Introduction
Neural stem cells (NSCs) are multipotent cells
capable of self-renewal and differentiation into neurons,
astrocytes and oligodendrocytes (1-8).
They have exhibited potential for the study of neural development,
disease modeling, and drug and toxin screening, and they have
therapeutic potential for neural disorders, including spinal cord
injury and a number of other neurodegenerative diseases (2-5,7-9).
As a complement to in vivo studies, the successful in
vitro primary culture of NSCs is important, as it provides a
powerful tool for determining the properties of NSCs under
controlled environmental conditions that may be modified and
monitored accurately (10).
However, the complicated procedures required for NSC isolation and
culture, and inconsistent operational details, not only restrict
the yield of viable NSCs but also impede the intra- and
inter-laboratory comparison and reproducibility of experimental
results (3,11,12).
Therefore, it is essential to provide a detailed and refined
protocol that may be readily reproduced for the study of embryonic
NSCs. The present study described a modified, detailed and feasible
protocol for the isolation, culture and cryopreservation of rat
embryonic NSCs. Compared with other previous protocols, the
colorless high-glucose medium and a sequential digestion strategy
were the primary modifications. In addition, the viability, nestin
expression, and capability for self-renewal and
multi-differentiation of NSCs cryopreserved for different time
periods (7 days, or 1, 6 or 12 months) were determined.
Materials and methods
Animals
All animal procedures were approved by the Ethics
Committee of Tianjin Medical University and complied with the
United States of America National Institutes of Health Guide for
the Care and Use of Experimental Animals and the Society for
Neuroscience Use of Animals in Neuroscience Research guidelines
(13,14). A specific-pathogen-free Sprague
Dawley female rat (age, 3-5 weeks; weight, 250±30 g; n=1), pregnant
at the embryonic age of 15.5 days (E15.5), was obtained from the
Radiation Study Institute Animal Center at Tianjin Medical
University (Tianjin, China), and was housed in a controlled
environment (23±1˚C) under a 12-h light/dark cycle with free access
to food and water. NSCs may be isolated as early as E10.5 in rats.
In the present study, E15.5 rats were selected as the sources for
NSC culture as NSCs were most abundant at E15.5, which is
approximately the onset of neurogenesis (15).
Uterus anatomy and isolation
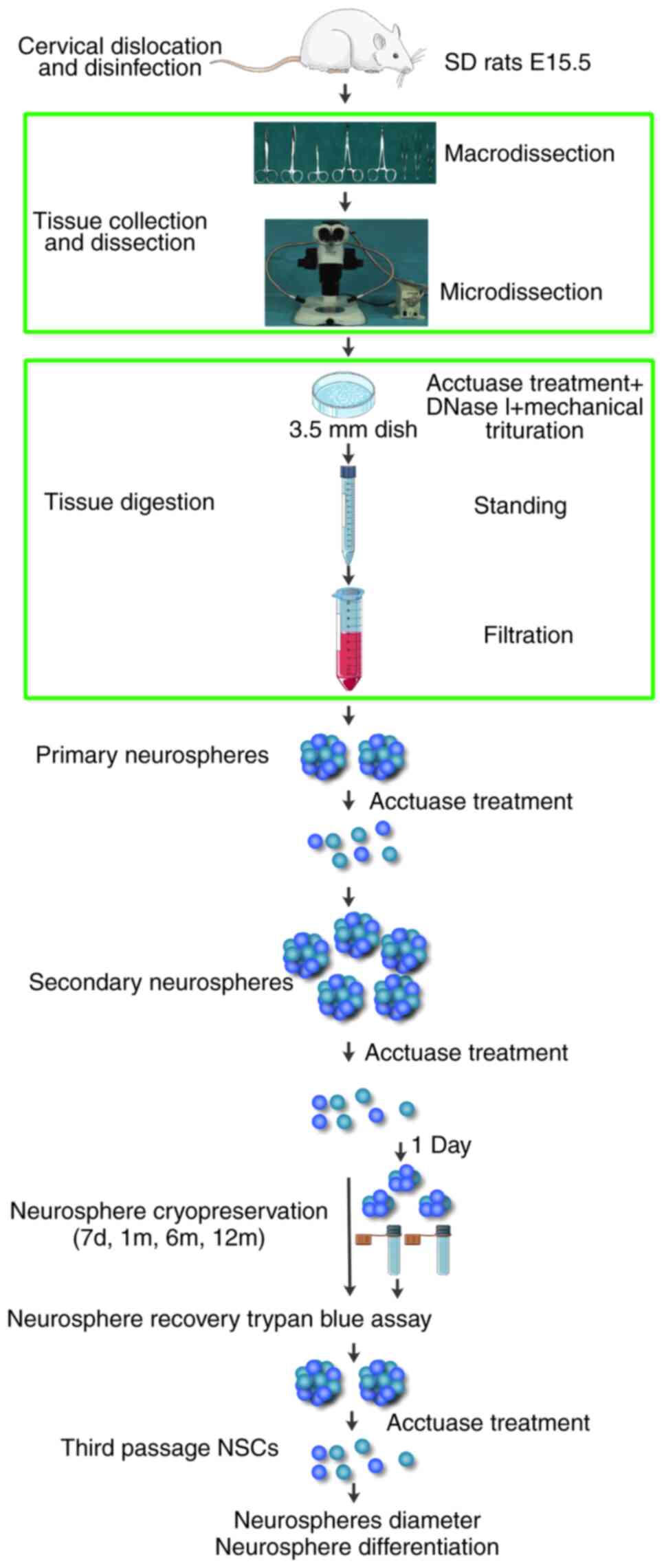
The major steps of the protocol modified in the
present study are summarized in the flow diagram presented in
Fig. 1. A set of microdissection
instruments were autoclaved prior to use for uterus removal, with
two sets of microdissection instruments for brain hemisphere
dissection. The pregnant rats were sacrificed by cervical
dislocation, immersed promptly into -20˚C 70% ethanol for 5 min for
disinfection, and positioned in a sterilized dissecting tray with
the abdomen facing upward. The abdominal fur of each rat was shaved
rapidly, and the skin was re-sterilized using povidone-iodine swabs
(Shandong Lierkang Medical Technology Co., Ltd.) to increase the
aseptic conditions; any loose fur was removed from the skin. A
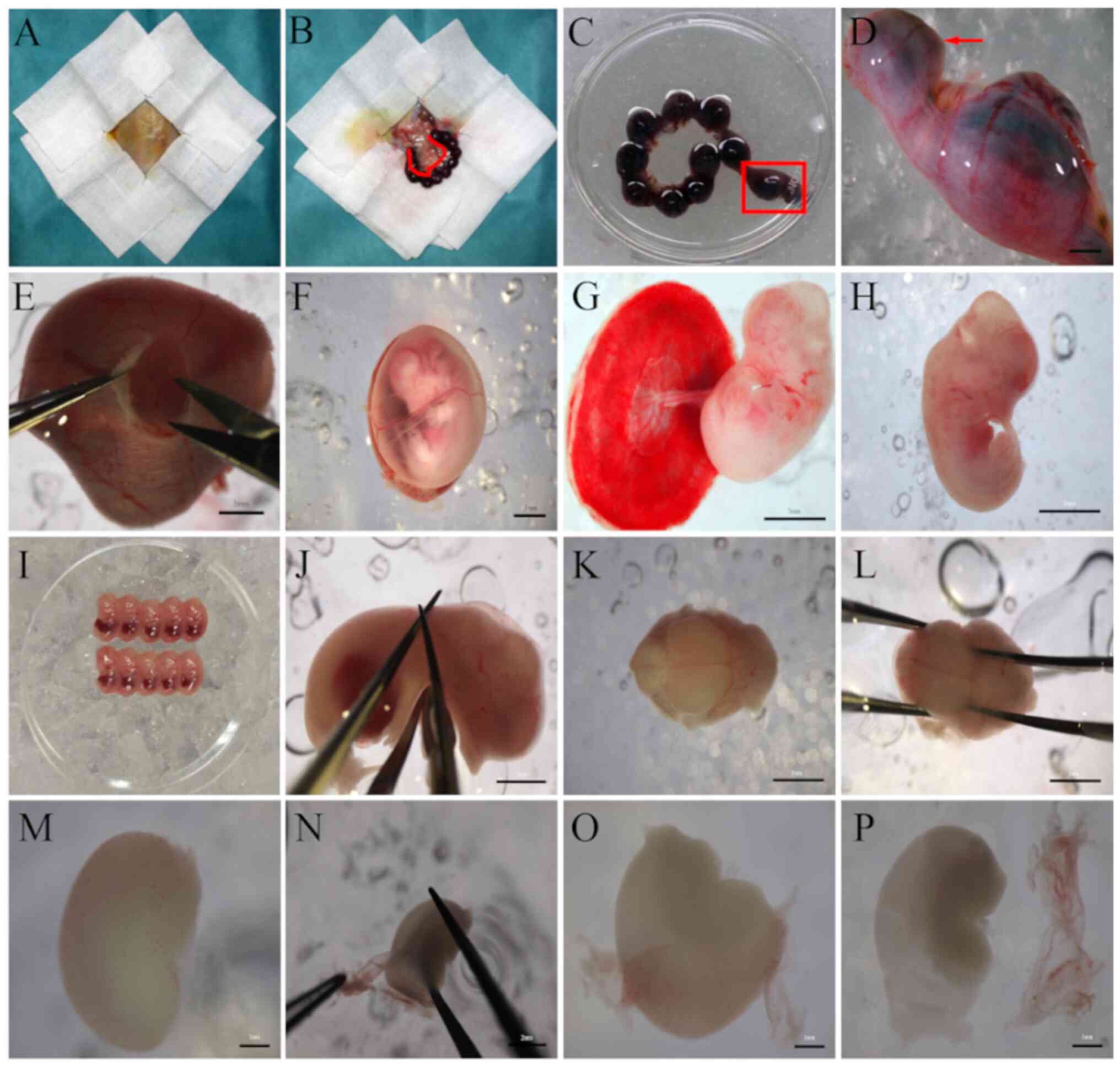
sterile drape was used to cover the rat (Fig. 2A), and the abdominal skin above the
genitalia was grasped using large straight forceps (Shanghai
Medical Instruments Co., Ltd.). Then, an incision from the lower
abdomen upward to the chest was made with large straight surgical
scissors (Shanghai Medical Instruments Co., Ltd.). The peritoneal
cavity was exposed sufficiently with hemostatic forceps (Shanghai
Medical Instruments Co., Ltd.) to view the uterus. The intact
uterus was held with a different pair of large straight forceps at
the junction with the vagina (Fig.
