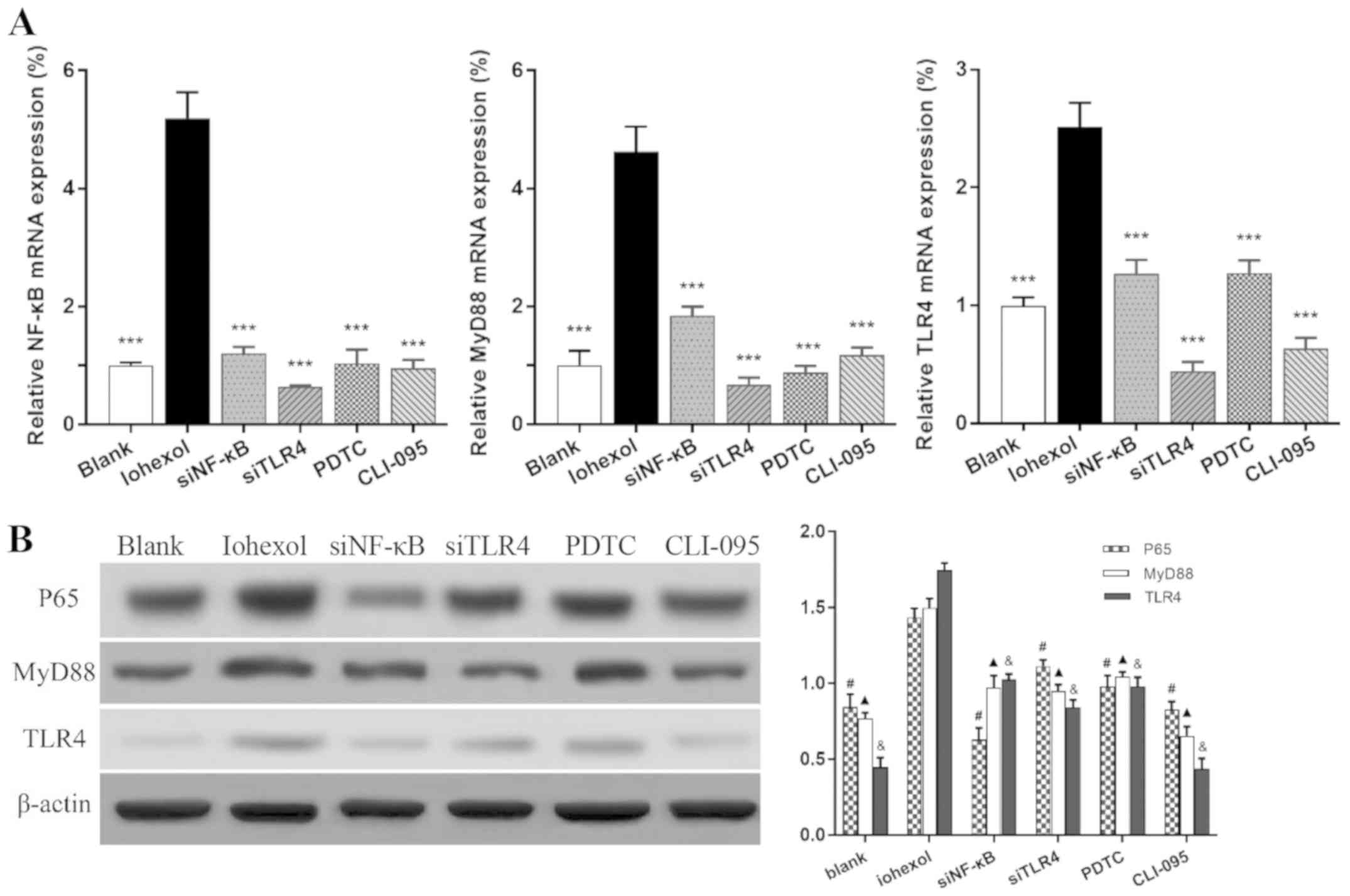
After 48 h of iohexol stimulation, the mRNA and
protein expression levels of TLR4, MyD88 and NF-κB were
significantly increased compared with those in the blank group
(Fig. 2A and B; P<0.001). The corresponding
expression levels were markedly lower following NF-κB or TLR4
silencing or blocking, respectively, compared with the iohexol
group (P<0.001).
