Introduction
Aortic dissection (AD) is a vascular disease
characterized by intramural hematoma following injury to the
innermost layer of the aorta. AD has a low incidence rate but a
high mortality rate. The mortality rate of patients with AD is 1-2%
per hour before treatment after the dissection occurs and the total
mortality rate is >90% (1).
Total aortic arch replacement and thoracic aortic intracavitary
repair have been widely used for the clinical treatment of Stanford
A type AD and Stanford B type AD, respectively; however, both
treatment strategies display a number of limitations, among them,
total aortic arch replacement is mostly applied to Stanford A type
AD, with relatively few complications, but large trauma and high
cost, which brings heavy psychological pressure and economic burden
to patients and families; While intracavitary thoracic aortic
repair is mostly applied to Stanford B type AD, which is relatively
inexpensive but can significantly reduce short-term mortality
(2,3). Therefore, further investigation into
the pathogenesis of AD, as well as the identification of additional
effective and safe targets are essential for the prevention and
treatment of AD.
Thrombospondin-2 (TSP-2) is a multifunctional
extracellular matrix protein that belongs to the TSP family
(4). TSP-2 is widely expressed in
organs and tissues in a variety of mammals, including humans and
mice (5). Numerous studies have
demonstrated that TSP-2 may be involved in the regulation of
angiogenesis, proliferation, apoptosis, tumor migration, autophagy
and transforming growth factor (TGF)-β activation (4-8).
Previous studies have also reported that TSP-2 can amplify the
inflammatory response (9-11).
Furthermore, the exact role of TSP-2 depends on the inflammatory
microenvironment. For example, TSP-2 displays an anti-inflammatory
role during chronic allograft nephropathy and viral myocarditis,
whereas proinflammatory effects of TSP-2 have been observed during
asthma (9-11).
Animal studies and clinical experiments have
reported that TSP-2 expression is closely associated with the
progression and development of a variety of cardiovascular
diseases. For instance, circulating TSP-2 levels are increased in
patients with chronic heart failure with or without preserved
ejection fraction, and higher TSP-2 levels are associated with an
increased frequency of cardiac events and poorer prognosis
(12,13). Elevated circulating TSP-2 levels are
also observed in elderly patients with aortic aneurysms and are
associated with increased mortality (14). Furthermore, TSP-2 knockout
aggravates doxorubicin-induced cardiac injury and dysfunction
(15) and prevents dilated
cardiomyopathy in elderly mice (16). However, the expression of TSP-2
during human AD has not yet been reported. The present study aimed
therefore to investigate TSP-2 expression during human AD and
determine the underlying mechanisms.
Materials and methods
Sample collection
Aortic tissue and blood samples were collected from
the Beijing Anzhen Hospital of Capital Medical University. The
present study was approved by the Ethics Committee of Beijing
Anzhen Hospital (approval no. 2017109H). Written informed consent
was obtained from all patients or their families.
Aortic tissue specimens (control group: Age, 3266;
median age, 47; 6 males and 3 females and AD group: Age, 3974;
median age 57; 12 males and 2 females) were collected by cardiac
surgeons in the operating room of the Department of Anesthesiology
of Anzhen Hospital between March 2016 and May 2018. Normal aortic
specimens (n=9) collected from brain dead donors who were involved
in traffic accidents served as the control group. Donors with a
history of significant cardiovascular diseases or aortic pathology
changes were excluded. Torn aortic samples (n=14) were obtained
from patients who had been admitted for acute AD, according to
clinical symptoms and results of computed tomography angiography
(CTA), and who underwent surgical treatment. Torn aortic samples
formed the AD group.
Blood samples of ~2 ml (control group: Age, 3365;
median age, 43; 42 males and 18 females and AD group: Age, 4177,
median age, 52; 132 males and 28 females) were obtained from
patients in the intensive care unit of Anzhen Hospital between May
2017 and September 2018. Patients (n=250) who were hospitalized due
to sudden chest pain were enrolled in the present study. A total of
28 patients were excluded due to a history of diseases that have
been reported to affect TSP-2 secretion, including chronic heart
failure (n=7), coronary artery disease (n=3), peripheral vascular
disease (n=11), chronic renal failure (n=4) and connective tissue
disease (n=3). A further two patients who were diagnosed with acute
left heart failure caused by the development of pulmonary edema and
dyspnea immediately after the onset of chest pain were also
excluded. According to clinical diagnoses, the remaining 220
patients were divided into the non-AD (NAD) control group (n=60)
and the AD group (n=160), which included the Stanford A group
(n=86) and the Stanford B group (n=74). The patients in the NAD
group were diagnosed with cervical spondylosis (n=18), gastric
erosion (n=9), gastric ulcer (n=9), cardiac neurosis (n=13) or
chest pain of unknown cause (n=11).
All blood samples were obtained after CTA but prior
to the administration of any treatment, as treatment, such as
lowering the heart rate, lowering blood pressure or/and surgery may
also affect TSP-2 secretion.
Western blotting
Total protein from each aortic specimen was
extracted using RIPA buffer (cat. no. G2002-30; Wuhan Servicebio,
Technology Co., Ltd.) and quantified using a bicinchoninic acid
Protein Assay kit (Thermo Fisher Scientific, Inc.). A total of 30
µg protein from each sample was separated by 8% SDS-PAGE and
transferred to PVDF membranes at 100 mA for 2 h. After blocking
with 5% nonfat milk at 25˚C for 90 min, the membranes were
incubated overnight at 4˚C with primary antibodies targeted against
TSP-2 (1 µg/ml; cat. no. MAB1635; R&D Systems, Inc.) and GAPDH
(1:1,000; cat. no. 97166; Cell Signaling Technology, Inc.) at 4˚C
for 12 h. Following primary incubation, the membranes were
incubated with a secondary IRDye 800CW-conjugated antibody
(1:12,500; cat. no. 925-32210; Li-Cor Biosciences) at room
temperature for 1 h. Protein bands were scanned and quantified
using an Odyssey Imaging system (LI-COR Biosciences).
Histological analysis
Aortic tissues were cut into appropriate sizes (1-3
mm3) immediately after dissection, and some specimens
were immediately fixed in 4% paraformaldehyde at 25˚C for 3 days.
After embedding in paraffin, tissues were cut into 4-5 µm slices
and placed on slides. After blocking with 5% goat serum (cat. no.
16210072; Gibco; Thermo Fisher Scientific, Inc.) at room
temperature for 5 min, the slides were incubated with a primary
anti-TSP-2 antibody (0.5 µg/ml; cat. no. BAF1635; R&D Systems,
Inc.). The sources of aortic TSP-2, including CD4+ T
lymphocytes (CD4+ TCs), macrophages (Møs), smooth muscle
cells (SMCs) and endothelial cells (ECs), were investigated by
double immunofluorescence staining. Slides were therefore incubated
with the following primary antibodies: Anti-CD4 (20 µg/ml; cat. no.