2B), pulled up slightly, and isolated using a pair of small
scissors (Shanghai Medical Instruments Co., Ltd.). The mesometrium
and blood vessels were removed from the uterus with care to avoid
bacterial contamination caused by damaging the intestines. Then,
the uterus was transferred quickly to a 100-mm dish (BD
Biosciences) and rinsed 3 times with a 4˚C PBS solution
(Sigma-Aldrich; Merck KGaA) supplemented with 1%
penicillin/streptomycin (P/S; Invitrogen; Thermo Fisher Scientific,
Inc.; cat no., 15140148; Fig. 2C
and D). The carcass was immediately
disposed following uterus removal. While grasping one horn of the
uterus in one hand using microforceps (Shanghai Medical Instruments
Co., Ltd.), the uterus was opened by careful incision of the top of
the uterus using microscissors (Shanghai Medical Instruments Co.,
Ltd.) placed in the other hand, under a SZX16 light
stereomicroscope (Olympus Corporation; Fig. 2E), and then the amniotic membrane
was exposed (Fig. 2F). Then, the
amniotic membrane was removed and the placenta and embryo was
exposed (Fig. 2G). Then the
placenta was cut and the embryos (Fig.
2H) were excised and rinsed 3 times with high-glucose
Dulbecco's Modified Eagle's Medium without phenol red (DMEM-HG;
Gibco; Thermo Fisher Scientific, Inc.; cat no., 31053028) with 1%
P/S. At this point, the embryos were assessed, and the color and
number of live births were recorded. Embryos that appear malformed,
too small or too large with respect to the expected gestational age
were discarded. Then, live embryos (5-7 embryos each; Fig. 2I) were transferred into several 50
ml centrifuge tubes (BD Biosciences) containing 4˚C DMEM-HG with 1%
P/S. These embryos were subjected to additional brain dissection on
ice, 1 centrifuge tube at a time, while the remaining embryos were
kept in the refrigerator.
Ethanol (-20˚C, 70%) was used to not only sterilize
the pregnant rats but also rapidly cool embryos and decrease the
robust consumption of oxygen and energy by tissues (7,10).
(2) Fur is a source of
contamination, and it may adhere to the peritoneal cavity, the
scissors or brain tissues (3);
therefore, some measures were taken to avoid this. For example, the
abdominal fur was thoroughly shaved, the abdominal skin was
carefully re-sterilized to remove any loose fur, and the rat was
covered by a sterile drape. These precautions decreased the chance
of fur contaminating the primary culture. However, if some fur
adhered to the scissors accidently, the surgical instruments were
rinsed using 70% ethanol and either allowed to dry or rinsed with
PBS before resuming the experiment, as ethanol would fix the
tissues (10,16). (3)
DMEM-HG without phenol red was selected as it provides a clear
operative view for tissue dissection (17).
Brain dissection
Straight microforceps (held in the non-dominant
hand) were used to grasp the body of each embryo, while a pair of
microscissors (held in the dominant hand) was used to cut the head
of each embryo at the level of the cervical spinal cord (Fig. 2J). All heads were collected and
transferred promptly into a 100-mm dish containing 4˚C DMEM-HG with
1% P/S on ice. Then, they were transferred and dissected
individually in a second dish containing 4˚C DMEM-HG with 1% P/S on
ice under a stereomicroscope (Fig.
2K). A pair of straight microforceps (held in the non-dominant
hand) was used to steadily grasp the head of each embryo by placing
the tip of the microforceps vertically along the caudal side of
each ear, while the other pair of forceps (held in the dominant
hand) was inserted into the nose (microforceps tip up) to peel off
the skin with an expansion force through simultaneous bilateral
tissue removal (Fig. 2L). The skull
and dura mater were dissected by inserting the straight
microforceps at the intersection of the lambda suture and then
removing layer by layer to expose the cerebral hemispheres. Then,
the entire cerebral hemispheres were excised and transferred gently
using curved microforceps, tip up, (Shanghai Medical Instruments
Co., Ltd.) to a 100-mm dish containing 4˚C DMEM-HG with 1% P/S on
ice. All heads were dissected until all cerebral hemispheres were
harvested.
Removal of the pia mater and blood
vessels
An additional set of microdissection instruments was
used to remove the pia mater and blood vessels of the cerebral
hemispheres. These samples were dissected and individually
transferred to another dish containing 4˚C DMEM-HG with 1% P/S on
ice under the stereomicroscope (Fig.
2M). A pair of straight microforceps (held in the non-dominant
hand) was used to steadily grasp the cerebral hemisphere, while the
other pair (held in the dominant hand) was used to carefully peel
off the pia mater and blood vessels (Fig. 2N-P). The dissected cerebral
hemisphere tissues were transferred to a separate dish containing
4˚C DMEM-HG with 1% P/S on ice until all cerebral hemispheres were
dissected. To facilitate the operation, it was necessary to
periodically replace the dish filled with the stripped pia mater
and blood vessels.
Sequential digestion of the dissected
tissues from each hemisphere and preparation of cell
suspensions
The dissected cerebral hemispheres were minced on
ice using a pair of microscissors into small, even pieces (~1
mm3) under the stereomicroscope for ~1 min. The minced
tissues were then carefully transferred into a 15-ml centrifuge
tube (BD Biosciences) using a pre-wetted, fire-polished glass
Pasteur pipette (Yangcheng Shuangfeng Glassware Co., Ltd), and then
centrifuged for 5 min at 200 x g (Heraeus Multifuge X1; Thermo
Scientific, Inc.) at 37˚C. The supernatant was then carefully
removed, and 3-5 ml pre-warmed accutase solution (Gibco: Thermo
Fisher Scientific, Inc.; cat no., 00-4555-56) with 20 units/ml
deoxyribonuclease I (DNase I; cat. no., LS002060; Worthington
Biochemical Corporation), was added to the precipitated tissues.
This was adjusted according to the number of pups used. Following
gentle trituration, the tissue was transferred to a 35 mm sterile
dish (BD Biosciences) using a pre-wetted, fire-polished glass
Pasteur pipette and incubated for 20 min at 37˚C. During the
incubation, the tissue was gently triturated with a glass Pasteur
pipette 7-8 times every 5 min. All suspended and undissociated
tissues were collected into a 15 ml centrifuge tube and held for 2
min; then, three-fourths of the supernatant was carefully aspirated
and passed through a 70 µm cell strainer (BD Biosciences) into a
50-ml centrifuge tube containing 30 ml DMEM-HG with 1% P/S to
remove any cell clumps and tissue pieces. The tube was kept at room
temperature (RT) until completion of the sequential digestion.
Following the initial digestion, the precipitated tissue in the 15
ml centrifuge tube was resuspended in 3 ml accutase solution with
20 units/ml DNase I for the second digestion. As with the first
digestion, the enzymatic mix was pipetted 7-8 times every 5 min
with a glass Pasteur pipette, and then aspirated and filtered
through a 70-µm cell strainer placed on the 50-ml centrifuge tube
containing the single-cell suspension from the first digestion.
Following centrifugation for 5 min at 200 x g at 37˚C, the
supernatant was discarded, and the cell pellet was resuspended in
20 ml fresh serum-free medium [SFM; DMEM/Ham's F-12 (DMEM/F-12);
Invitrogen: Thermo Fisher Scientific, Inc.; cat no., 11330032)
containing 20 ng/ml basic fibroblast growth factor (bFGF; cat. no.,
450-33; PeproTech, Inc.), 20 ng/ml epidermal growth factor (EGF;
cat. no., 400-25; PeproTech, Inc.), 2.5 µg/ml heparin (cat. no.,
2812; Tocris Bioscience), 2% B-27 supplement (cat. no. 17504044), 1
mM L-glutamine (cat. no., 25030149) and 1% P/S (all from
Invitrogen; Thermo Fisher Scientific, Inc.). Suspension aliquots
(10 µl) were mixed with 10 µl Trypan blue (Sigma-Aldrich; Merck
KGaA) at room temperature for 3 min to calculate cell viability
using a hemocytometer and phase-bright IX71 microscope with a x10
objective (Olympus Corporation). The dissociated cells were seeded
in uncoated 15 ml T75 culture flasks (Corning Life Sciences) at a
density of 2x105 cells/ml and incubated at 37˚C with 5%
CO2.
Compared with a glass Pasteur pipette, 1,000 µl
plastic pipette tips are considered superior for the discharge of
cells, leading to lower cell viability; in addition, cells adhere
to plastic tips more easily, leading to increased cell loss
(2,7,18,19).
Therefore, a glass Pasteur pipette was used, and it was pre-wetted
with a small amount of medium to prevent tissue from sticking to
the glass surface. The total digestion time should not be >40
min; excessive and prolonged digestion may lead to fewer viable
cells (7,16,20).
Fresh SFM could be kept at 4˚C for a maximum of 1 week to avoid
significant decreases in the biological effect of critical growth
factors, including bFGF, EGF and B-27, which were added to the
medium over time (16,20-22).