Blocking or silencing the
TLR4/MyD88/NF-κB signaling pathway alleviates the inhibition of
HK-2 cell proliferation induced by iohexol
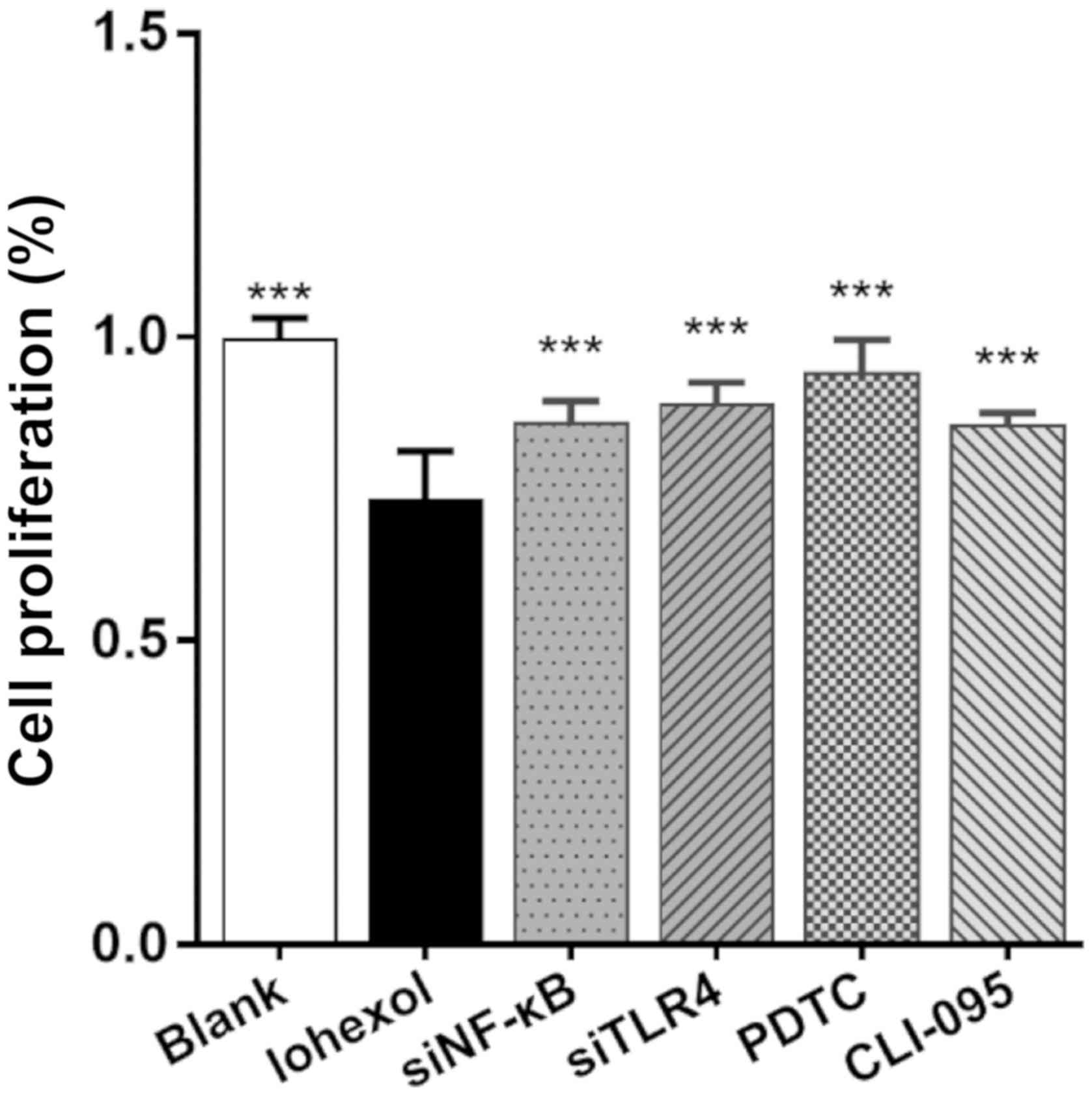
The CCK-8 assay was used to evaluate HK-2 cell
proliferation. As shown in Fig. 3,
the proliferation rate of the blank group was considered to be
100%, and the proliferation rates of the treatment groups
(%)=(Ax-A0)/(Ac-A0)*100%
(Ax, absorbance value of other groups; A0,
absorbance value of the A0 group; Ac, absorbance value
of the blank group). After a 48-h incubation period with iohexol,
the proliferation rate of the iohexol group was significantly
reduced (P<0.001). Following NF-κB- or TLR4-RNAi inhibition, or
treatment with PDTC or CLI-095, the proliferation rates were found
to be higher compared with those of the iohexol treatment group
(P<0.001), albeit lower compared with those of the blank control
group (PDTC group: P<0.05; CLI-095, NF-κB RNAi and TLR4 RNAi
groups: P<0.001, compared with the blank group).
Blocking or silencing the
TLR4/MyD88/NF-κB signaling pathway inhibits iohexol-induced HK-2
cell apoptosis
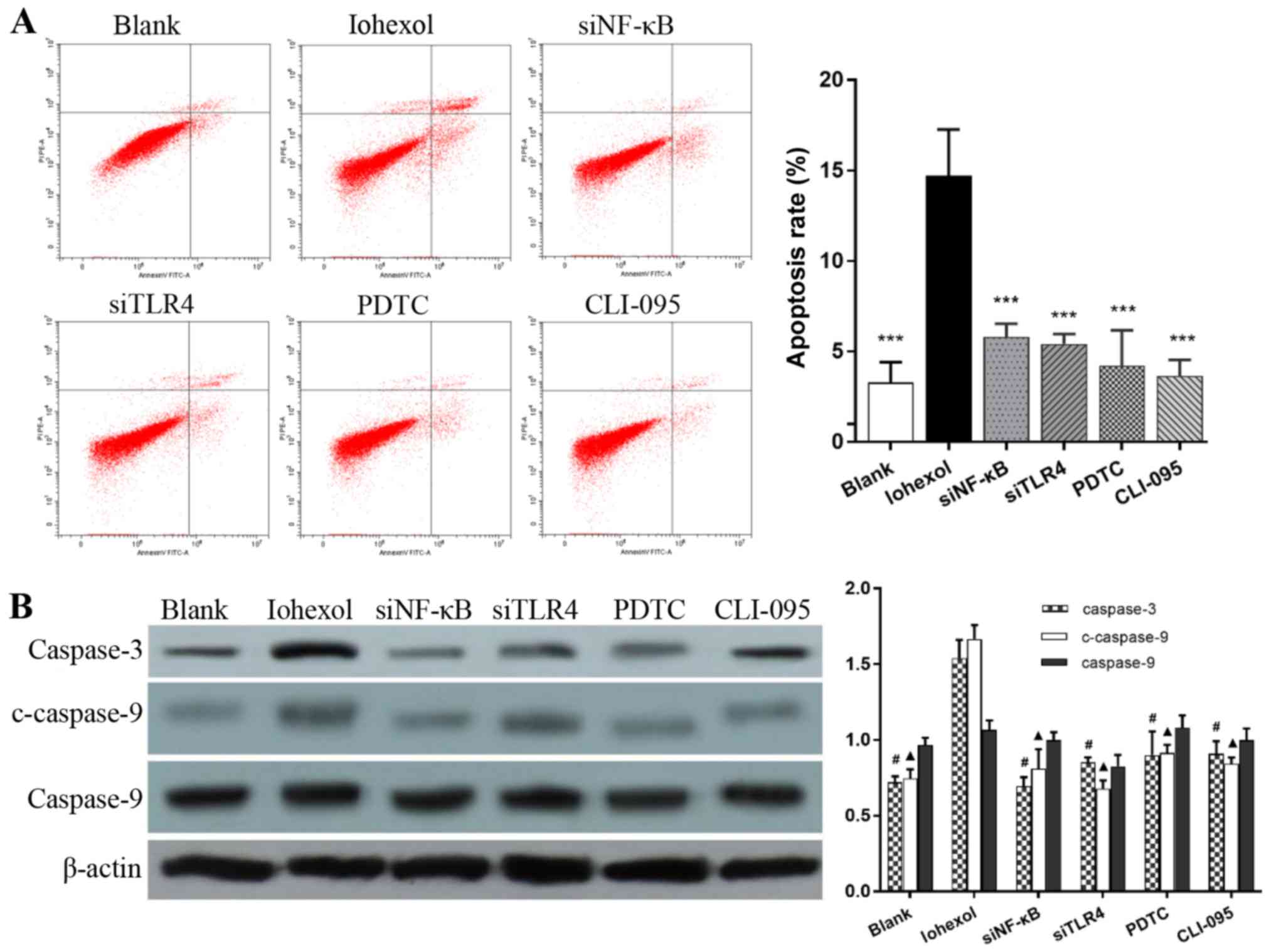
The apoptotic rate of the iohexol group
