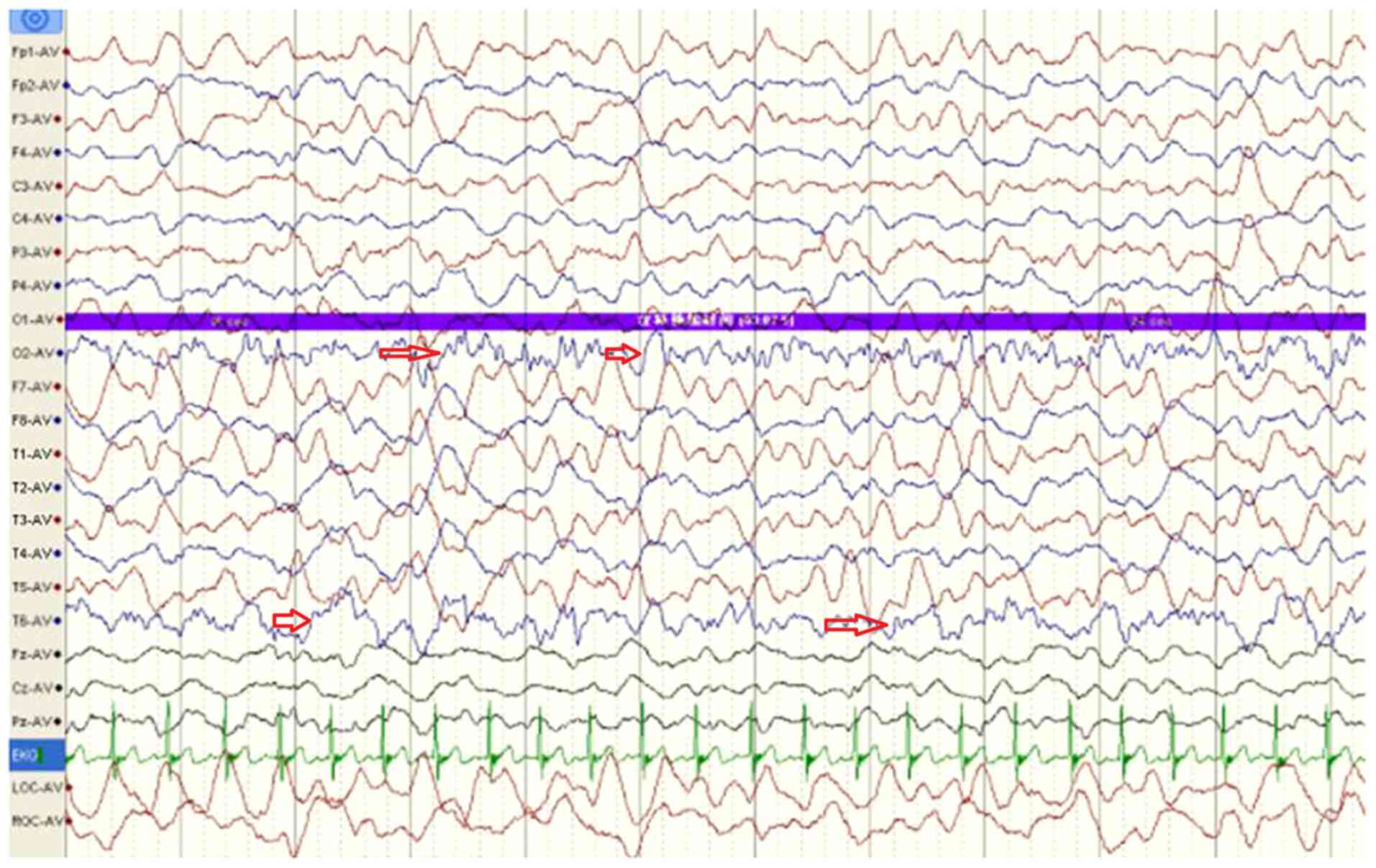
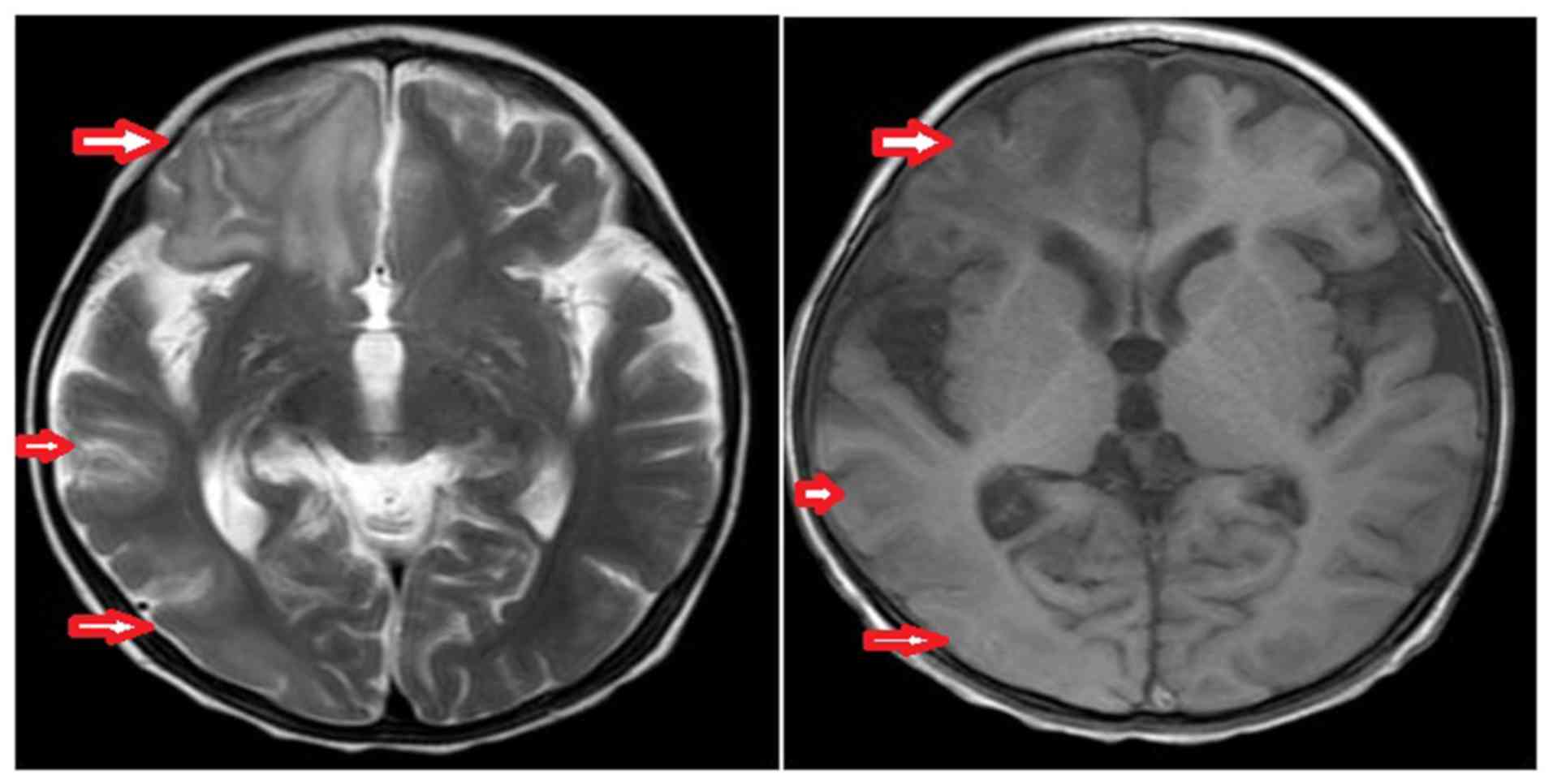
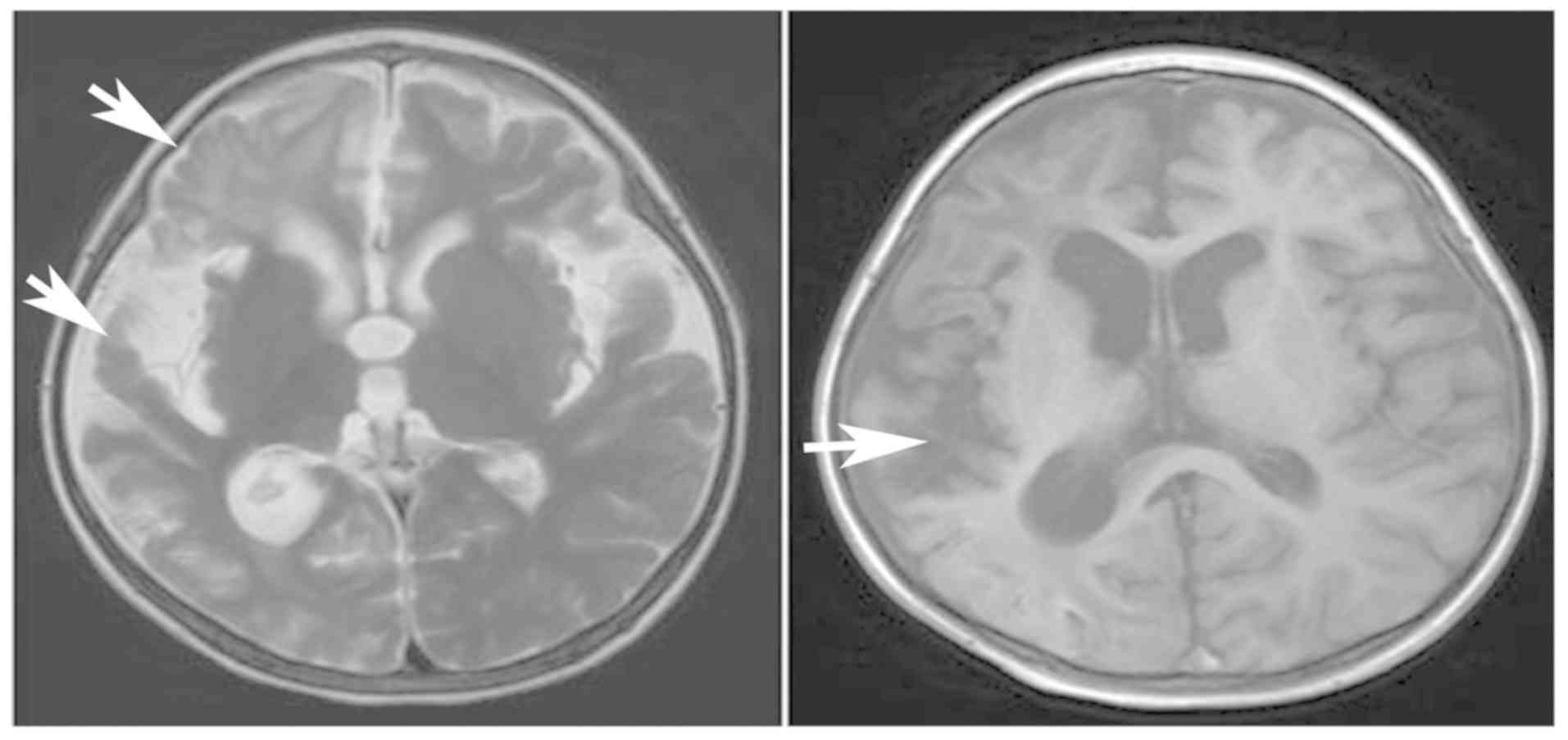
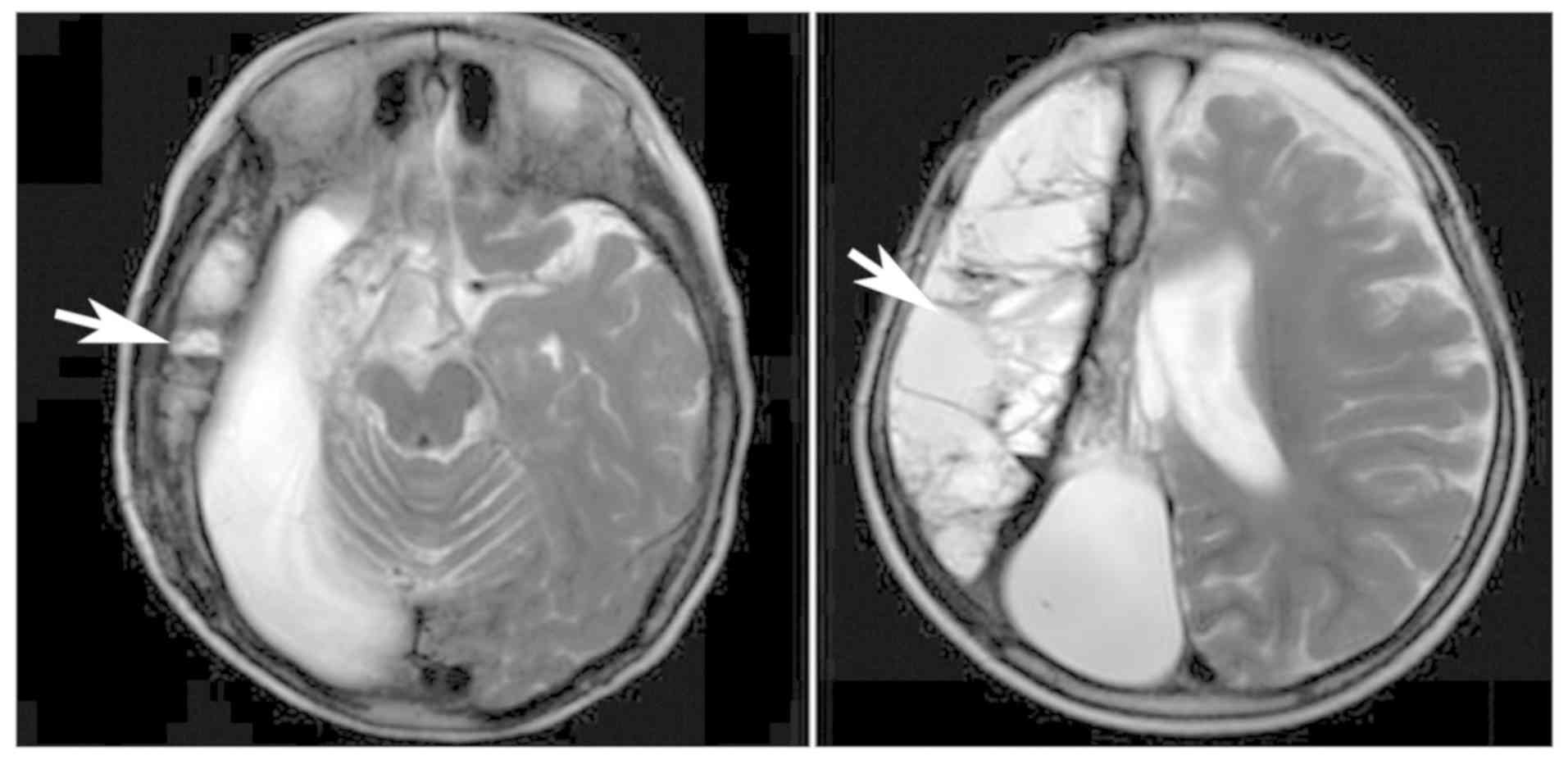
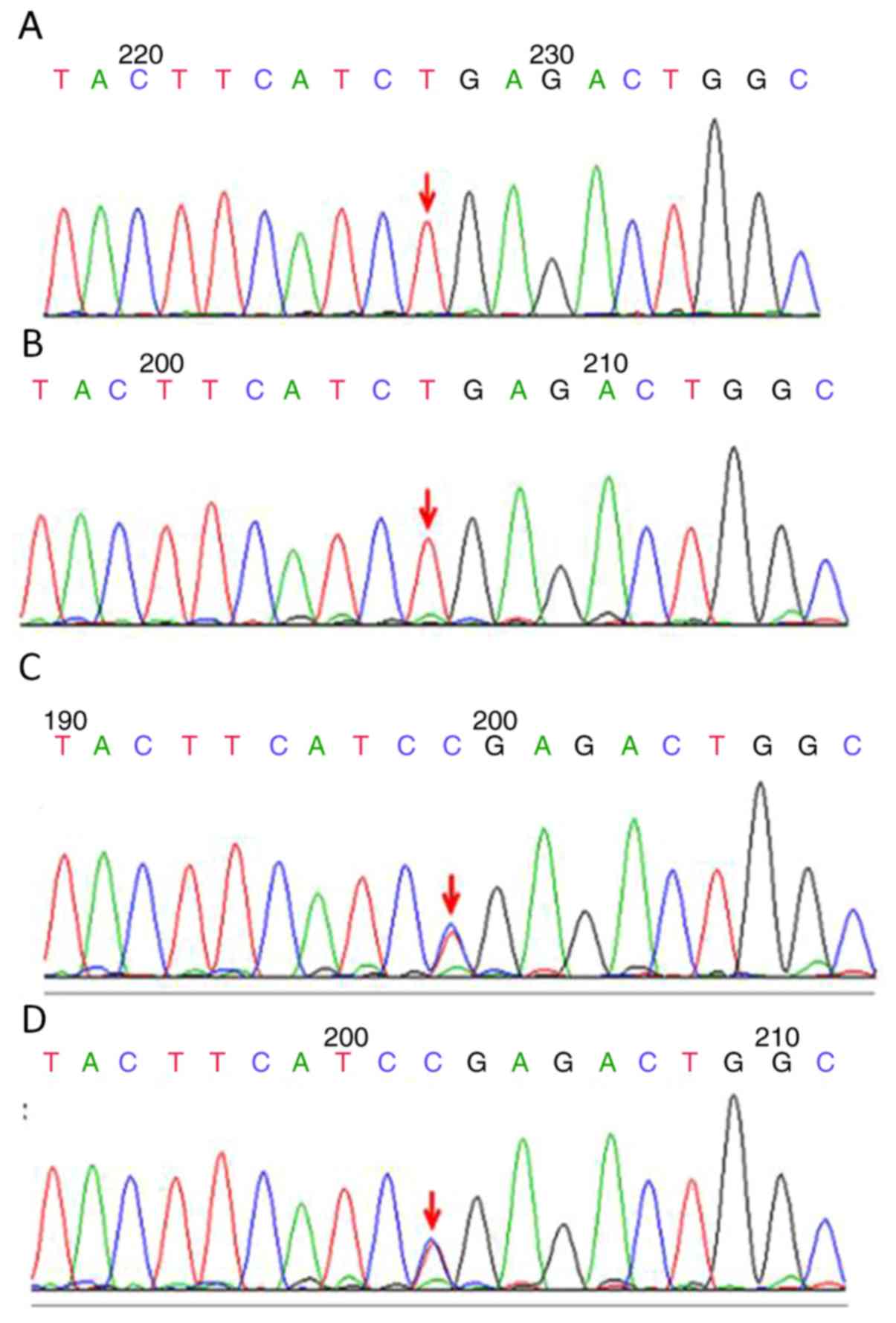
|
1
|
Tan JQ, Chen DY, Li ZT, Yan TZ, Huang JW
and Cai R: Genetic diagnosis of 10 neonates with primary carnitine
deficiency. Zhongguo Dang Dai Er Ke Za Zhi. 19:1150–1154.
2017.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
2
|
Buist NR: Historical perspective on
clinical trials of carnitine in children and adults. Ann Nutr