Therefore, bFGF, EGF and B-27 were added to the medium on the day
of the experiments to obtain optimal results. In addition, the SFM
was pre-warmed prior to use, to avoid the cell damage and low cell
viability generated by sudden temperature change (16). (4)
To prevent neurospheres from adhering to the bottom of the culture
flasks, ultra-low attachment flasks (Corning Inc.) were used, and
tapped them several times every week, with the exception of the
first 3 days after initial plating (19,23).
Medium replacement and cell
passaging
After 3 days of culture, medium containing
neurospheres was transferred to a 50 ml centrifuge tube using a
glass Pasteur pipette. Following centrifugation for 5 min at 200 x
g at 37˚C, the supernatant was transferred into a new 50 ml
centrifuge tube. A total of one-half of the supernatant was
replaced with fresh SFM, and the resulting mixture was used to
resuspend the pelleted cells, which were then seeded into new
culture flasks and incubated at 37˚C with 5% CO2.
Passaging was performed after 7-8 days in culture.
Neurospheres were handled similarly as aforementioned, with the
exception of the resuspension of cell pellets in pre-warmed
accutase solution. The enzyme-cell suspension was incubated at 37˚C
for 10 min, and then triturated 5 times using a 2 ml disposable
needle (23 Gauge, 0.6x32 mm; BD Biosciences). These enzymatically
and mechanically dissociated cells were then transferred to a 50 ml
centrifuge tube containing 30 ml pre-warmed DMEM-HG. Following
centrifugation for 5 min at 200 x g at 37˚C, the cell pellets were
resuspended in fresh SFM. An aliquot of cell suspension (10 µl) was
mixed with 10 µl Trypan blue at room temperature for 3 min to
calculate cell viability using a hemocytometer under bright-phase
microscopy with a x10 objective. Cells were plated into culture
flasks at a density of 2x105 cells/ml and incubated at
37˚C with 5% CO2. A total of one-half of the medium was
replaced with fresh SFM 3 days after passaging, and the cells were
sub-cultured after a total of 7-8 days in culture. The majority of
the second-passage neurospheres were cryopreserved, as described
subsequently, and the third-passage neurospheres (non-cryopreserved
or cryopreserved) were used in the subsequent experiments.
During the first 3 days after initial plating, cells
adjust to their new environment, start communicating with
neighboring cells and begin to form neurospheres. Therefore, the
culture flasks could not be moved during the first 3 days of
culture (17,21,22).
If neurospheres became attached to the culture flasks during
harvest, they were detached by either tapping on the sides of the
flasks or aspirating them with medium. For cell dissociation, it
was critical to aspirate all the enzymatic suspension into the
syringe and discharge it slowly through the needle to ensure
uniform and sufficient trituration (21).
Cryopreservation and recovery of
neurospheres
The primary neurospheres were dissociated with
pre-warmed accutase, resuspended in fresh SFM, and cultured at 37˚C
with 5% CO2 for 24 h prior to cryopreservation upon the
formation of small cellular spheres. A freezing container (Nalgene™
Cryo; Thermo Fisher Scientific, Inc.) was placed at RT and filled
with 100% isopropyl alcohol (Tianjin Wanding Chemicals Co., Ltd.);
freezing medium was prepared [90% SFM and 10% dimethyl sulfoxide
(DMSO); cat. no., D2650; Sigma-Aldrich; Merck KGaA] protected from
light sources. Culture medium containing small neurospheres was
transferred into a 50 ml centrifuge tube. Following centrifugation
for 5 min at 200 x g at 37˚C, the supernatant was decanted, and the
cell pellet was gently resuspended in freshly prepared freezing
medium at a concentration of 5-10x106 cells/ml using a
glass Pasteur pipette. The suspension was immediately aliquoted (1
ml) into labeled cryovials, and the vials were immediately placed
into the container. Following storage at -80˚C for 24 h, the cell
aliquots were transferred into liquid nitrogen for storage for 7
days, or 1, 6 or 12 months.
For the recovery of cryopreserved NSCs, 1 cryovial
per time point (7 days, or 1, 6 or 12 months) was removed from the
liquid nitrogen as quickly as possible, promptly placed into a
water bath at 37˚C, and swirled quickly to accelerate the thawing
process. The entire cryovial was sprayed with 70% ethanol and moved
to a clean bench; then, the cell suspension was immediately
aspirated in a controlled manner and added dropwise to a 50 ml
centrifuge tube containing 40 ml pre-warmed DMEM-HG. Following
centrifugation for 5 min at 200 x g at 37˚C, the cell pellet was
resuspended in 40 ml pre-warmed DMEM-HG for a second rinse and
centrifugation (5 min at 200 x g at 37˚C). The cell pellet was then
resuspended in 20 ml fresh, pre-warmed SFM. The cell suspension was
transferred into culture flasks and incubated at 37˚C with 5%
CO2. A total of one-half of the medium was replaced
after 3 days of culture, and cells were sub-cultured after 7-8 days
of culture for additional experiments.
During the thawing process, the cryovials should not
be not completely submerged in water, and care must be taken when
swirling the cryovial to prevent contamination. The DMEM-HG and SFM
were all pre-warmed to protect the NSCs from damage and low cell
viability caused by the sudden temperature change that occurs when
vulnerable post-thaw cells are added to the medium.
Differentiation of dissociated
neurospheres
Induced differentiation is commonly applied to
examine the multipotential differentiation capability of
non-cryopreserved and cryopreserved NSCs (16,23).
Firstly, sufficient poly-L-lysine (PLL; molecular weight
150,000-300,000, 0.01%; cat. no., P4832; Sigma-Aldrich; Merck KGaA)
was added to coat the surface of 12-well culture plates for 24 h in
a 37˚C incubator. Following removal of the PLL, the plates were
washed 3 times with PBS and air-dried in the hood prior to use. The
non-cryopreserved and cryopreserved secondary neurospheres (stored
for 7 days, or 1, 6 or 12 months) were collected, dissociated, and
seeded on PLL-coated 12-well culture plates at a density of
5x104/cm2 in differentiation medium
(DMEM/F-12 medium containing B-27 supplement and 1% FBS; cat. no.,
16000044; Invitrogen; Thermo Fisher Scientific, Inc.) at 37˚C with
5% CO2 for up to 7 days. The differentiation medium was
changed every 3 days. To detect astrocyte, neuron and
oligodendrocyte differentiation, the cells were immunostained with
glial fibrillary acidic protein (GFAP), neuron-specific class III
beta-tubulin (β-III-tubulin) and cyclic nucleotide
phosphodiesterase (CNPase) antibodies, respectively (see details in
the Immunocytochemistry section).
PLL coating may be performed for 2-24 h at 37˚C, and
PLL-coated plates may be stored at RT for at least 1 week (7,10,15,23).
In the present study, to guarantee the maximum consistency of
experimental conditions, the plates were coated with PLL at 37˚C
for 24 h prior to commencing the experiment.
Immunocytochemistry
Nestin
Non-cryopreserved and cryopreserved secondary
neurospheres were dissociated and seeded on PLL-coated 12-well
culture plates in a monolayer (1x105 cells/ml) and
maintained for 3 days in fresh SFM. Non-cryopreserved and
cryopreserved secondary neurospheres were collected and plated on
PLL-coated 12-well culture plates and cultured for 24 h. Both the
monolayer cells and neurospheres were fixed with 4%
paraformaldehyde (cat. no., P1110; Beijing Solarbio Science &
Technology Co., Ltd.) for 20 min at RT, washed with PBS and
permeabilized with 0.3% Triton X-100 (cat. no., T8787;
Sigma-Aldrich; Merck KGaA) for 5 min. They were rinsed 3 times with
PBS and blocked with 10% normal goat serum (cat. no., PCN5000;
Invitrogen; Thermo Fisher Scientific, Inc.) in PBS for 1 h at RT.
Cells were then incubated with nestin primary antibody (cat. no.,
N5413; Sigma-Aldrich; Merck KGaA) at a 1:200 dilution in PBS
overnight at 4˚C. After 3 washes, the cells were subsequently
incubated with Alexa Fluor 555-conjugated goat anti-rabbit
secondary antibody (cat. no., A-21429; Invitrogen: Thermo Fisher
Scientific, Inc.) at a 1:500 dilution in PBS in the dark for 1 h.
Then, the cells were rinsed 3 times with PBS and incubated with
DAPI (1 mg/ml diluted in PBS; Cat No. D9542; Sigma-Aldrich; Merck
KGaA) for 5 min at RT.
GFAP, β-III-Tubulin and CNPase
Non-cryopreserved and cryopreserved secondary
neurospheres were passaged and seeded on PLL-coated 12-well culture
plates in a monolayer and maintained for 7 days in fresh
differentiation medium, as aforementioned. The protocols used for
GFAP, β-III-tubulin, and CNPase immunostaining were similar to the
aforementioned method for nestin immunolabeling, with the exception
that differentiated cells were incubated with rabbit anti-GFAP
(1:1,000; cat. no., ab7260; Abcam), anti-β-III-tubulin (1:500; cat.
no., PRB-435P; Covance, Inc.), and anti-CNPase (1:100; cat. no.,
5664; Cell Signaling Technology, Inc.).
All immunolabeled cells were examined under a
fluorescence microscope (IX71). Images of 5 randomly selected
fields were acquired at magnification, x20. Image-Pro plus 6.0
software (Media Cybernetics, Inc.) was used for the data analysis,
and quantification of immunolabeled cells was presented as the
percentage of specific marker-positive cells in comparison to the
total number of DAPI-stained cells.
Trypan blue exclusion assay
Viable cells were counted using trypan blue
exclusion assays in a hemocytometer (16,17,21,23,24).