MAB379; R&D Systems, Inc.), anti-CD68 (20 µg/ml; cat. no.
MAB20401; R&D Systems, Inc.), anti-α-SMA (15 µg/ml; cat. no.
MAB1420; R&D Systems, Inc.) and anti-CD31 (20 µg/ml; cat. no.
BBA7; R&D Systems, Inc.). After incubation with anti-Mouse IgG
H&L fragment (Alexa Fluor® 647 Conjugate) secondary
antibody (1:1,000; cat. no. 4410; Cell Signaling Technology, Inc.)
at room temperature for 90 min, samples were observed using a
fluorescence microscope (magnification, x200). The localization of
TSP-2 expression could be observed by assessing whether the green
and red signals merge together (yellow signals).
TSP-2, TNF-α and IL-6
measurements
Blood samples were centrifuged at 5,000 x g at 4˚C
for 30 min and the the plasma of each sample collected and stored
at -80˚C until further analysis. Prior to the measurement of
cytokine levels, the supernatant samples were thawed and a
preliminary test (ELISA) for dilution of different multiples was
performed to determine the dilution ratio of the supernatant. After
5-fold dilution with deionized water, 100 µl plasma solution was
added to the plate and the TSP-2 (cat. no. DTSP20, R&D Systems,
Inc.), TNF-α (cat. no. KHC3014C; Thermo Fisher Scientific, Inc.)
and IL-6 (cat. no. EH2IL6; Thermo Fisher Scientific, Inc.) ELISA
kits were used according to the manufacturer's protocol.
SMC isolation, culture and
treatment
A total of 60 male C57BL/6 mice (age, 9-10 weeks;
weight, 23-26 g) were purchased from the Institute of Model Zoology
of Nanjing University and housed in a pathogen-free mouse room on a
12 h light/12 dark cycle at a temperature of 22-24˚C and a humidity
of 50 ± 5% at Anzhen Hospital and received water ad libitum.
Mice were anesthetized with 2% isoflurane and subsequently
euthanized by cervical dislocation. The spleens, femurs and aortas
were immediately isolated. CD4+ TCs (17,18),
bone marrow-derived Møs (18,19),
aortic SMCs (20,21) and aortic ECs were isolated as
previously described (22).
Briefly, to obtain CD4+ TCs, a spleen cell suspension
was prepared in complete RPMI-1640 culture medium (cat. no.
22400105; Gibco; Thermo Fisher Scientific, Inc.) and
CD4+ TCs were positively selected using CD4 magnetic
beads (Miltenyi Biotec, Inc.) and an auto-magnetic actived cell
soring (MACS) separator. Furthermore, to obtain bone marrow-derived
Møs, both ends of the femurs were cut and RPMI-1640 culture medium
was used to flush out the cells from the femoral canal.
Subsequently, red blood cells were lysed using ACK lysis buffer
(cat. no. G2015-250; Wuhan Servicebio Technology Co., Ltd.), and
the remaining cells were plated in 6-well plates (106
cells per well) and treated with 50 ng/ml murine macrophage colony
stimulating factor (cat. no. 315-02; PeproTech, Inc.) for 5 days at
37˚C to promote Mø differentiation. For the isolation of aortic
SMCs, the isolated aorta was treated with 0.1% (37˚C) type II
collagenase to remove the adventitia, and a sterile cotton-tipped
applicator was used to gently remove the endothelium of the intima.
The remaining aortic tissue was digested at 37˚C with a mixture of
0.1% collagenase and 100 U/ml elastase to generate SMCs.
Subsequently, a caliber 24 cannula was inserted into the proximal
end of the ligated aorta and the inside of the aorta was washed
with sterile PBS. The distal end was bound and the aorta was filled
with 0.1% type II collagenase for 45 min at 37˚C. The inside of the
aorta was washed with RPMI-1640 medium, and the medium was
collected and centrifuged at 1000 x g at 4˚C for 15 min to obtain
ECs. All isolated cells were cultured in RPMI-1640 medium
supplemented with 10% FBS (cat. no. 16140071; Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin-streptomycin and placed at 37˚C
in a humidified incubator containing 5% CO2.
Subsequently, 106 CD4+ TCs,
106 Møs, 106 SMCs and 106 ECs were
treated with 100 nM Ang II for 12 h. Control cells were incubated
with saline. Additionally, different doses of Ang II (25, 50 or 100
nM; diluted in saline; cat. no. 05-23-0101; Sigma-Aldrich; Merck
KGaA) or indomethacin (10 µM, cat. no. I7378; Sigma-Aldrich; Merck
KGaA) were added to the 106 SMC culture at 37˚C for 12
h, as previously described (23).
Groups were as follows: i) Saline; ii) 25 nM Ang II; iii) 50 nM Ang
II; iv) 100 nM Ang II; v) 100 nM Ang II + 10 µM indomethacin.
Furthermore, SMCs were grouped and treated as
follows: i) Vehicle (0.1% DMSO); ii) recombinant mouse TSP-2
(rmTSP-2; 30 ng/ml; R&D Systems, Inc.); iii) TSP-2 + JSH-23 (15
µM; NF-κB p65 signaling pathway inhibitor; Sigma-Aldrich; Merck
KGaA); iv) TSP-2 + JSH-23 + mouse anti-TNF-α neutralizing antibody
(TNF-α nAb; 1 µg/ml; R&D Systems, Inc.); v) TSP-2 + JSH-23 +
mouse anti-IL-6 neutralizing antibody (IL-6 nAb; 1 µg/ml; R&D
Systems, Inc.); vi) Ang II (100 nM); vii) Ang II + TSP-2; viii) Ang
II + TSP-2 + JSH-23; ix) Ang II + TSP-2 + JSH-23 + TNF-α nAb; and
x) Ang II + TSP-2 + JSH-23 + IL-6 nAb. After incubation at 37˚C for
12 h, mRNA expression levels in SMCs were measured.
TSP-2, Bax and Bcl2 mRNA expression
detection
Total RNA was extracted from CD4+ TCs,
Møs, SMCs and ECs using TRIzol® reagent (Invitrogen;
Thermo Fisher Scientific, Inc.). Total RNA was reverse transcribed
to cDNA using a high-capacity cDNA reverse transcription kit (cat.
no. 4368813; Thermo Fisher Scientific, Inc.) at 92˚C for 5 min
according to the manufacturer's protocol. Subsequently, qPCR was
performed using LightCycler® 480 SYBR-Green Master Mix
(Roche Diagnostics). The following thermocycling conditions were
used for the PCR: 35 cycles at 92˚C for 30 sec and 58˚C for 40 sec,
and 72˚C for 35 sec. The primer pairs used for qPCR were validated
in previous studies, and were as follows: TSP-2, forward:
5'-TGAGTTCCAGGGCACACCA-3'; reverse: 5'-GGCTTTCTGGGCAATGGTA-3'; Bax,
forward: 5'-TTGCTGATGGCAACTTCAAT-3'; reverse:
5'-GATCAGCTCGGGCACTTTAG-3'; Bcl2, forward:
5'-CAGAAGATCATGCCGTCCTT-3'; reverse: 5'-CTTTCTGCTTTTTATTTCATGAGG-3'
(24,25). mRNA levels were normalized to the
internal reference gene GAPDH levels using the
2-ΔΔCq method (26).