Metab. 68:1–4. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ravindranath A, Pai G, Srivastava A,
Poddar U and Yachha SK: Infant with hepatomegaly and hypoglycemia:
A setting for fatty acid oxidation defects. Indian J Gastroenterol.
36:429–434. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Belousova ED: The decreased level of
plasma carnitine in patients with epilepsy. Zh Nevrol Psikhiatr Im
S S Korsakova. 117:106–110. 2017.PubMed/NCBI View Article : Google Scholar : (In Russian).
|
|
5
|
Greenberg DA, Aminoff MJ and Simon RP:
Clinical Neurogy (5th edition). The United States: Mc Graw-Hill,
New York, NY, pp157-158, 2002.
|
|
6
|
Ferdinandusse S, Brinke HT, Ruiter JP,
Haasjes J, Oostheim W, Ven Lenthe H, IJlst L, Ebberink MS, Wanders
RJ, Vaz FM and Waterham HR: Mutation creating an upstream
translation initiation codon in SLC22A5 5'UTR is a frequent cause
of primary carnitine deficiency. Hum Mutat. 40:1899–1904.
2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Koizumi A, Nozaki J, Ohura T, Kayo T, Wada
Y, Nezu J, Ohashi R, Tamai I, Shoji Y, Takada G, et al: Genetic
epidemiology of the carnitine transporter OCTN2 gene in a Japanese
population and phenotypic characterization in Japanese pedigrees
with primary systemic carnitine deficiency. Hum Mol Genet.
8:2247–2254. 1999.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hassan FA, El Mougy F, Sharaf SA, Mandour
I, Morgan MF, Selim LA, Hassan SA, Salem F, Oraby A, Girgis MY, et
al: Inborn errors of metabolism detectable by tandem mass
spectrometry in Egypt: The first newborn screening pilot study. J
Med Screen. 23:124–129. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Nałęcz KA and Nałęcz MJ:
Carnitine-mitochondria and beyond. Postepy Biochem. 62:85–93.
2016.PubMed/NCBI
|
|
10
|
Lindner M, Gramer G, Haege G,
Fang-Hoffmann J, Schwab KO, Tacke U, Trefz FK, Mengel E, Wendel U,
Leichsenring M, et al: Efficacy and outcome of expanded newborn
screening for metabolic diseases-report of 10 years from South-West
Germany. Orphanet J Rare Dis. 6(44)2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Lehotay DC, Hall P, Lepage J, Eichhorst
JC, Etter ML and Greenberg CR: LC-MS/MS progress in newborn
screening. Clin Biochem. 44:21–31. 2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Longo N, Frigeni M and Pasquali M:
Carntiin transport and fatty acid oxidation. Biochim Biophys Acta.