Necrotic cells with compromised plasma membranes permit dye entry,
whereas viable cells exclude the dye (24). Therefore, the proportion of necrotic
cells is the percentage of trypan blue-positive cells, whereas cell
viability is the ratio of the number of trypan blue-impermeable
cells to the total number of cells (24). In brief, non-cryopreserved and
cryopreserved secondary neurospheres were dissociated and
resuspended, and then suspension aliquots (10 µl) were mixed with
10 µl of trypan blue solution (cat. no., T8154; Sigma-Aldrich;
Merck KGaA) for cell counting using a hemocytometer (room
temperature) under a light microscope with x20 magnification. The
total and trypan blue-positive cell numbers were then determined by
counting at least 200 cells per sample in a hemocytometer.
Determination of neurosphere
diameter
Non-cryopreserved and cryopreserved secondary
neurospheres were passaged and cultured in flasks with fresh SFM.
NSCs (single and spheres) were observed by bright-phase microscopy,
and images were captured from 5 randomly selected fields with a x20
objective on the first, second, third and fourth days after
passaging. The number and diameter of the spheres were determined
using Image-Pro plus 6.0 software.
Statistical analysis
GraphPad Prism 5.0 (GraphPad Software, Inc.) was
used for the statistical analysis. All data are presented as the
mean ± standard error of the mean of experimental replicates.
Statistical differences between the non-cryopreserved (the control
groups) and cryopreserved (the experimental groups) cells were
determined using a one-way analysis of variance followed by
Bonferroni post-hoc test. P<0.05 were considered to indicate a
statistically significant difference.
Results
Isolation and characterization of rat
embryonic NSCs
A single-cell suspension was obtained directly
following isolation, and these cells began to proliferate to form
small, round cell clusters with clear boundaries, which loosely
adhered to the bottom of the flask as observed under phase contrast
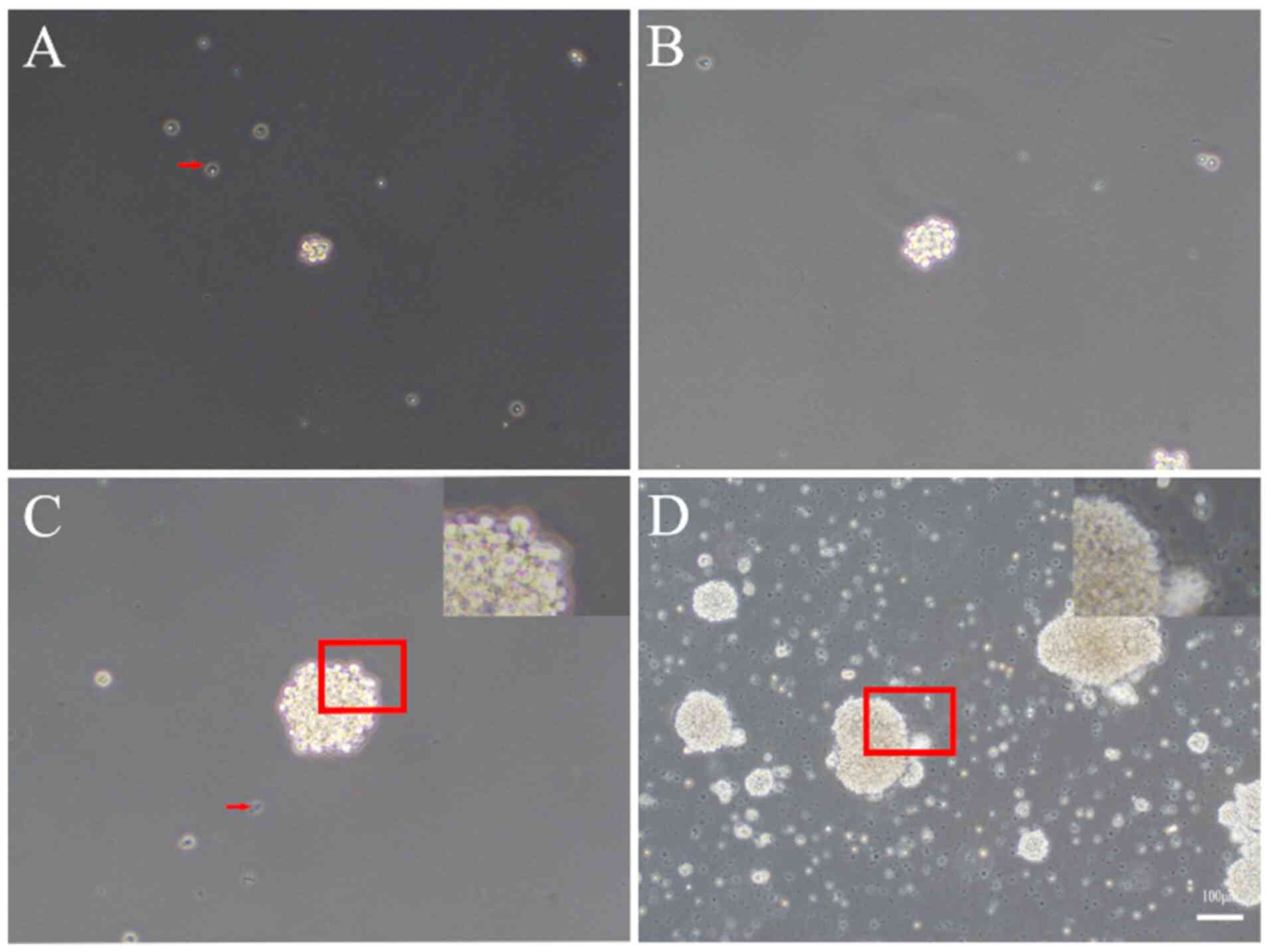
microscopy. The diameter of the cell clusters ranged from 30-50 µm
(Fig. 3A). On day 3, the cell
clusters grew larger, detached from the bottom of the flask and
became suspended in the medium, forming neurospheres of 50-80 µm in
diameter. These neurospheres were phase-bright and nearly perfectly
spherical in shape, with sharp, phase-bright outer edges (Fig. 3B). On day 7, these neurospheres
exhibited a translucent center and small cilia-like projections
around the periphery with a diameter of 130-150 µm (Fig. 3C), and were ready for dissociation
and passaging. If allowed to proliferate further, the center of the
spheres turned dark, and the spheres began to attach to the bottom
of the flask on day 9 (Fig. 3D).
Notably, numerous dead cells and debris were present in the primary
culture, and they aggregated and formed pseudo-spheres, which were
not spheroid in shape but were instead composed of small,
phase-dark, irregularly shaped cells. However, following 2
passages, the majority of dead cells and debris were effectively
eliminated.
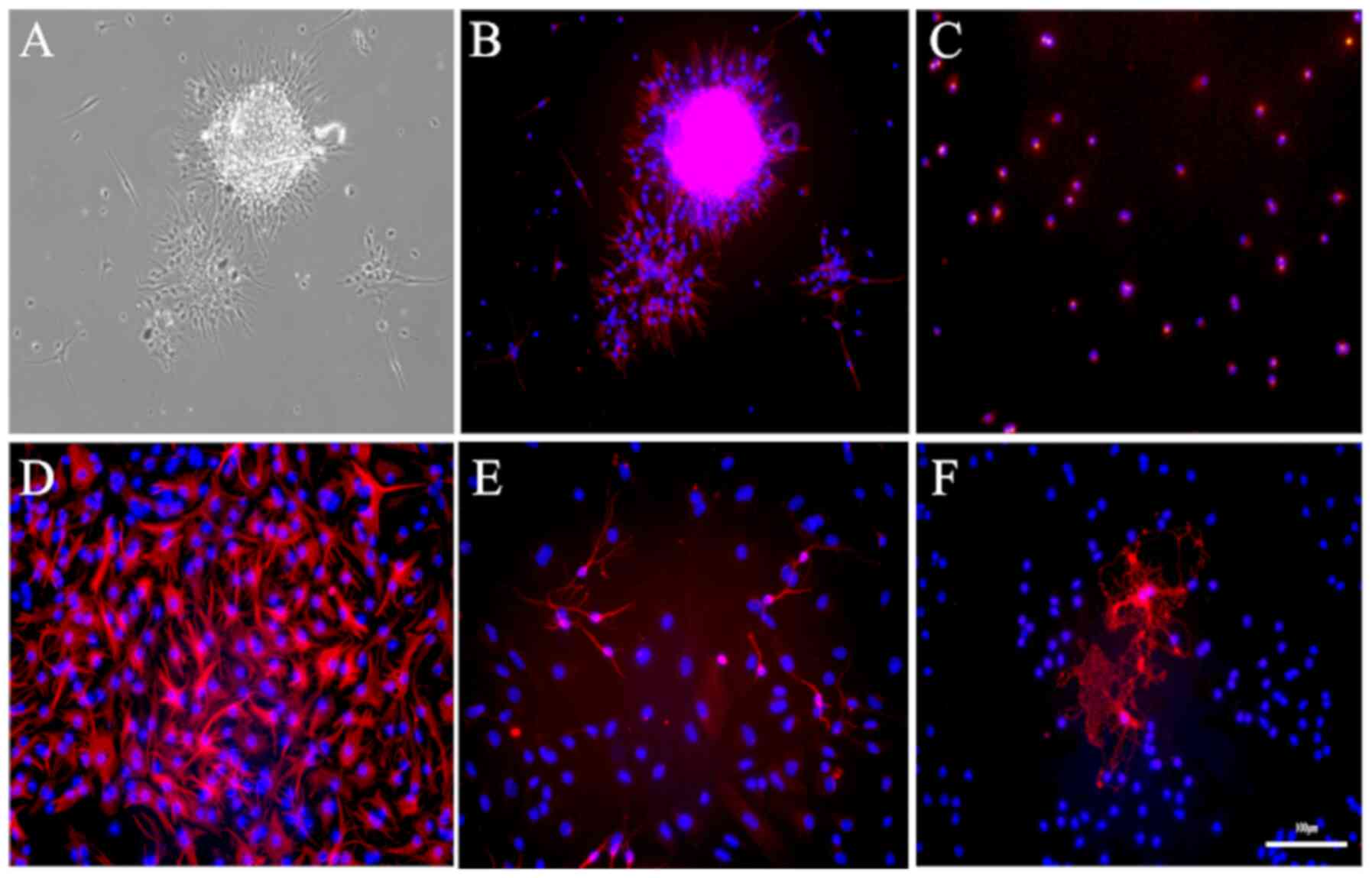
To characterize these rat embryonic NSCs, the
secondary neurospheres were first immunostained with nestin, a
specific marker for NSCs. A total of two-thirds of the cells in the
outer zone of the neurospheres were positive for nestin, whereas
cells in the center were nestin-negative (Fig. 4A and B). To obtain a more accurate estimation of
the proportion of nestin-positive NSCs in these 3D neurospheres,
the neurospheres were dissociated into single cells, seeded on
PLL-plated plates in SFM, and immunostained with nestin. As
demonstrated in Fig. 4C,
94.40±0.63% of cells in the monolayer expressed nestin.