Statistical analysis
Statistical analyses were performed using SPSS
software (version 23.0; IBM Corp.). Categorical variables were
presented as the count (%) and continuous variables were presented
as the median (interquartile range). Categorical data were analyzed
using χ2 test and continuous variables were analyzed
using Mann-Whitney U test. The Student's t-test was used to analyze
differences between two groups. Comparisons among multiple groups
were analyzed using Kruskal-Wallis followed by Dunn's post hoc
test. Spearman's correlation analysis was used to identify
correlations between TSP-2 expression level and TNF-α and IL-6
expression levels in patients with AD. Multivariate regression
analysis was used to assess the association between TSP-2
expression level and AD occurrence. P<0.05 was considered to
indicate a statistically significant difference.
Results
Comparison of clinical characteristics
between the normal group and the AD group
Compared with the normal group, patients in the AD
group displayed significantly higher ages, D-dimer levels, white
blood cell (WBC) counts and C-reactive protein (CRP) levels, but
lower fasting glucose (Glu) levels. No significant differences were
observed for the number of men, high blood pressure (HBP), smoking,
lipid levels or creatinine (CREA) levels. In certain patients in
the normal group, true blood pressure and heart rate (HR)
measurements were not obtained as vital signs were being maintained
with vasoactive drugs following severe traumatic brain injury. The
clinical characteristics of the individuals in the normal and AD
groups are presented in Table
I.
 | Table IClinical characteristics of subjects
in the normal and AD groups. |
Table I
Clinical characteristics of subjects
in the normal and AD groups.
| Characteristic | Normal | AD | P-value |
|---|
| Gender
(male/female) | 6/3 | 12/2 | 0.343 |
| Age (years) | 46 (34-58) | 57 (43-65) | 0.004 |
| HBP (n, %) | 5 (55.6%) | 11 (78.6%) | 0.363 |
| Smoking (n, %) | 2 (22.2%) | 6 (42.9%) | 0.400 |
| Glu (mmol/l) | 5.9 (4.6-6.9) | 5.9 (4.6-6.9) | 0.028 |
| SBP (mmHg) | - | 157 (132-178) | - |
| DBP (mmHg) | - | 94 (78-101) | - |
| TC (mmol/l) | 4.6 (4.2-5.1) | 4.4 (3.9-5.2) | 0.278 |
| TG (mmol/l) | 1.2 (0.9-1.7) | 1.3 (1.0-1.7) | 0.872 |
| HDL-C (mmol/l) | 1.5 (1.0-1.8) | 1.4 (0.9-1.6) | 0.509 |
| LDL-C (mmol/l) | 2.1 (1.5-2.5) | 2.2 (1.7-2.8) | 0.315 |
| HR (bpm) | - | 78 (67-89) | - |
| CREA (µmol/l) | 74 (62-85) | 79 (68-91) | 0.078 |
| D-dimer
(µg/ml) | 1.4 (0.8-2.1) | 4.9 (2.1-8.5) | 0.008 |
| WBC
(x109/l) | 6.1 (4.9-7.6) | 12.2
(8.9-15.7) | <0.001 |
| CRP (mg/l) | 0.5 (0.2-2.2) | 11.9
(2.7-17.9) | <0.001 |
Comparison of clinical characteristics
between the NAD and AD groups
The AD group displayed an increased number of men,
as well as higher ages, smoking incidence, HBP incidence, systolic
blood pressure, WBC counts, CRP levels and D-dimer levels, compared
with the NAD group. Furthermore, the AD group displayed lower Glu