1863:2422–2435. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Gallant NM, Leydiker K, Wilnai Y, Lee C,
Lorey F, Feuchtbaum L, Tang H, Carter J, Enns GM, Packman S, et al:
Biochemical characteristics of newborns with carnitine transporter
defect identified by newborn screening in California. Mol Genet
Metab. 122:76–84. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Lin Y, Lin W, Yu K, Zheng F, Zheng Z and
Fu Q: Mutational analysis of SLC22A5 gene in eight patients with
systemic primary carnitine deficiency. Zhonghua Yi Xue Yi Chuan Xue
Za Zhi. 34:35–39. 2017.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
15
|
Frigeni M, Balakrishnan B, Yin X, Calderon
FR, Mao R, Pasquali M and Longo N: Functional and molecular studies
in primary carnitine deficiency. Hum Mutat. 38:1684–1699.
2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li FY, El-Hattab AW, Bawle EV, Boles RG,
Schmitt ES, Scaglia F and Wong LJ: Molecular spectrum of SLC22A5
(OCTN2) gene mutations detected in 143 subjects evaluated for
systemic carnitine defciency. Hum Mutat. 31:E1632–E1651.
2010.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Angelini C: Letter: Carnitine Deficiency.
Lancet. 2(554)1975.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Longo N: Primary carnitine deficiency and
newborn screening for disorders of the carnitinecycle. Ann Nutr
Metab. 68:5–9. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zheng J, Zhang Y, Hong F, Yang J, Tong F,
Mao H and Huang X, Zhou X, Yang R, Zhao Z and Huang X: Screening
for fatty acid oxidation disorders of newborns in Zhejiang
province: prevalence, outcome and follow-up. Zhejiang Da Xue Xue
Bao Yi Xue Ban. 46:248–255. 2017.PubMed/NCBI(In Chinese).
|
|
20
|
Deswal S, Bijarnia-Mahay S, Manocha V,
Hara K, Shigematsu Y, Saxena R and Verma IC: Primary carnitine
deficiency-a rare treatable cause of cardiomyopathy and massive
hepatomegaly. Indian J Pediatr. 84:83–85. 2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Lahrouchi N, Lodder EM, Mansouri M, Tadros
R, Zniber L, Adadi N, Clur SA, van Spaendonck-Zwarts KY, Postma AV,
Sefiani A, et al: Exome sequencing identifies primary carnitine
deficiency in a family with cardiomyopathy and sudden death. Eur J
Hum Genet. 25:783–787. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Madsen KL, Preisler N, Rasmussen J,
Hedermann G, Olesen JH, Lund AM and Vissing J: L-Carnitine improves
skeletal muscle fat oxidation in primary carnitine deficiency. J
Clin Endocrinol Metab. 103:4580–4588. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Jun JS, Lee EJ, Park HD and Kim HS:
Systemic primary carnitine deficiency with hypoglycemic
encephalopathy. Ann Pediatr Endocrinol Metab. 21:226–229.
2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Perin F, Rodríguez-Vázquez Del Rey MD,
Carreras-Blesa C, Arrabal-Fernández L, Jiménez-Jáimez J and
Tercedor L: Dilated cardiomyopathy with short QT interval suggests
primary carnitine deficiency. Rev Esp Cardiol (Engl Ed).
71:1074–1075. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Papadopoulou-Legbelou K, Gogou M, Dokousli
V, Eboriadou M and Evangeliou A: Dilated cardiomyopathy as the only
clinical manifestation of carnitine transporter deficiency. Indian
J Pediatr. 84:231–233. 2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Morrison-Levy N, Go C, Ochi A, Otsubo H,
Drake J, Rutka J and Weiss SK: Children with autism spectrum
disorders and drug-resistant epilepsy can benefit from epilepsy
surgery. Epilepsy Behav. 85:200–204. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Hirsch E and Arzimanoglou A: Children with
drug-resistant partial epilepsy: Criteria for the identification of
surgical candidates. Rev Neurol (Paris). 160:5S210–5S219.
2004.PubMed/NCBI(In French).
|
|
28
|
Roth J, Nagar S, Constantini S and Fried
I: Hemispherotomy for treatment of refractory epilepsy in children.
Harefuah. 156:482–485. 2017.PubMed/NCBI(In Hebrew).
|