Next, the multipotent differentiation capability of
NSCs was determined. According to the immunofluorescence staining,
66.45±4.52, 25.74±2.31 and 1.62±0.42% of the cells differentiated
into GFAP-positive astrocytes, β-III-tubulin-positive neurons and
CNPase-positive oligodendrocytes, respectively (Fig. 4D and F).
Effects of the cryopreservation
duration on rat embryonic NSCs
To examine the effects of cryopreservation duration
on rat embryonic NSCs, the cell viability, nestin expression,
multipotentiality and diameter of non-cryopreserved NSCs and
cryopreserved NSCs (7 days, or 1, 6 or 12 months) were further
analyzed and compared.
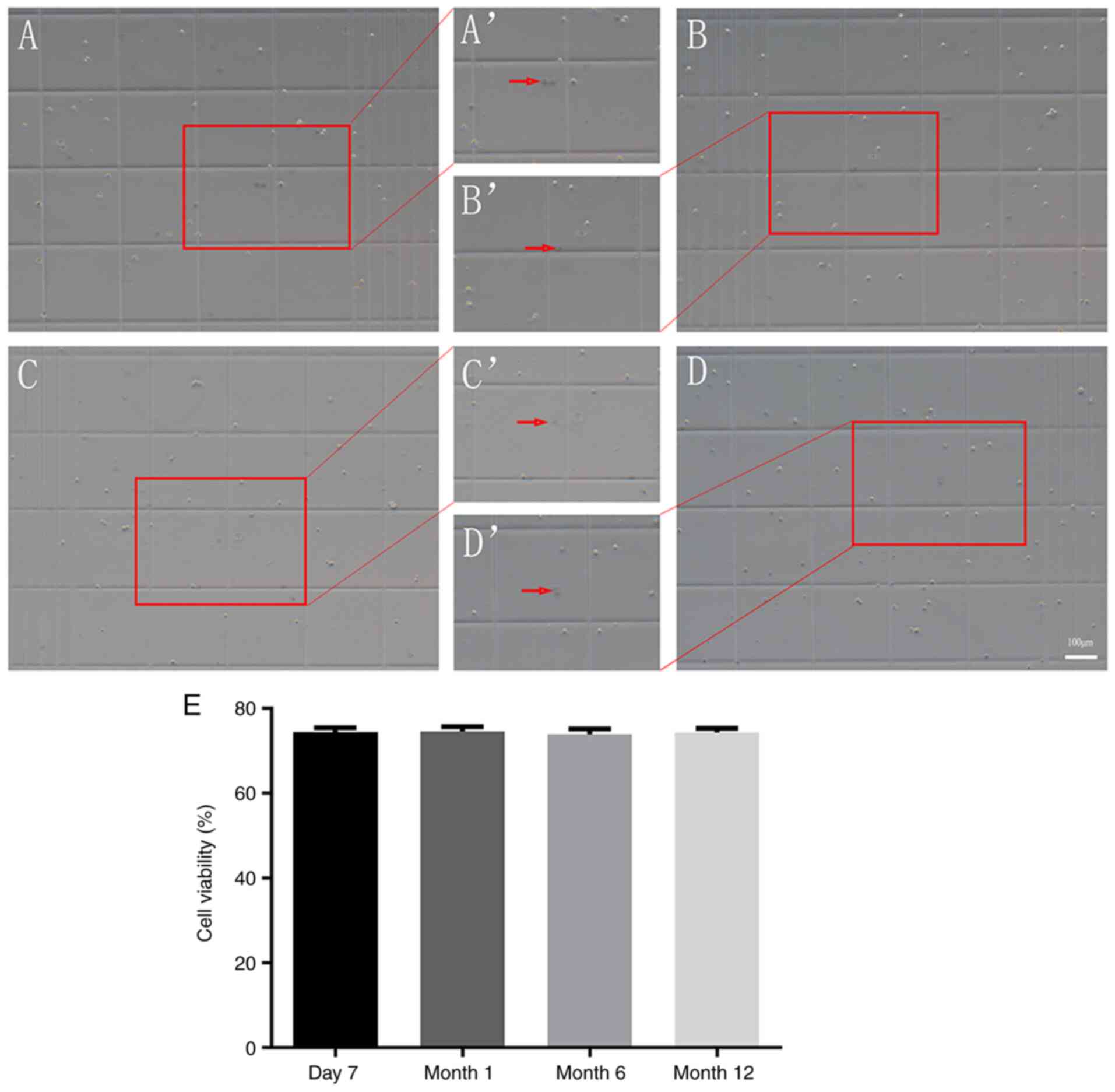
Cell viability following
cryopreservation
The trypan blue exclusion assay was performed to
assess cell viability (16,17,21,23,17,24).
As indicated in Fig. 5, 73.99±1.14,
74.23±1.29, 73.85±1.26 and 73.96±1.05% of the cells survived from
cryopreservation for 7 days, or 1, 6 or 12 months, respectively.
These data indicated that NSCs cryopreserved for 7 days, or 1, 6 or
12 months did not exhibit differences in cell viability following
recovery (all P>0.05).
Expression of the NSC marker nestin
following cryopreservation
To investigate the stem state of NSCs following
different durations of cryopreservation, non-cryopreserved or
cryopreserved secondary neurospheres were either directly
immunostained for nestin or dissociated into single cells and then
subjected to nestin immunostaining. Immunostaining of neurospheres
indicated that all neurospheres expressed nestin (Fig. 6). Single-cell immunostaining
additionally confirmed that 94.01±0.71, 94.36±0.68, 93.51±0.56 and
94.27±0.63% of the cells expressed nestin in the recovered NSCs
cryopreserved for 7 days, or 1, 6 or 12 months, respectively. No
significant differences were observed in the percentage of
nestin-positive cells between non-cryopreserved and cryopreserved
NSCs (all P>0.05) or among NSCs cryopreserved for 7 days, or 1,
6 or 12 months (all P>0.05).
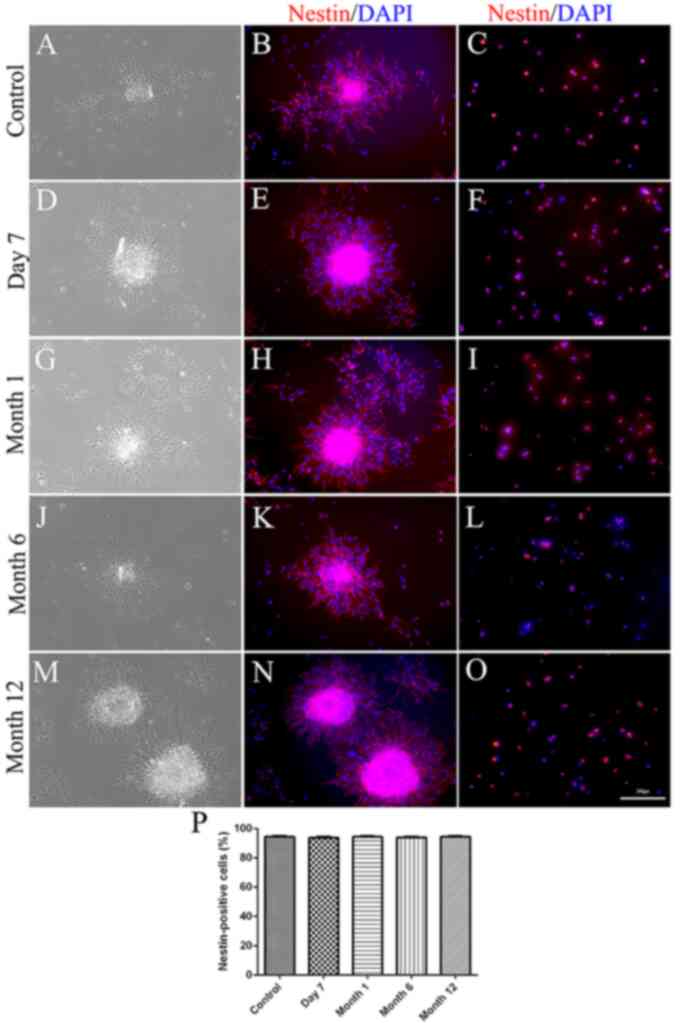 | Figure 6(A-P) Nestin expression in
neurospheres. (A, D, G, J and M) Phase contrast micrograph of
non-cryopreserved and cryopreserved secondary neurospheres. (B, E,
H, K and N) Expression of nestin in non-cryopreserved and
cryopreserved secondary neurospheres. (C, F, I, L and O) Expression
of nestin in dissociated neurospheres. (P) Quantification of
immunostaining data. No significant differences were observed in
the percentage of nestin-positive cells between non-cryopreserved
and cryopreserved secondary neurospheres (all P>0.05) or among
NSCs cryopreserved for 7 days, or 1, 6 or 12 months (all
P>0.05). Scale bar=100 µm. |
Multipotent differentiation after
cryopreservation
The differentiation potential of NSCs following
different durations of cryopreservation was analyzed. As
demonstrated in Fig. 7, all
non-cryopreserved and cryopreserved NSCs were able to differentiate
into astrocytes, neurons and oligodendrocytes. In addition, there
were no significant differences between non-cryopreserved and
cryopreserved NSCs (all P>0.05) or among NSCs cryopreserved for
7 days, or 1, 6 or 12 months (all P>0.05).