levels compared with the NAD group. No significant differences were
identified for other clinical characteristics, including diastolic
blood pressure, lipid levels, HR, CREA levels and time, which was
defined as the time interval between chest pain onset and
collection of blood samples. In addition, no significant
differences for the clinical characteristics were observed between
the Stanford A and Stanford B groups. The clinical characteristics
of patients in the NAD and AD groups are presented in Table II.
| Table IIClinical characteristics of patients
in the NAD and AD groups. |
Table II
Clinical characteristics of patients
in the NAD and AD groups.
| | | AD |
|---|
| Characteristic | NAD | Total | Stanford A | Stanford B |
|---|
| Sex
(male/female) | 42/18 (70.0) | 132/28a | 74/10a | 58/18
(78.4%)a |
| Age (years) | 44 (38-59) | 56
(46-71)a | 54
(44-69)a | 57
(47-73)a |
| Smoking (n, %) | 14 (23.3) | 62
(38.8)a | 35
(41.7)a | 27 (36.5%) |
| HBP (n, %) | 35 (58.3) | 132
(82.5)a | 71
(84.5%)a | 61
(82.4%)a |
| Glu (mmol/l) | 5.9 (4.9-7.3) | 5.3
(4.1-6.7)a | 5.4
(4.2-6.6)a | 5.2 (4.0,
6.9)a |
| SBP (mmHg) | 149 (132-163) | 159
(146-170)a | 160
(147-173)a | 158
(145-168)a |
| DBP (mmHg) | 84 (75-101) | 88 (82-105) | 87 (84-104) | 88 (80-105) |
| WBC
(x109/l) | 6.4 (5.2-7.7) | 10.9
(8.5-13.1)a | 10.7
(8.2-13.7)a | 11.2
(8.9-12.8)a |
| TC (mmol/l) | 4.4 (3.8-5.4) | 4.5 (4.0-5.5) | 4.4 (4.1-5.4) | 4.5 (4.0-5.4) |
| TG (mmol/l) | 1.0 (0.8-1.3) | 1.1 (0.9-1.4) | 1.1 (0.9-1.5) | 1.0 (0.9-1.3) |
| HDL-C (mmol/l) | 1.1 (0.8-1.4) | 1.2 (0.9-1.5) | 1.1 (0.9-1.4) | 1.2 (0.8-1.5) |
| LDL-C (mmol/l) | 1.6 (1.1-1.9) | 1.5 (1.1-2.0) | 1.6 (1.1-2.1) | 1.5 (1.0-2.0) |
| HR (bpm) | 74 (69-82) | 77 (72-89) | 80 (74-90) | 76 (71-86) |
| CREA (µmol/l) | 85 (72-96) | 90 (68-100) | 89 (71-104) | 91 (65, 98) |
| CRP (mg/l) | 1.0 (0.5-1.7) | 11.5
(5.4-39.5)a | 14.6
(3.9-32.5)a | 10.2
(6.3-42.7)a |
| D-dimer
(µg/ml) | 0.8 (0.2-1.2) | 5.9
(3.9-9.5)a | 5.2
(3.0-10.5)a | 6.4
(4.5-8.5)a |
| Time (h) | 10 (7-14) | 12 (6-16) | 11 (6-16) | 12 (7-15) |
Aortic TSP-2 expression is increased
in patients with AD
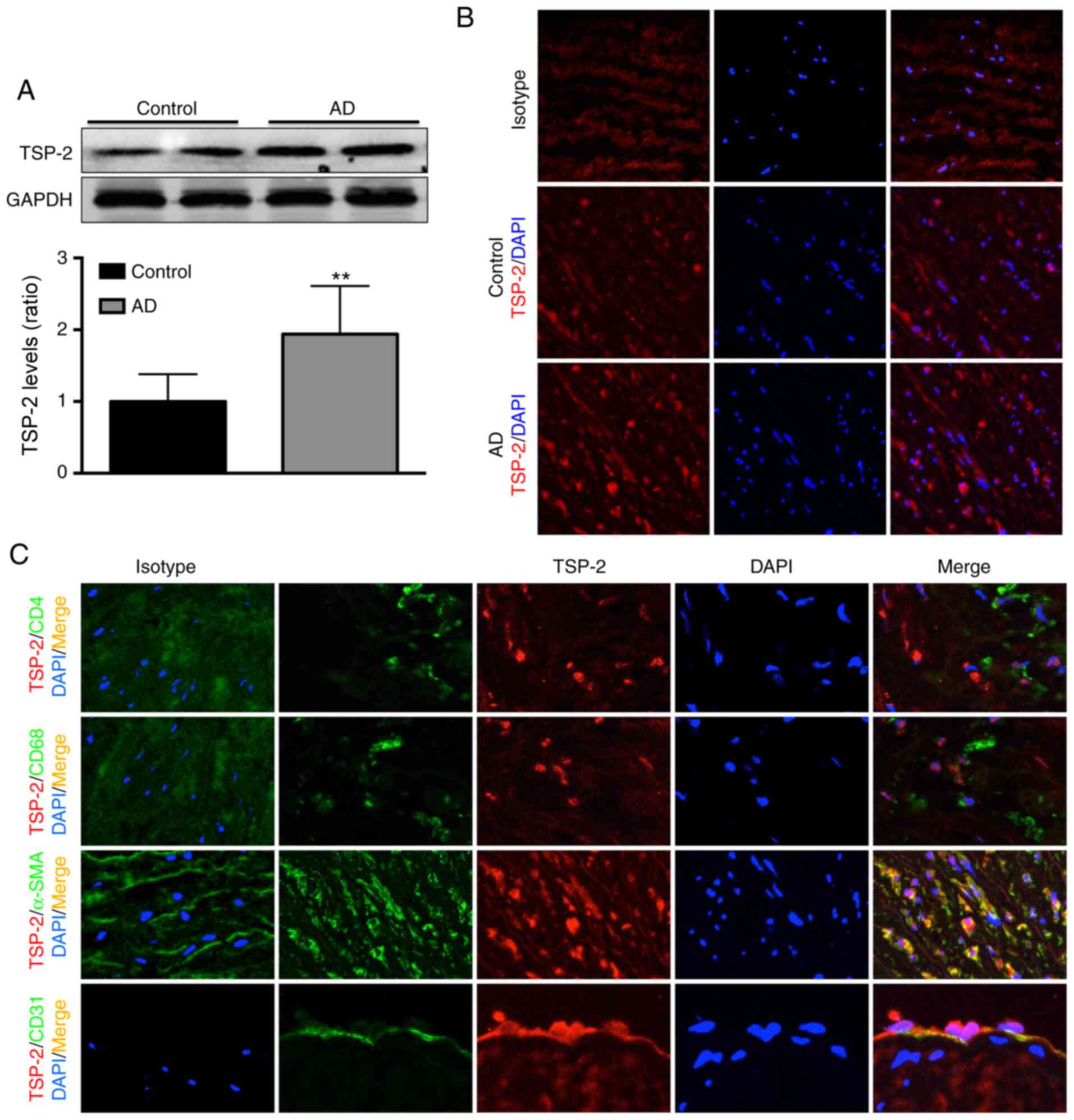
Aortic TSP-2 expression was detected by western
blotting, and the results suggested that TSP-2 expression was
~0.9-fold higher in the AD group compared with the normal group
(Fig. 1A). The results from the
histological analysis displayed a similar trend, and TSP-2
expression in the AD group was increased compared with the control
group (Fig. 1B). In addition, the
results from double immunofluorescence staining indicated that
aortic TSP-2 was localized in SMCs and at lower levels in ECs but
was not expressed by CD4+ TCs or Møs (Fig. 1C).
Circulating TSP-2, TNF-α and IL-6
levels are increased in patients with AD
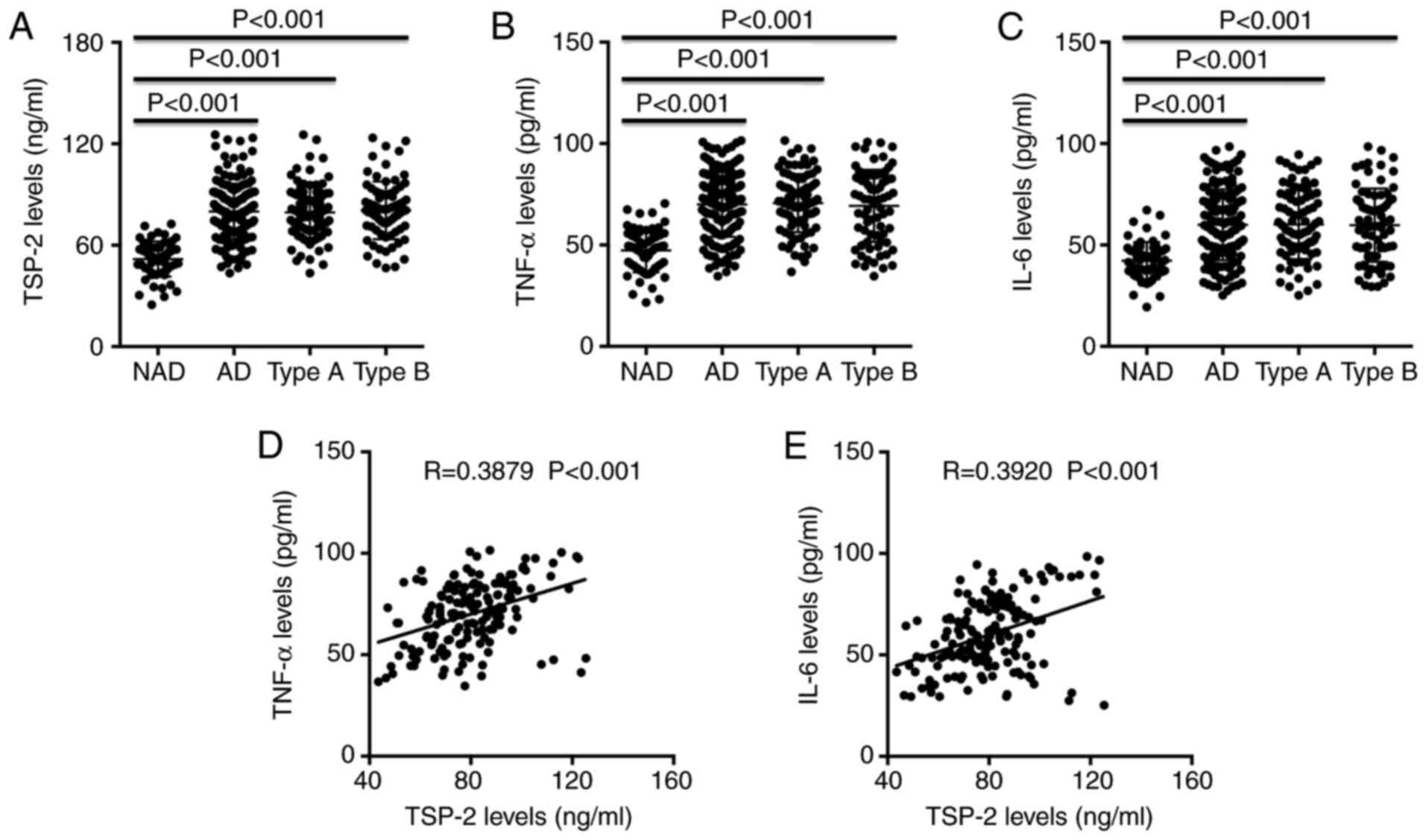
Plasma TSP-2, TNF-α and IL-6 levels of each patient
were measured by ELISA. The results demonstrated that TSP-2, TNF-α