(14.71±2.55%) was significantly higher compared with that of the
other groups (P<0.001), and there were no significant
differences in the apoptotic rates of the PDTC (4.21±1.93%),
CLI-095 (3.64±0.90%), NF-κB RNAi (5.80±0.72%) or TLR4 RNAi groups
(5.42±0.54%) compared with that of the blank group (3.29±1.11%;
P>0.05) (Fig. 4A). The
expression levels of apoptosis-related proteins were directly
associated with the corresponding apoptotic rates. Although there
was no significant difference in the expression of caspase-9
amongst the groups, the expression levels of caspase-3 and
c-caspase-9 were significantly increased in the iohexol group,
compared with the blank group (P<0.001), and decreased in the
other groups compared with the iohexol group (P<0.001; Fig. 4B).
Blocking or silencing TLR4/MyD88/NF-κB
signaling attenuates iohexol-induced inflammation
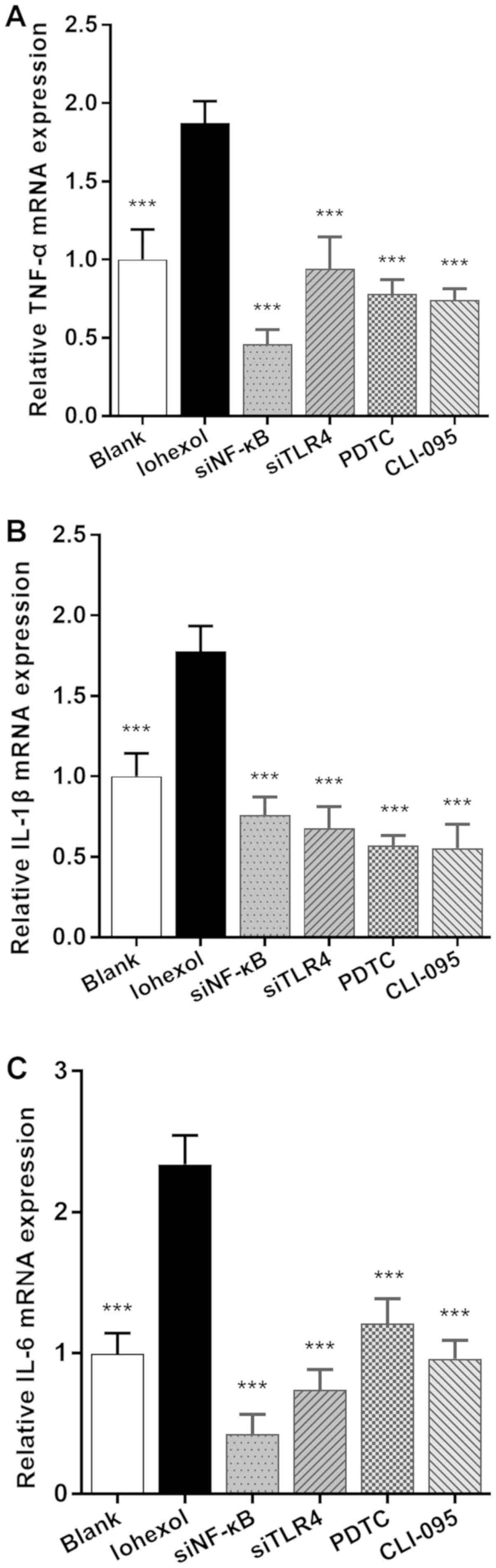
As presented in Fig.
5, the mRNA expression levels of TNF-α, IL-1β and IL-6 in the
iohexol group were significantly higher compared with those in the
blank group (P<0.001). After blocking or silencing of the TLR4
or NF-κB loci, the expression levels were significantly decreased