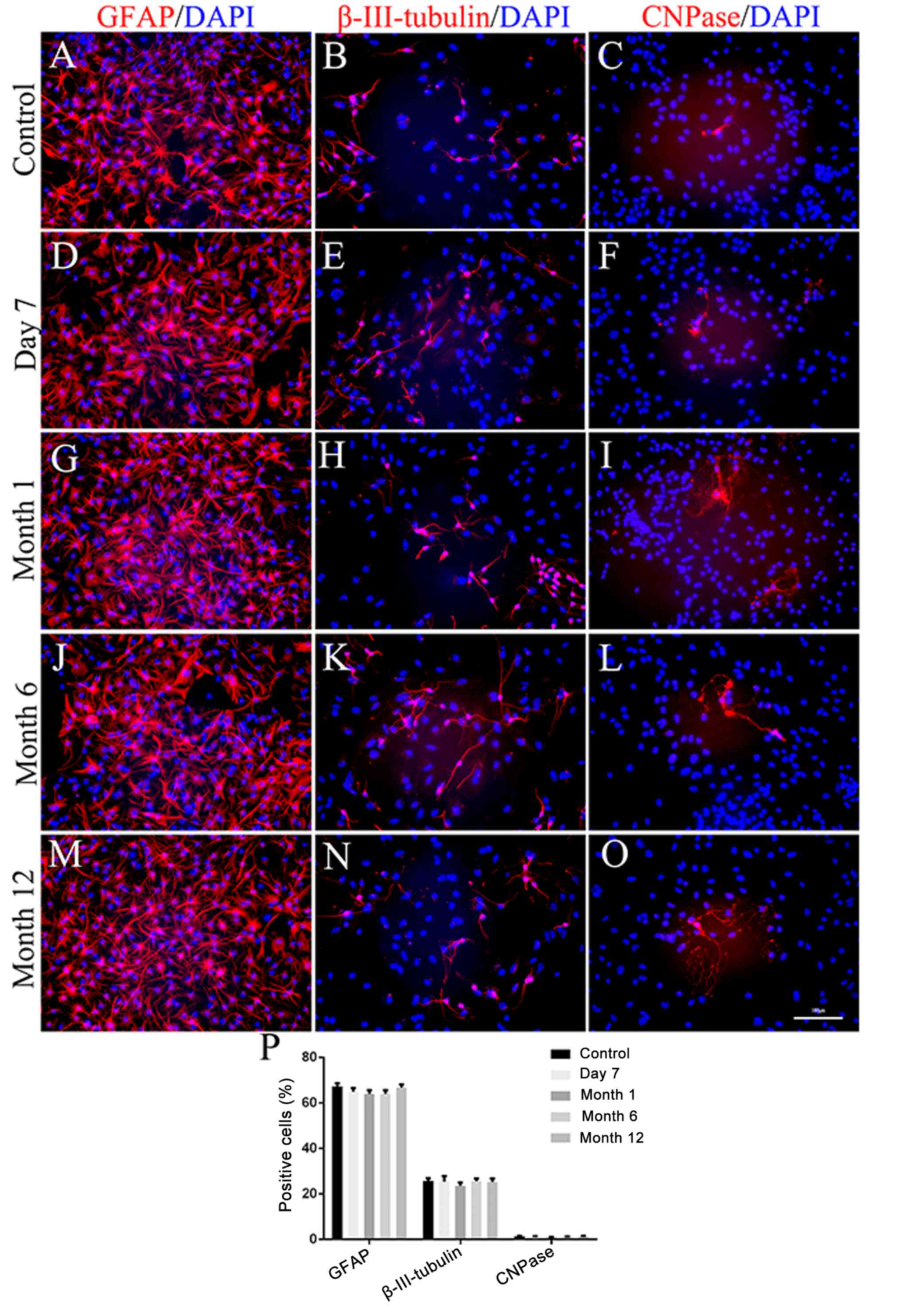 | Figure 7(A-P) Differentiation capacity of
neurospheres. (A, D, G, J and M) Representative image
immunocytochemical staining for GFAP (red) and DAPI (blue) in
non-cryopreserved and cryopreserved neurospheres-derived
astrocytes. (B, E, H, K and N) Representative image of
immunocytochemical staining for β-III-tubulin (red) and DAPI (blue)
in non-cryopreserved and cryopreserved neurospheres-derived
neurons. (C, F, I, L and O) Representative image of
immunocytochemical staining for CNPase (red) and DAPI (blue) in
non-cryopreserved and cryopreserved neurospheres-derived
oligodendrocytes. (P) Quantification of immunostaining data. No
significant differences were observed between non-cryopreserved and
cryopreserved secondary neurospheres (all P>0.05) or among NSCs
cryopreserved for 7 days, or 1, 6 or 12 months (all P>0.05).
Scale bar=100 µm. GFAP, glial fibrillary acidic protein;
β-III-tubulin, neuron-specific class III beta-tubulin; CNPase,
cyclic nucleotide phosphodiesterase. |
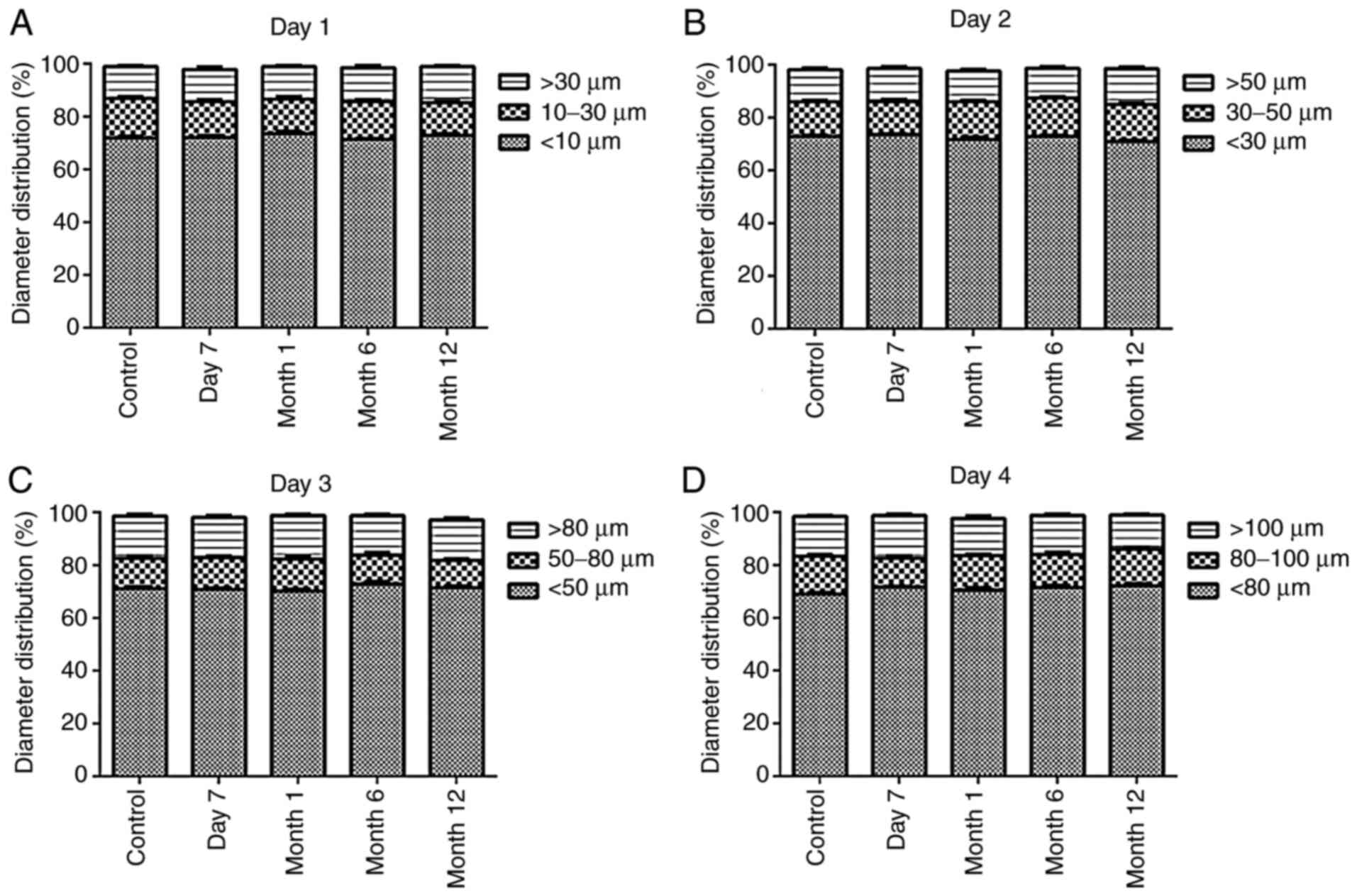
Determination of the cell diameter
following cryopreservation
To assess the self-renewal capability of NSCs
following different cryopreservation durations, non-cryopreserved
and cryopreserved secondary NSCs were dissociated and cultured in
SFM. As indicated in Fig. 8,
cryopreserved NSCs formed neurospheres identical to those of
non-cryopreserved NSCs (all P>0.05). In addition, NSCs
cryopreserved for 7 days, or 1, 6 or 12 months did not show
significant differences in cell diameter (all P>0.05).
Discussion
The present study detailed a modified technique for
the isolation, culture and cryopreservation of rat embryonic NSCs.