and IL-6 levels were significantly increased in the AD groups
compared with the NAD group; however, no significant difference was
observed between the Stanford A and Stanford B groups (Fig. 2A-C). In addition, Spearman's
correlation analysis indicated that TSP-2 levels were positively
correlated with TNF-α and IL-6 levels in patients with AD (Fig. 2D and E).
TSP-2 is independently associated with
AD occurrence
To investigate whether TSP-2 was associated with AD
occurrence, univariate and multivariate linear regression analyses
were performed. The results suggested that TSP-2 levels (β=0.245;
95% CI, 0.178-0.312; P=0.006), TNF-α levels (β=0.178; 95% CI,
0.139-0.217; P=0.014), IL-6 levels (β=0.142; 95% CI, 0.107-0.177;
P=0.026), HBP incidence (β=0.133; 95% CI, 0.098-0.168; P=0.039),
Glu levels (β=-0.131; 95% CI, -0.358- -0.096; P=0.032) and D-dimer
levels (β=0.121; 95% CI, 0.091-0.151; P=0.047) were independently
associated with AD occurrence. The results of the univariate and
multivariate analyses are presented in Table III.
| Table IIIUnivariate and multivariate linear
regression analysis. |
Table III
Univariate and multivariate linear
regression analysis.
| | Univariate | Multivariate |
|---|
| Variable | β | 95% CI | P-value | β | 95% CI | P-value |
|---|
| TSP-2 (ng/ml) | 0.464 | 0.294-0.634 | <0.001 | 0.245 | 0.178-0.312 | 0.006 |
| TNF-α (pg/ml) | 0.313 | 0.237-0.389 | <0.001 | 0.178 | 0.139-0.217 | 0.014 |
| IL-6 (pg/ml) | 0.287 | 0.216-0.358 | <0.001 | 0.142 | 0.107-0.177 | 0.026 |
| Male (n, %) | 0.192 | 0.089-0.295 | 0.042 | 0.084 | 0.038-0.130 | 0.102 |
| Age (years) | 0.204 | 0.102-0.306 | 0.158 | - | - | - |
| Smoking (n, %) | 0.377 | 0.262-0.492 | 0.098 | - | - | - |
| HBP (n, %) | 0.109 | 0.058-0.160 | 0.026 | 0.133 | 0.098-0.168 | 0.039 |
| Glu (mmol/l) | -0.218 | -0.312- -0.124 | 0.012 | -0.131 | -0.358- -0.096 | 0.032 |
| SBP (mmHg) | 0.098 | 0.069-0.127 | 0.518 | - | - | - |
| WBC
(x109/l) | 0.074 | 0.055-0.093 | 0.414 | - | - | - |
| CRP (mg/l) | 0.215 | 0.124-0.306 | 0.029 | 0.089 | 0.042-0.136 | 0.374 |
| D-dimer
(µg/ml) | 0.128 | 0.094-0.162 | 0.009 | 0.121 | 0.091-0.151 | 0.047 |
TSP-2 treatment increases Ang
II-induced SMC apoptosis
Compared with saline treatment, Ang II treatment did
not significantly alter TSP-2 mRNA expression levels in
CD4+ TCs or Møs. However, Ang II treatment significantly
increased TSP-2 mRNA expression levels by 5.5-fold in SMCs and
0.8-fold in ECs (Fig. 3A). In
addition, Ang II treatment increased TSP-2 mRNA expression levels
in SMCs in a dose-dependent manner, which was significantly
inhibited by indomethacin (Fig.
3B). Furthermore, rmTSP-2 treatment significantly increased Ang
II-induced Bax mRNA expression levels and decreased Bcl2 mRNA
expression levels in SMCs. In addition, the rmTSP-2-induced effects
on SMCs were significantly reversed by JSH-23 treatment alone, and
further reversed by JSH-23 treatment combined with TNF-α nAb or
IL-6 nAb (Fig. 3C).