compared with those of the iohexol group (P<0.001).
Discussion
CIN is defined as an increase in serum creatinine of
>25% or 44 µmol/l, compared with the baseline value, within 3
days of using CM and in the absence of any other causes of renal
injury (13). The specific
pathogenesis of CIN has not been completely elucidated, although it
is widely accepted that immune inflammatory responses play an
important role in the occurrence and development of CIN. Numerous
studies have confirmed that the expression of specific cytokines is
elevated during CIN, and it was reported that immune inflammatory
responses in a CIN rat model were alleviated by NF-κB silencing
(14).
TLR4, which is expressed in renal intrinsic cells
(such as epithelial cells), endothelial and mesangial cells, is one
of the key factors of the inflammatory response. TLR4 activation
transduces transmembrane signals via the MyD88-dependent pathway,
activating transcription factors such as NF-κB, and promoting the
subsequent release of a variety of cytokines and inflammatory
factors (15). TLR4 signaling is
initiated by the recognition and binding of pathogen-associated
molecular patterns and other specific ligands, including
lipopolysaccharide, taxol, fusion protein, envelope proteins, heat
shock proteins, oligosaccharides of hyaluronic acid, polysaccharide
fragments of heparan sulfate and fibrinogen (16). A previous report verified that, in
an animal model of ischemia-reperfusion injury, the gene expression
and protein synthesis of TLR4 in renal tubular epithelial cells was
significantly upregulated (17).
Additionally, Pulskens et al (18) reported that, in an animal model of
acute ischemic kidney injury, the inflammatory response and tubular
damage were alleviated in TLR4-knockout mice compared with
wild-type mice.
PDTC is a commonly-used NF-κB inhibitor that reduces
the expression and transcriptional activity of all NF-κB subunits
(19). It was previously reported
that PDTC prevented the degradation of inhibitory factor B-α and
inhibited the nuclear translocation of NF-κB, thus reducing the
occurrence of acute and chronic inflammation (20). Borghi et al (21) found that PDTC reduced the levels of
serum urea and creatinine, and mitigated diclofenac-induced AKI.
CLI-095 (also known as TAK-242), potently inhibits TLR4 signaling
and downregulates the production of nitric oxide and
pro-inflammatory cytokines (22).