The viability, nestin expression, multipotentiality and diameter of
non-cryopreserved NSCs and NSCs cryopreserved for 7 days, or 1, 6
or 12 months were evaluated. Rat embryonic NSCs were successfully
obtained, and they maintained the capability for self-renewal and
multipotent differentiation even after long-term cryopreservation
(up to 12 months).
Compared with other protocols, 2 important skills
were modified in the protocol of the present study: Colorless
high-glucose medium and a sequential digestion strategy. Firstly,
NSCs are cells with a high metabolism, and they are easily damaged
upon exposure to a sugar-free environment, which may not provide
sufficient energy and therefore result in decreased cell viability.
In previous studies, PBS (21,25),
Hank's balanced salt solution (HBSS) (7,17) or
Leibovitz's L-15 (10,15) were used, but in the present study,
DMEM-HG was chosen as a temporary storage solution to provide
energy. Furthermore, DMEM-HG is colorless (without phenol red) and
may provide a clear operative view for tissue dissection (17), which is particularly important
during the process of pia mater and blood vessel removal. In
addition, 1% P/S was added to the DMEM-HG to decrease potential
contamination during the relatively long dissection procedure
(10,26). Secondly, a strategy of sequential
digestion was applied in the present study. Following the initial
digestion (a combination of enzymatic dissociation by
accutase/DNase I and mechanical pipetting during enzymatic
dissociation), undissociated tissues settled at the bottom of the
tube while single cells were suspended in the upper phase after
standing for 2 min. Suspended cells were collected for temporary
storage to prevent over-digestion, and the remaining undissociated
tissues were collected for a secondary digestion to ensure the
maximal harvest of NSCs (11,12,19).
The sequential digestion method minimizes the probability of
excessive dissociation and thereby promotes NSCs survival and
viability compared with the previous one-step digestion protocols
used by a number of previous studies (23,27).
In addition, a 35 mm Petri dish was applied for tissue digestion
rather than a centrifuge tube or a microtube, which was used by
numerous previous studies (23,27,28).
This approach allowed tissue pieces to be well-distributed in the
enzymatic solution, which not only facilitated close contact
between the tissues and enzymes but also allowed for improved
monitoring of the digestion status under a microscope (12).
To obtain high-viability NSCs, tissue collection and
cell isolation should be performed as quickly as possible (within 2
h), not only because tissues become viscous and pulpy, leading to
increased difficulty of dissection, but also due to the fact that
the viability of NSCs decreases over time (2,11,25,29,30).
In addition, low-temperature strategies were applied throughout the
dissection process to enhance cell viability (8,10,12,21,23,31).
It was identified to decrease the tissue consumption of oxygen and
energy, which is crucial for the survival of the isolated NSCs
(7,10,12,23).
Therefore, the temperature was maintained at a low level during
tissue dissection; for example, 70% ethanol at -20˚C was used for
disinfection and to rapidly decrease the embryo temperature. Dishes
containing 4˚C DMEM-HG with 1% P/S were placed on ice for tissue
dissection, and centrifuge tubes and dishes containing 4˚C DMEM-HG
with 1% P/S were placed on ice or under refrigeration for tissue
preservation. In addition to the aforementioned noes for the
dissection procedure, complete and thorough removal of the pia
mater and blood vessels is important (10,12,16).
The presence of the pia mater and blood vessels may increase the
force required for pipetting, ultimately increasing the mechanical
damage to the NSCs (10,12,16).
In addition, pia mater and blood vessels are the primary sources of
contaminating cells, for example endothelial cells, fibroblasts and
blood cells (10,12,16).
If they are not completely removed, these cells may overgrow and
decrease the purity of the NSC culture.
The tissue digestion procedure is one of the most
critical steps for obtaining a high yield of NSCs. If not handled
properly, this process may result in excessive cell death (7,10,16,30).
Notably, smaller tissue pieces increase the cell dissociation
efficiency, primarily due to the increased contact area between the
tissue and dissociation enzyme solutions. In addition, the
potential for mechanical damage by increased pipetting force is
decreased (7,10,16,30).
Therefore, dissected tissues were minced into small pieces with
microscissors prior to commencing the digestion procedure. However,
the mincing step should not be performed for extended periods of
time; the optimal duration is ~1 min. The efficiency of mincing may
be improved with practice and the aid of a stereomicroscope. Tissue
digestion involves enzymatic and mechanical methods (30,31).
The enzymatic treatment is the application of proteolytic enzymes,
including trypsin-EDTA and accutase, which degrade the highly
structured extracellular matrix surrounding NSCs to isolate them
from the rest of the tissue (30).
Trypsin-EDTA has been employed in several studies (17,21-23,26,31),
However, it is difficult to control the enzymatic reaction time,
which may affect sphere formation, cause cells to attach to the
substrate and differentiate, or result in massive cell death.
Accutase is a less aggressive enzyme (17,19,21-23)
that has been demonstrated to increase cell survival compared with
that associated with trypsin-EDTA and does not require FBS. By
contrast, trypsin-EDTA is usually inactivated by adding FBS; the
latter may affect NSC differentiation and introduce contaminants
(26,30). In addition to accutase, DNase I is
always added to break down the accumulated mass of sticky DNA
released from damaged cells, which prevents subsequent dissociation
(18,30).
During or following enzymatic dissociation,
mechanical dissociation is applied. This relatively aggressive
method is accomplished using either a fire-polished bevel tip glass
pipette, a commercially available disposable plastic pipette or a
sterile syringe needle that breaks up tissue pieces by pushing them
against the bottom/base or side of the tube/plate (19,30).
Of note, during mechanical digestion, the formation of air bubbles
should be avoided as an excess of bubbles decreases the cell
viability and trituration efficiency and increases the risk of
contamination (2,7,10,16-19,21,23).
Compared with enzymatic digestion, mechanical dissociation alone
takes more time, requires an increased level of experimental
skills, induces more variability (digestion efficiency depends on
an experimenter's experience), and causes more cell aggregation and
cell death (11,19,21,22,30).
Therefore, a combination of enzymatic dissociation by
accutase/DNase I and mechanical pipetting during enzymatic
dissociation is considered o be one of the best procedures to
achieve sufficient tissue dissociation with minimal cell death.
Following tissue digestion and cell plating, the
growth of NSCs was closely monitored. A total of one-half of the
medium was replaced with new medium after 3 days of culture. The
mixed medium contained pro-survival factors produced by cells and
released into the conditioning medium (10). A complete medium change was
performed during cell passaging after 7-8 days of culture, when the
majority of neurospheres were <150-200 µm in diameter. Larger
neurospheres are difficult to dissociate and eventually begin to
differentiate (2,11,16,30,32).
In addition, certain cells in the core of excessively large
neurospheres are susceptible to death due to a lack of access to
oxygen and nutrients, as they are not in direct contact with the
culture medium (2,11,16,30,32).
To characterize the multipotent ability of the
NSCs, the cells were exposed to 1 type of medium (DMEM/F-12 medium
containing B-27 supplement and 1% FBS), not 3 different media, for
example: Retinoic acid plus forskolin for neurons induction;
insulin-like growth factor-1 for oligodendrocytes induction; and
leukemia inhibitory factor with bone morphogenic protein-2 for
astrocytes induction, as described previously (33,34).
The difference between the methods involving 1 medium and 3 media
lies in the final different differentiation rate of neurons,
oligodendrocytes and astrocytes; the 1 medium method has been
applied by a number of previous studies to illustrate the different
function of drugs or materials on NSCs (35,36).
In addition, if 3 different media are used, more stimulation
factors, for example retinoic acid and forskolin, are added in the
media; and different time periods are required for neuron (4 days),
astrocyte (4 days), and oligodendrocyte (6 days) differentiation
(33,34). Consequently, incubation time becomes
an influencing factor; the results of the present study may have
been affected by these additional factors. For the present study,
the multipotent character and recovery ability of the cryopreserved
NSCs, not the different effect of stimulation factors and time
period, were the focus. Therefore, the 1 medium method was chosen
to examine our hypothesis. Future studies should also apply the 3
media protocol to characterize the multipotent ability of NSCs in
more detail.
Using the procedure described, the present study
successfully obtained highly viable rat embryonic NSCs that
proliferated, expressed nestin and differentiated into neurons,
astrocytes and oligodendrocytes. However, these cells have a
limited expansion period in vitro and frequently exhibit
spontaneous differentiation. Therefore, NSCs are commonly
cryopreserved for future research and transplantation (24,37).
The NSC state (single-cell suspension or NSC spheres), freezing
medium composition and freezing cell density are the major
determinants of NSC survival and viability following the thawing
process (15,16). In the present study, small spheres
in the logarithmic phase 24 h after the secondary passage were
collected for cryopreservation. A high sphere density
(5-10x106 cells/ml) was used during freezing to enhance
sphere re-establishment following cryopreservation, based on data
from previous studies (7,16,30).
SFM supplemented with 10% DMSO was used as the freezing medium, and
FBS was not added to the freezing medium considering its induction
effect on NSC differentiation and the chance of introducing
contaminants (4,15,16,30).
Furthermore, it is critical to remove all the freezing solution
from the frozen pellet during recovery and re-rinse the cells using
DMEM-HG, as the introduction of DMSO into the culture medium is
cytotoxic (7,16,30).
Therefore, the thawed NSCs were rinsed twice using 40 ml DMEM-HG,
and the entire supernatant was discarded following centrifugation.
In addition, the effect of different cryopreservation durations on
rat embryonic NSCs was explored. Cells were thawed after 7 days, or
1, 6 or 12 months of storage in liquid nitrogen, and the viability,
stemness state, self-renewal ability and multipotency of
differentiation were determined. The results suggested that a
number of the cells retained their viability following frozen
storage and that the viability of NSCs cryopreserved for different
time periods was similar. NSCs cryopreserved for different periods
of time maintained stemness and the ability to self-renew and
differentiate into astrocytes, neurons and oligodendrocytes.