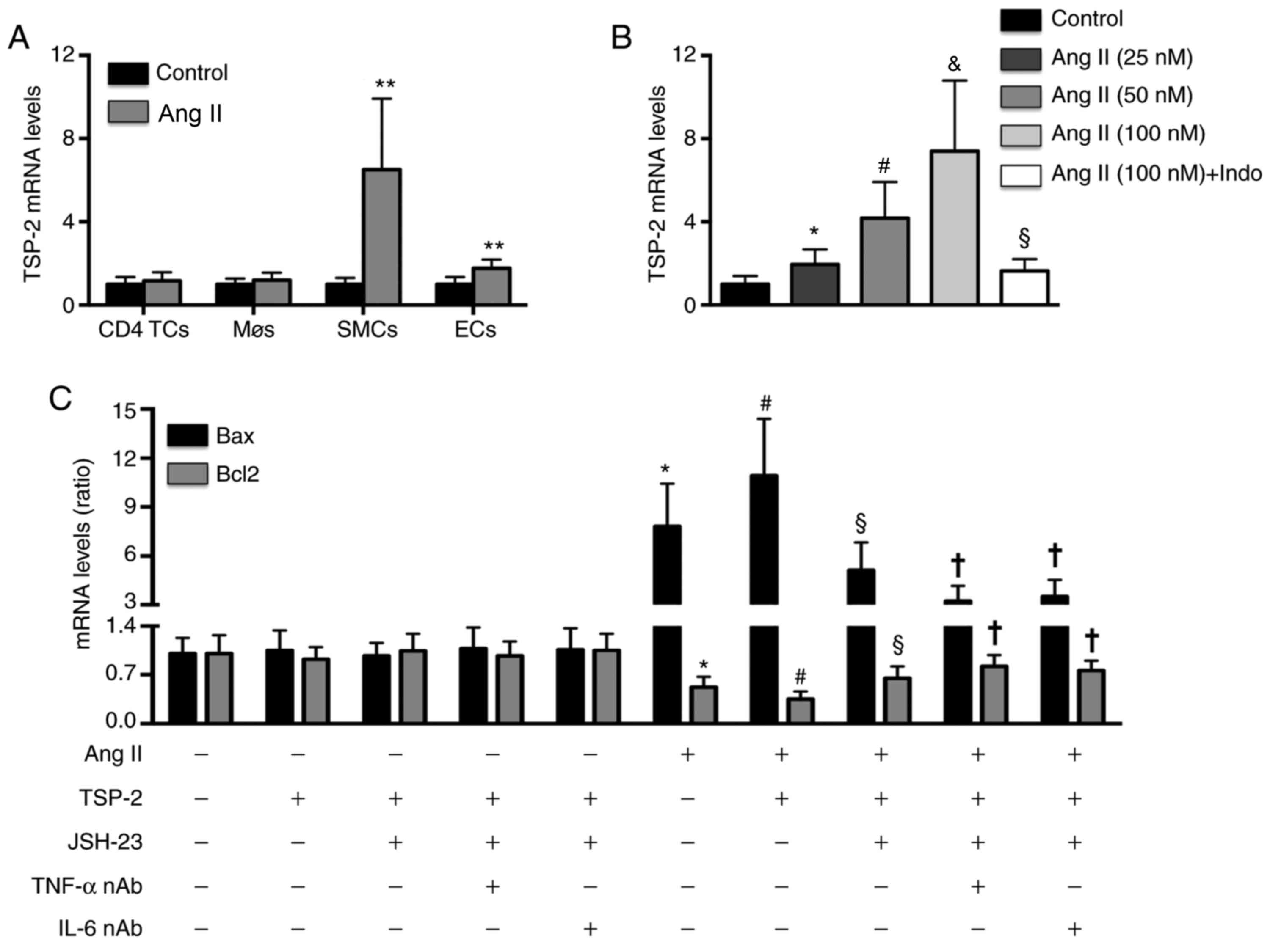 | Figure 3Effects of TSP-2 on Ang II-induced
SMC apoptosis. (A) Effect of Ang II treatment on TSP-2 mRNA
expression in CD4+ TCs, Møs, SMCs and ECs.
**P<0.01 vs. the control group. (B) Effects of
different doses of Ang II and Indo on TSP-2 mRNA expression in
SMCs. *P<0.05 vs. the negative control group.
#P<0.05 vs. the Ang II (25 nM) group.
&P<0.05 vs. the Ang II (50 nM) group.
§P<0.05 vs. the Ang II (100 nM) group. (C) Effects of
TSP-2, JSH-23, anti-TNF-α nAb and anti-IL-6 nAb on Bax and Bcl2
mRNA expression levels in SMCs. n=5 per group.
*P<0.05 vs. the negative control group.
#P<0.05 vs. the Ang II group. §P<0.05
vs. the Ang II + TSP-2 group. †P<0.05 vs. the Ang II
+ TSP-2 + JSH-23 group. TSP-2, thrombospondin-2; Ang II,
angiotensin II; TCs, T lymphocytes; Møs, macrophages; SMCs, smooth
muscle cells; ECs, endothelial cells; Indo, indomethacin; TNF-α,
tumor necrosis factor-α; IL-6, interleukin-6; nAb, neutralizing
antibody; AD, aortic dissection. |
Discussion
In the present study, TSP-2 expression was
significantly higher in aortic tissues derived from patients with
AD compared with healthy subjects. In addition, aortic SMCs and
ECs, especially SMCs, were identified as the main sources of
secreted TSP-2. Furthermore, circulating TSP-2 levels were higher
in patients with AD compared with NAD subjects, and circulating
TSP-2 levels were positively correlated with the circulating levels
of the proinflammatory cytokines TNF-α and IL-6 in patients with
AD. Furthermore, circulating TSP-2 levels were independently
associated with the occurrence of AD. Ang II treatment increased
TSP-2 mRNA levels in SMCs in a dose-dependent manner in
vitro, and Ang II-induced effects were reversed by
indomethacin. By contrast, Ang II treatment displayed no effects on
TSP-2 secretion in CD4+ TCs and Møs. TSP-2 treatment
enhanced Ang II-induced SMC apoptosis, and JSH-23, TNF-α nAb and
IL-6 nAb treatment reversed the effects of TSP-2.
Clinical experiments and animal studies have
demonstrated that TSP-2 expression is increased in a variety of
cardiovascular diseases. For example, numerous studies have
reported that serum TSP-2 levels are increased in patients with
heart failure with preserved or reduced ejection fraction, elderly
patients with aortic aneurysm and patients with aortic stenosis
(12-14,27).
In addition, TSP-2 expression levels are increased by 17-fold in
the heart tissue of patients with viral myocarditis compared with
healthy heart tissue (10).
Coxsackie virus exposure, doxorubicin treatment and aging also
promote cardiac TSP-2 expression (10,15,16).
In the present study, TSP-2 expression was increased in plasma and
aortic samples from patients with AD compared with healthy
subjects, which suggested that TSP-2 was involved in AD
occurrence.
TSP-2 is an extracellular matrix protein that,
unlike inflammatory cytokines secreted by the immune system, is
secreted primarily by innate cells in blood vessels and is also
involved in the regulation of inflammatory responses (5). The immunofluorescence assay in the
present study indicated that TSP-2 was primarily derived from SMCs
and partly from ECs, but was not derived from CD4+ TCs
or Møs, and Ang II-treated cells displayed similar trends in
vitro, which was consistent with previous studies (9-11).
Coxsackie virus and doxorubicin can induce strong cardiac immune
and inflammatory responses. Cardiac TSP-2 expression is closely
associated with the dose of Coxsackie virus, which suggests that
the level of TSP-2 secretion is associated with the strength of the
inflammatory response (10,15). Ang II can mediate a strong
inflammatory response and can be used to induce AD in mouse
(28). In the present study, the
effect of Ang II treatment on TSP-2 expression in SMCs was
investigated, and the results suggested that Ang II increased TSP-2
mRNA expression in a dose-dependent manner. The addition of
indomethacin, a non-specific and anti-inflammatory substance,
reversed Ang II-induced effects on TSP-2 expression. The results
were consistent with the aforementioned previous studies and
suggested that in the AD microenvironment, the inflammatory
response may serve as an important factor mediating TSP-2
secretion.