TAK-242 binds directly to a specific amino acid, Cys747, in the
intracellular domain of TLR4, which inhibits TLR4 signaling and, in
turn, inhibits MyD88-dependent and -independent signaling (23). In mouse models, TAK-242 was also
shown to alleviate renal inflammation, tubulointerstitial damage,
fibrosis and the loss of renal function induced by cyclosporin A
(24). In a randomized,
double-blinded, controlled trial of patients with severe sepsis,
the 28-day all-cause mortality rate was lower in patients receiving
TAK-242 compared with that in the placebo treatment group (25), suggesting the potential inhibition
of TLR4-mediated inflammatory responses in clinical practice.
In the present study, NF-κB and TLR4 were directly
inhibited by PDTC and CLI-095, and silenced using specific siRNAs.
Treated HK-2 cells were incubated with 100 mg/ml iohexol as an
in vitro model of CIN. Notably, the mRNA and protein
expression levels of TLR4, MyD88 and NF-κB in the iohexol group
were significantly higher compared with those in the blank group,
while even with the stimulation of iohexol, these levels were still
decreased following the blocking or silencing of the
TLR4/MyD88/NF-κB signaling pathway. These results indicate that
iohexol is able to activate TLR4/MyD88/NF-κB signaling in CIN, and
that this activation can be effectively counteracted by blocking or
silencing NF-κB and TLR4. Furthermore, inhibiting TLR4/MyD88/NF-κB
signaling significantly reduced the mRNA expression levels of
TNF-α, IL-1β and IL-6, thereby delaying the inflammatory response.
However, the changes in the expression of these inflammatory
factors following blocking or silencing were not completely
consistent with those in the blank group, suggesting that other
pathways may be involved in the inflammation-associated injury of
renal tubular epithelial cells.
In CIN, caspase-9 is activated through the
mitochondrial pathway, which subsequently activates the downstream
caspase-3 and induces apoptosis (26). Previous studies have demonstrated
that, in hypotonic or isotonic CM-induced in vitro models,
caspase-9 and -3 were activated, while caspase-8 and -10 were not
(27). In another study, specific
inhibitors of caspase-3 and -9 attenuated the CM-induced injury of
LLC-PK1 cells, while specific inhibitors of caspase-8 did not
(28), indicating that
contrast-induced apoptosis occurs primarily through the
mitochondrial pathway. In the present study, the expression levels
of various apoptosis-associated proteins were investigated,
demonstrating that the expression of caspase-3 and c-caspase-9 were
markedly higher following iohexol treatment, compared with the
blank group, which supports previous findings (24-26).
However, no significant differences were observed in the expression
levels of caspase-9 between the two groups. After inhibiting
TLR4/MyD88/NF-κB signaling, the elevated expression levels of
caspase-3 and c-caspase-9 were downregulated, and no significant
differences were observed when compared with the blank group.
Furthermore, the apoptotic rates of HK-2 cells were assessed by
flow cytometry, the results of which confirmed the elevated protein
expression levels of caspase-3 and c-caspase-9. These results
indicate that the TLR4/MyD88/NF-κB signaling pathway is involved in
the iohexol-induced apoptosis of renal tubular epithelial
cells.
There were several limitations to the present study.
First, the conclusions were drawn from in vitro
experimentation only, and have not been confirmed in in vivo
experiments or clinical studies. Second, although its mRNA and
protein levels were determined, the expression of MyD88 was not
experimentally inhibited. Finally, only a 48-h time point was
selected, which was insufficient to elucidate the role of
TLR4/MyD88/NF-κB signaling in the entire process of CIN
development. In future studies, the establishment of CIN mouse
models of TLR4, MyD88 and NF-κB gene-silencing may further
elucidate the role of TLR4/MyD88/NF-κB signaling in CIN.
To conclude, the results of the present study
suggested that the TLR4/MyD88/NF-κB signaling pathway is involved
in the development of contrast-induced nephropathy by promoting
inflammatory responses and apoptosis. These findings may enable a
deeper understanding of the pathogenesis of CIN, and highlight the
TLR4/MyD88/NF-κB signaling pathway as a potential target for the
clinical prevention of CIN.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Sichuan
Science and Technology Agency (grant no. 0040205301F93).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XW performed the experiments and was a major
contributor to the writing of the manuscript. JZ and LY were
responsible for the experimental design and statistical analysis.