The present study contained a few limitations that
should be addressed. Firstly, more immunostaining markers of NSCs,
including transcription factor SOX-2 (SOX2), RNA-binding protein
Musashi homolog 1 (MUSASHI-1) and prominin-1 (CD133+), were not
used to verify their nature. Subsequent studies should perform
SOX2, MUSASHI-1 and CD133+ immunostaining. Secondly, only 1 medium
was used to characterize the multipotent ability of NSCs; the use
of 3 media concomitantly would address this point in greater detail
in future studies.
In conclusion, the in vitro culture of rat
embryonic NSCs has been widely applied for the investigation of the
cellular and molecular mechanisms underlying neurogenesis and brain
regeneration. The present study described a modified primary
culture protocol for successfully obtaining rat embryonic NSCs that
maintain their ability to self-renew and multipotency after
different time periods of cryopreservation.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant no., 81501899),
the State Key Program of the National Natural Science Foundation of
China (grant nos., 81330042 and 81620108018), the Special Program
for Sino-Russian Joint Research Sponsored by the Ministry of
Science and Technology, China (grant no., 2014DFR31210), the Key
Program Sponsored by the Tianjin Science and Technology Committee,
China (grant nos., 13RCGFSY19000 and 14ZCZDSY00044), the Science
Foundation of Tianjin Medical University for Young Scholars (grant
no., 2014KYQ01), and the Science Foundation of Tianjin Medical
University General Hospital for Young Scholars (grant no.,
ZYYFY2014037).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
HZ, HY, LLu and XL designed and performed the
experiments, analyzed the data and co-wrote the article. BP, ZF,
TC, JL, YK and LLiu analyzed the data and edited the manuscript.
GN, WYD and PW designed the study and critically revised the
article for important intellectual content. XK and SF designed the
experiments, provided materials and funding and co-wrote the
article. All authors agree that all the questions related to the
accuracy or integrity of the paper have been appropriately
investigated and resolved and have given final approval of the
version to be published.
Ethics approval and consent to
participate
The present study was performed with the approval
of the Ethics Committee of Tianjin Medical University and complied
with the U.S. National Institutes of Health Guide for the Care and
Use of Experimental Animals and the Society for Neuroscience Use of
Animals in Neuroscience Research Guidelines.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Schiefer HB: Guide to the care and use of
experimental animals, volume 2. Can Vet J. 26(59)1985.
|
|
2
|
OlferdB E, Cross B, McWillian D and Mc
Willian A: Guidelines for the use of animal in neuroscience
research. Canadian Council on Care (CCAC), Ottawa, Canada 163-165,
1997.
|
|
3
|
Reynolds BA, Tetzlaff W and Weiss S: A
multipotent EGF-responsive striatal embryonic progenitor cell
produces neurons and astrocytes. J Neurosci. 12:4565–4574.
1992.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Rietze RL and Reynolds BA: Neural stem
cell isolation and characterization. Methods Enzymol. 419:3–23.
2006.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Chojnacki A and Weiss S: Production of
neurons, astrocytes and oligodendrocytes from mammalian CNS stem
cells. Nat Protoc. 3:935–940. 2008.
|
|
6
|
Gritti A, Galli R and Vescovi AL: Clonal
analyses and cryopreservation of neural stem cell cultures. Methods
Mol Biol. 438:173–184. 2008.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Revishchin AV, Korochkin LI, Okhotin VE
and Pavlova GV: Neural stem cells in the mammalian brain. Int Rev
Cytol. 265:55–109. 2008.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ahmed S: The culture of neural stem cells.
J Cell Biochem. 106:1–6. 2009.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Guo W, Patzlaff NE, Jobe EM and Zhao X:
Isolation of multipotent neural stem or progenitor cells from both
the dentate gyrus and subventricular zone of a single adult mouse.
Nat Protoc. 7:2005–2012. 2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kumar K, Singh R, Kumar M, Agarwal P,
Mahapatra PS, Kumar A, Malakar D and Bag S: Isolation and
characterization of neural stem cells from buffalo. Int J Neurosci.
124:450–456. 2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Huang H, Mao G, Chen L and Liu A: Progress
and challenges with clinical cell therapy in neurorestoratology. J
Neurorestoratol. 3:91–95. 2015.
|
|
12
|
Hameed LA and Simon A: Isolation and
culture of neurospheres from the adult newt brain. Methods Mol
Biol. 1290:197–204. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Louis SA and Reynolds BA: Generation and
differentiation of neurospheres from murine embryonic day 14
central nervous system tissue. Methods Mol Biol. 290:265–280.
2005.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Xu SY, Wu YM, Ji Z, Gao XY and Pan SY: A
modified technique for culturing primary fetal rat cortical
neurons. J Biomed Biotechnol. 2012(803930)2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Ahlenius H and Kokaia Z: Isolation and
generation of neurosphere cultures from embryonic and adult mouse
brain. Methods Mol Biol. 633:241–252. 2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Pacey L, Stead S, Gleave J, Tomczyk K and
Doering L: Neural stem cell culture: Neurosphere generation,
microscopical analysis and cryopreservation. Nat Protoc. 1:215–222.
2006.
|
|
17
|
Bonner JF, Haas CJ and Fischer I:
Preparation of neural stem cells and progenitors: Neuronal
production and grafting applications. Methods Mol Biol. 1078:65–88.
2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Brewer GJ and Torricelli JR: Isolation and
culture of adult neurons and neurospheres. Nat Protoc. 2:1490–1498.
2007.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Louis SA, Mak CK and Reynolds BA: Methods
to culture, differentiate, and characterize neural stem cells from
the adult and embryonic mouse central nervous system. Methods Mol
Biol. 946:479–506. 2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ortega F, Berninger B and Costa MR:
Primary culture and live imaging of adult neural stem cells and
their progeny. Methods Mol Biol. 1052:1–11. 2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Bizy A and Ferrón SR: Isolation, long-term
expansion, and differentiation of murine neural stem cells. Methods
Mol Biol. 1212:103–112. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Lopez-Ramirez MA, Calvo CF, Ristori E,
Thomas JL and Nicoli S: Isolation and culture of adult zebrafish
brain-derived neurospheres. J Vis Exp. 53617:2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Aligholi H, Hassanzadeh G, Gorji A and
Azari H: A novel biopsy method for isolating neural stem cells from
the subventricular zone of the adult rat brain for autologous
transplantation in CNS injuries. Methods Mol Biol. 1462:711–731.
2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Hinsch K and Zupanc GK: Isolation,
cultivation, and differentiation of neural stem cells from adult
fish brain. J Neurosci Methods. 158:75–88. 2006.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Belenguer G, Domingo-Muelas A, Ferrón SR,
Morante-Redolat JM and Fariñas I: Isolation, culture and analysis
of adult subependymal neural stem cells. Differentiation. 91:28–41.
2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Hitoshi S, Kippin T and van der Kooy D:
Culturing adult neural stem cells: Application to the study of
neurodegenerative and neuropsychiatric pathology. In: Neurogenesis
in the adult brain II: Clinical implications. Seki T, Sawamoto K,
Parent JM and Alvarez-Buylla A (eds.) Springer, Tokyo, pp189-207,
2011.
|
|
27
|
Annese T, Corsi P, Ruggieri S, Tamma R,
Marinaccio C, Picocci S, Errede M, Specchia G, De Luca A,
Frassanito MA, et al: Isolation and characterization of neural stem
cells from dystrophic mdx mouse. Exp Cell Res. 343:190–207.
2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Hugnot JP: Isolate and culture neural stem
cells from the mouse adult spinal cord. Methods Mol Biol.
1059:53–63. 2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ferrari D, Binda E, De Filippis L and
Vescovi AL: Isolation of neural stem cells from neural tissues
using the neurosphere technique. Curr Protoc Stem Cell Biol.
2(2D.6)2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Oliver-De la Cruz J and Ayuso-Sacido A:
Neural stem cells from mammalian brain: Isolation protocols and
maintenance conditions. In: Neural Stem Cells and Therapy. Sun T
(ed.) Intech, Rijeka, pp3-30, 2012.
|
|
31
|
Azari H, Rahman M, Sharififar S and
Reynolds BA: Isolation and expansion of the adult mouse neural stem
cells using the neurosphere assay. J Vis Exp 2393, 2010.
|
|
32
|
Siebzehnrubl FA, Vedam-Mai V, Azari H,
Reynolds BA and Deleyrolle LP: Isolation and characterization of
adult neural stem cells. Methods Mol Biol. 750:61–77.
2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Hsieh J, Nakashima K, Kuwabara T, Mejia E
and Gage FH: Histone deacetylase inhibition-mediated neuronal
differentiation of multipotent adult neural progenitor cells. Proc
Natl Acad Sci USA. 101:16659–16664. 2004.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Hsieh J, Aimone JB, Kaspar BK, Kuwabara T,
Nakashima K and Gage FH: IGF-I instructs multipotent adult neural
progenitor cells to become oligodendrocytes. J Cell Biol.
164:111–122. 2004.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Li JY, Liu J, Manaph NPA, Bobrovskaya L
and Zhou XF: ProBDNF inhibits proliferation, migration and
differentiation of mouse neural stem cells. Brain Res. 1668:46–55.
2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Shirazi HA, Rasouli J, Ciric B, Rostami A
and Zhang GX: 1,25-Dihydroxyvitamin D3 enhances neural stem cell
proliferation and oligodendrocyte differentiation. Exp Mol Pathol.
98:240–245. 2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Ma XH, Shi Y, Hou Y, Liu Y, Zhang L, Fan
WX, Ge D, Liu TQ and Cui ZF: Slow-freezing cryopreservation of
neural stem cell spheres with different diameters. Cryobiology.
60:184–191. 2010.PubMed/NCBI View Article : Google Scholar
|