Swinnen et al (16) reported that downregulation of TSP-2
expression by adeno-associated virus-9 aggravates the inflammatory
response in elderly mice. A subsequent study reported that TSP-2
downregulation regulates CD4+ TC differentiation and
increases proinflammatory factor expression, while decreasing
anti-inflammatory cytokine levels (15). Another study demonstrated that TSP-2
knockdown promotes inflammatory factor secretion in mice treated
with doxorubicin (10). The results
of the aforementioned studies indicated that TSP-2 could
participate in cardiovascular diseases by regulating the
inflammatory response. AD is a chronic inflammatory disease, which
is characterized by increases in proinflammatory factor expression
and decreases in anti-inflammatory factor expression in the aortic
wall and plasma (29,30). TNF-α and IL-6 are two inflammatory
cytokines that have been reported to promote the progression of AD
(30). To explore whether TSP-2
participated in AD by regulating the inflammatory response,
circulating TNF-α and IL-6 levels were measured in the present
study, and the correlation between TNF-α, IL-6 and TSP-2 expression
levels in patients with AD were analyzed. The results indicated
that TNF-α and IL-6 levels were positively correlated with TSP-2
levels, which supported the proposed hypothesis that TSP-2 may
regulate the inflammatory response during AD; however, the role of
TSP-2 during AD remains unclear.
Vascular SMCs are important components of the aorta,
accounting for >90% of the total number of inherent cells.
Extracellular matrix material secretion is critical for the
maintenance of the normal structure and function of the aorta and
for the dynamic balance of the matrix (31). It has been reported that patients
with AD display excessive SMC apoptosis in aortic tissues, which
releases myosin heavy chains into the blood to significantly
increase the level of circulating myosin heavy chains (21,32).
The excessive loss of SMCs leads to the destruction of the dynamic
balance of the extracellular matrix, resulting in structural and
functional destruction of the aorta, which leads to increased
susceptibility to AD (33,34). Therefore, excessive loss of SMCs is
a fundamental factor of AD occurrence. In a recent study, Ye et
al (29) reported that
treatment of SMCs with plasma from patients with AD or patients
with anti-inflammatory and proinflammatory factor imbalance
significantly increased Ang II-induced SMC apoptosis, suggesting
that the inflammatory response is a key mechanism regulating
excessive SMC apoptosis. To further explore the possible mechanisms
underlying the involvement of TSP-2 during AD, Ang II-treated cells
were also treated with rmTSP-2. The NF-κB p65 signaling pathway is
closely related to inflammatory regulation, and previous studies
have confirmed that TSP-2 regulates downstream inflammatory signals
by activating the NF-κB p65 signaling pathway (28,35);
therefore, JSH-23 was used to inhibit the NF-κB p65 signaling
pathway in SMCs in the present study. The results indicated that
TSP-2 treatment significantly increased Ang II-induced SMC
apoptosis. Furthermore, the proapoptotic effect of TSP-2 was
blocked by JSH-23 and further prevented by TNF-α or IL-6
neutralization. The results suggested that TSP-2 may promote the
expression of inflammatory factors and amplify their inflammatory
effects by activating the NF-κB p65 signaling pathway, thereby
enhancing SMC apoptosis and positively regulating AD development.
However, the aforementioned hypotheses require further
investigation in vivo.
In conclusion, the present study suggested that
TSP-2 may be closely related to the occurrence of AD, and TSP-2
downregulation may serve as a novel strategy for the prevention of
AD.
Acknowledgements
Not applicable
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81770472).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QB conceived and designed the study; LQ and KW
collected the samples; LQ, KW, SS and HM performed the experiments;
QJ analyzed the data; LQ and KW were involved in drafting the
manuscript and revising it critically for important intellectual
content; QB reviewed and edited the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Both animal and human experiments in the present
study were approved by the Ethics Committee of Beijing Anzhen
Hospital (approval no. 2017109H). Written informed consent was
obtained from all patients or their families.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hagan PG, Nienaber CA, Isselbacher EM,
Bruckman D, Karavite DJ, Russman PL, Evangelista A, Fattori R,
Suzuki T, Oh JK, et al: The international registry of acute aortic
dissection (IRAD): New insights into an old disease. JAMA.
283:897–903. 2000.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Chen LW, Wu XJ, Lu L, Zhang GC, Yang GF,
Yang ZW, Dong Y, Cao H and Chen Q: Total arch repair for acute type
A aortic dissection with 2 modified techniques: Open
single-branched stent graft placement and reinforcement of the
dissected arch vessel stump with stent graft. Circulation.
123:2536–2541. 2011.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Fattori R, Cao P, De Rango P, Czerny M,
Evangelista A, Nienaber C, Rousseau H and Schepens M:
Interdisciplinary expert consensus document on management of type B
aortic dissection. J Am Coll Cardiol. 61:1661–1678. 2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Hugo C and Daniel C: Thrombospondin in
renal disease. Nephron Exp Nephrol. 111:e61–e66. 2009.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Calabro NE, Kristofik NJ and Kyriakides
TR: Thrombospondin-2 and extracellular matrix assembly. Biochim
Biophys Acta. 1840:2396–2402. 2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Rusnati M, Borsotti P, Moroni E, Foglieni
C, Chiodelli P, Carminati L, Pinessi D, Annis DS, Paiardi G,
Bugatti A, et al: The calcium-binding type III repeats domain
ofthrombospondin-2 binds to fibroblast growth factor 2 (FGF2).
Angiogenesis. 22:133–144. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Morris AH, Stamer DK, Kunkemoeller B,
Chang J, Xing H and Kyriakides TR: Decellularized materials derived
from TSP2-KO mice promote enhanced neovascularization and
integration in diabetic wounds. Biomaterials. 169:61–71.
2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Helkin A, Maier KG and Gahtan V:
Thrombospondin-1, -2 and -5 have differential effects on vascular
smooth muscle cell physiology. Biochem Biophys Res Commun.