JY and SW participated in the experiments and data analysis. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nash K, Hafeez A and Hou S:
Hospital-acquired renal insufficiency. Am J Kidney Dis. 39:930–936.
2002.PubMed/NCBI View Article : Google Scholar
|
|
2
|
McCullough PA, Choi JP, Feghali GA,
Schussler JM, Stoler RM, Vallabahn RC and Mehta A: Contrast-induced
acute kidney injury. J Am Coll Cardiol. 68:1465–1473.
2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Pickering JW, Blunt IRH and Than MP: Acute
kidney injury and mortality prognosis in acute coronary syndrome
patients: A meta-analysis. Nephrology (Carlton). 23:237–246.
2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Narula A, Mehran R, Weisz G, Dangas GD, Yu
J, Genereux P, Nikolsky E, Brener SJ, Witzenbichler B, Guagliumi G,
et al: Contrast-induced acute kidney injury after primary
percutaneous coronary intervention: Results from the HORIZONS-AMI
substudy. Eur Heart J. 35:1533–1540. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Persson PB, Hansell P and Liss P:
Pathophysiology of contrast medium-induced nephropathy. Kidney Int.
68:14–22. 2005.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Heyman SN, Rosen S and Rosenberger C:
Renal parenchymal hypoxia, hypoxia adaptation, and the pathogenesis
of radiocontrast nephropathy. Clin J Am Soc Nephrol. 3:288–296.
2008.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Sadat U, Usman A, Boyle JR, Hayes PD and
Solomon RJ: Contrast medium-induced acute kidney injury.
Cardiorenal Med. 5:219–228. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kawai T and Akira S: The role of
pattern-recognition receptors in innate immunity: Update on
toll-like receptors. Nat Immunol. 11:373–384. 2010.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Cook DN, Pisetsky DS and Schwartz DA:
Toll-like receptors in the pathogenesis of human disease. Nat
Immunol. 5:975–979. 2004.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Lee JW, Kim SC, Ko YS, Lee HY, Cho E, Kim
MG, Jo SK, Cho WY and Kim HK: Renoprotective effect of paricalcitol
via a modulation of the TLR4-NF-κB pathway in
ischemia/reperfusion-induced acute kidney injury. Biochem Biophys
Res Commun. 444:121–127. 2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zhang D, Li Y, Liu Y, Xiang X and Dong Z:
Paclitaxel ameliorates lipopolysaccharide-induced kidney injury by
binding myeloid differentiation protein-2 to block toll-like
receptor 4-mediated nuclear factor-κB activation and cytokine
production. J Pharmacol Exp Ther. 345:69–75. 2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Stacul F, van der Molen AJ, Reimer P, Webb
JA, Thomsen HS, Morcos SK, Almén T, Aspelin P, Bellin MF and
Clement O: Contrast induced nephropathy: Updated ESUR contrast
media safety committee guidelines. Eur Radiol. 21:2527–2541.