464:1022–1027. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Daniel C, Vogelbacher R, Stief A, Grigo C
and Hugo C: Long-term gene therapy with thrombospondin 2 inhibits
TGF-β activation, inflammation and angiogenesis in chronic
allograft nephropathy. PLoS One. 8(e83846)2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Papageorgiou AP, Swinnen M, Vanhoutte D,
VandenDriessche T, Chuah M, Lindner D, Verhesen W, de Vries B,
D'hooge J, Lutgens E, et al: Thrombospondin-2 prevents cardiac
injury and dysfunction in viral myocarditis through the activation
of regulatory T-cells. Cardiovasc Res. 94:115–124. 2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Li K, Huang EP, Su J, Zhu P, Lin J, Luo SQ
and Yang CL: Therapeutic role for TSP-2 antibody in a murine asthma
model. Int Arch Allergy Immunol. 175:160–170. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kimura Y, Izumiya Y, Hanatani S, Yamamoto
E, Kusaka H, Tokitsu T, Takashio S, Sakamoto K, Tsujita K, Tanaka
T, et al: High serum levels of thrombospondin-2 correlate with poor
prognosis of patients with heart failure with preserved ejection
fraction. Heart Vessels. 31:52–59. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hanatani S, Izumiya Y, Takashio S, Kimura
Y, Araki S, Rokutanda T, Tsujita K, Yamamoto E, Tanaka T, Yamamuro
M, et al: Circulating thrombospondin-2 reflects disease severity
and predicts outcome of heart failure with reduced ejection
fraction. Circ J. 78:903–910. 2014.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Golledge J, Clancy P, Hankey GJ and Norman
PE: Relation between serum thrombospondin-2 and cardiovascular
mortality in older men screened for abdominal aortic aneurysm
aneurysm. Am J Cardiol. 111:1800–1804. 2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
van Almen GC, Swinnen M, Carai P, Verhesen
W, Cleutjens JP, D'hooge J, Verheyen FK, Pinto YM, Schroen B,
Carmeliet P and Heymans S: Absence of thrombospondin-2 increases
cardiomyocyte damage and matrix disruption in doxorubicin-induced
cardiomyopathy. J Mol Cell Cardiol. 51:318–328. 2011.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Swinnen M, Vanhoutte D, Van Almen GC,
Hamdani N, Schellings MW, D'hooge J, Van der Velden J, Weaver MS,
Sage EH, Bornstein P, et al: Absence of thrombospondin-2 causes
age-related dilated cardiomyopathy. Circulation. 120:1585–1597.
2009.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L,
Wang F, Takahashi T, Loperena R, Foss JD, Mernaugh RL, et al: Renal
denervation prevents immune cell activation and renal inflammation
in angiotensin II-induced hypertension. Circ Res. 117:547–557.
2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Li Y, Zhang C, Wu Y, Han Y, Cui W, Jia L,
Cai L, Cheng J, Li H and Du J: Interleukin-12p35 deletion promotes
CD4 T-cell-dependent macrophage differentiation and enhances
angiotensin II-Induced cardiac fibrosis. Arterioscler Thromb Vasc
Biol. 32:1662–1674. 2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Lake FR, Noble PW, Henson PM and Riches
DW: Functional switching of macrophage responses to tumor necrosis
factor-alpha (TNF alpha) by interferons. Implications for the
pleiotropic activities of TNF alpha. J Clin Invest. 93:1661–1669.
1994.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Golovina VA and Blaustein MP: Preparation
of primary cultured mesenteric artery smooth muscle cells for
fluorescent imaging and physiological studies. Nat Protoc.
1:2681–2687. 2006.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Jia LX, Zhang WM, Zhang HJ, Li TT, Wang
YL, Qin YW, Gu H and Du J: Mechanical stretch-induced endoplasmic
reticulum stress, apoptosis and inflammation contribute to thoracic
aortic aneurysm and dissection. J Pathol. 236:373–383.
2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kobayashi M, Inoue K, Warabi E, Minami T
and Kodama T: A simple method of isolating mouse aortic endothelial
cells. J Atheroscler Thromb. 12:138–142. 2005.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ye J, Que B, Huang Y, Lin Y, Chen J, Liu
L, Shi Y, Wang Y, Wang M, Zeng T, et al: Interleukin-12p35 knockout
promotes macrophage differentiation, aggravates vascular
dysfunction, and elevates blood pressure in angiotensin II-infused
mice. Cardiovasc Res. 115:1102–1113. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Bae ON, Wang JM, Baek SH, Wang Q, Yuan H
and Chen AF: Oxidative stress-mediated thrombospondin-2
upregulation impairs bone marrow-derived angiogenic cell function
in diabetes mellitus. Arterioscler Thromb Vasc Biol. 33:1920–1927.
2013.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Xiao T, Zhang L, Huang Y, Shi Y, Wang J,
Ji Q, Ye J, Lin Y and Liu H: Sestrin2 increases in aortas and
plasma from aortic dissection patients and alleviates angiotensin
II-induced smooth muscle cell apoptosis via the Nrf2 pathway. Life
Sci. 218:132–138. 2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Pohjolainen V, Mustonen E, Taskinen P,
Näpänkangas J, Leskinen H, Ohukainen P, Peltonen T, Aro J, Juvonen
T, Satta J, et al: Increased thrombospondin-2 in human
fibrosclerotic and stenotic aortic valves. Atherosclerosis.
220:66–71. 2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Ortega R, Collado A, Selles F,
Gonzalez-Navarro H, Sanz MJ, Real JT and Piqueras J: SGLT-2
(Sodium-Glucose Cotransporter 2) inhibition reduces Ang II
(Angiotensin II)-induced dissecting abdominal aortic aneurysm in
ApoE (Apolipoprotein E) knockout mice. Arterioscler Thromb Vasc
Biol. 39:1614–1628. 2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ye J, Wang Y, Wang Z, Ji Q, Huang Y, Zeng
T, Hu H, Ye D, Wan J and Lin Y: Circulating Th1, Th2, Th9, Th17,
Th22, and treg levels in aortic dissection patients. Mediators
Inflamm. 2018(5697149)2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zeng T, Shi L, Ji Q, Shi Y, Huang Y, Liu
Y, Gan J, Yuan J, Lu Z, Xue Y, et al: Cytokines in aortic
dissection. Clin Chim Acta. 486:177–182. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
El-Hamamsy I and Yacoub MH: Cellular and
molecular mechanisms of thoracic aortic aneurysms. Nat Rev Cardiol.
6:771–786. 2009.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Suzuki T, Katoh H, Watanabe M, Kurabayashi
M, Hiramori K, Hori S, Nobuyoshi M, Tanaka H, Kodama K, Sato H, et
al: Novel biochemical diagnostic method for aortic dissection.
Results of a prospective study using an immunoassay of smooth
muscle myosin heavy chain. Circulation. 93:1244–1249.
1996.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Michel JB, Jondeau G and Milewicz DM: From
genetics to response to injury: Vascular smooth muscle cells in
aneurysms and dissections of the ascending aorta. Cardiovasc Res.
114:578–589. 2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Ye P, Chen W, Wu J, Huang X, Li J, Wang S,
Liu Z, Wang G, Yang X, Zhang P, et al: GM-CSF contributes to aortic
aneurysms resulting from SMAD3 deficiency. J Clin Invest.
123:2317–2331. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
35
|
De Stefano D, Nicolaus G, Maiuri MC,
Cipolletta D, Galluzzi L, Cinelli MP, Tajana G, Iuvone T and
Carnuccio R: NF-kappaB blockade upregulates Bax, TSP-1, and TSP-2
expression in rat granulation tissue. J Mol Med (Berl). 87:481–492.
2009.PubMed/NCBI View Article : Google Scholar
|