2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Machado RA, Constantino de Souza L, Tomasi
CD, Rojas HA, Vuolo FS, Vitto MF, Cesconetto PA, de Souza CT,
Ritter C and Dal-Pizzol F: Sodium butyrate decreases the activation
of NF-κB reducing inflammation and oxidative damage in the kidney
of rats subjected to contrast-induced nephropathy. Nephrol Dial
Transplant. 27:3136–3140. 2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
O'Neill LA and Bowie AG: The family of
five: TIR-domain-containing adaptors in Toll-like receptor
signalling. Nat Rev Immunol. 7:353–364. 2007.PubMed/NCBI View
Article : Google Scholar
|
|
16
|
Takeda K, Kaisho T and Akira S: Toll-like
receptors. Annu Rev Immunol. 21:335–376. 2003.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Mudaliar H, Pollock C, Komala MG, Chadban
S, Wu H and Panchapakesan U: The role of Toll-like receptor
proteins (TLR) 2 and 4 in mediating inflammation in proximal
tubules. Am J Physiol Renal Physiol. 305:F143–F154. 2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Pulskens WP, Teske GJ, Butter LM, Roelofs
JJ, van der Poll T, Florquin S and Leemans JC: Toll-like receptor-4
coordinates the innate immune response of the kidney to renal
ischemia/reperfusion injury. PLoS One. 3(e3596)2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Morais C, Gobe G, Johnson DW and Healy H:
Anti-angiogenic actions of pyrrolidine dithiocarbamate, a nuclear
factor kappa B inhibitor. Angiogenesis. 12:365–379. 2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Cuzzocrea S, Chatterjee PK, Mazzon E, Dugo
L, Serraino I, Britti D, Mazzullo G, Caputi AP and Thiemermann C:
Pyrrolidine dithiocarbamate attenuates the development of acute and
chronic inflammation. Br J Pharmacol. 135:496–510. 2002.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Borghi SM, Fattori V, Ruiz-Miyazawa KW,
Bertozzi MM, Lourenco-Gonzalez Y, Tatakihara RI, Bussmann AJC,
Mazzuco TL, Casagrande R and Verri Jr WA: Pyrrolidine
dithiocarbamate inhibits mouse acute kidney injury induced by
diclofenac by targeting oxidative damage, cytokines and NF-κB
activity. Life Sci. 208:221–231. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ii M, Matsunaga N, Hazeki K, Nakamura K,
Takashima K, Seya T, Hazeki O, Kitazaki T and Iizawa Y: A novel
cyclohexene derivative, ethyl
(6R)-6-[N-(2-Chloro-4-fluorophenyl)sulfamoyl]cyclohex-1-ene-1-carboxylate
(TAK-242), selectively inhibits toll-like receptor 4-mediated
cytokine production through suppression of intracellular signaling.
Mol Pharmacol. 69:1288–1295. 2006.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Takashima K, Matsunaga N, Yoshimatsu M,
Hazeki K, Kaisho T, Uekata M, Hazeki O, Akira S, Iizawa Y and Ii M:
Analysis of binding site for the novel small-molecule TLR4 signal
transduction inhibitor TAK-242 and its therapeutic effect on mouse
sepsis model. Br J Pharmacol. 157:1250–1262. 2009.PubMed/NCBI View Article : Google Scholar
|
|
24
|
González-Guerrero C, Cannata-Ortiz P,
Guerri C, Egido J, Ortiz A and Ramos AM: TLR4-mediated inflammation
is a key pathogenic event leading to kidney damage and fibrosis in
cyclosporine nephrotoxicity. Arch Toxicol. 91:1925–1939.
2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Rice TW, Wheeler AP, Bernard GR, Vincent
JL, Angus DC, Aikawa N, Demeyer I, Sainati S, Amlot N, Cao C, et
al: A randomized, double-blind, placebo-controlled trial of TAK-242
for the treatment of severe sepsis. Crit Care Med. 38:1685–1694.
2010.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Stewart JD, Hengstler JG and Bolt HM:
Contrast agent-induced nephrotoxicity: Role of oxidative stress and
apoptosis through the mitochondrial pathway. Arch Toxicol.
85:163–164. 2011.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Romano G, Briguori C, Quintavalle C, Zanca
C, Rivera NV, Colombo A and Condorelli G: Contrast agents and renal
cell apoptosis. Eur Heart J. 29:2569–2576. 2008.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Yano T, Itoh Y, Sendo T, Kubota T and
Oishi R: Cyclic AMP reverses radiocontrast media-induced apoptosis
in LLC-PK1 cells by activating A kinase/PI3 kinase. Kidney Int.
64:2052–2063. 2003.PubMed/NCBI View Article : Google Scholar
|