Introduction
Mitochondria generates ATP via oxidative
phosphorylation (OXPHOS) mediated by OXPHOS complexes I, II, III,
IV and V, and the dysfunction of the mitochondrial protein
synthesis system is involved in OXPHOS deficiency, leading to
mitochondrial disease with early-onset and fatal phenotypes
(1). Complexes I, III, IV and V all
contain ≥1 mitochondrial DNA (mtDNA) encoded subunit, while complex
II is encoded completely by nuclear DNA (1). The G elongation factor mitochondrial 1
(GFM1) gene encodes the nuclear-encoded elongation factor G,
mitochondrial (EFG1) protein, which is one of the mitochondrial
elongation factors that enforces the elongation-dependent movement
of mtDNA encoded transfer (t)RNAs through the mitochondrial
ribosomes by removing the de-acetylated tRNA and replacing it with
the peptidyl-tRNA as a GTPase (2).
In 2004, an association was identified between the mutation of the
GFM1 gene and the occurrence of early hepatic encephalopathy, a
fatal mitochondrial disease, in two siblings with poor prognosis
(3). At present, the total number
of GFM1 mutations reported in families is 25 (3-13).
In the present study, the clinical, laboratory and molecular
results in two siblings with heterozygous GFM1 gene defects, one a
novel mutation and the other a previously reported mutation
(7), are described.
Materials and methods
Patients
A patient with suspected mitochondrial disease and
his asymptomatic parents, aged 33 years, were asked to participate
in the present study in February 2019. Written informed consent and
consent for publication to use their samples and clinical data were
obtained from the parents and the present study was formally
approved by the Ethics Committee of The Women's Hospital, Zhejiang
University School of Medicine, Zhejiang, China.
Whole-exome and Sanger sequencing
The blood and the amniotic fluid samples were
collected and stored at -80˚C until further use. Whole-exome
sequencing was performed as previously described by Simon et
al (11). Single nucleotide
variants were confirmed using Sanger sequencing, as previously
described (14).
Protein structure analysis
The three-dimensional structure of the EFG1 protein
was predicted using the Iterative Threading ASSEmbly Refinement
computer modeling program (15). An
elimination of part of domain IV and the whole of domain V was
predicted with the R526 premature stop codon. The multiple sequence
alignment between diverse species was performed using the T-Coffee
program (16).
Histological analysis
The hematoxylin and eosin staining procedure was
performed. In brief, the collected hepatic and renal tissue samples
were fixed for 24 h in 4% paraformaldehyde at 4˚C and embedded in
paraffin. Following this, samples were sliced into 4-µm thick
sections. For hematoxylin and eosin staining, the slides were
stained with hematoxylin for 5 min at room temperature (RT),
alcohol hydrochloric acid for 1 sec and eosin for 1 sec. The tissue
was observed under an optical microscope (Olympus Corporation;
magnification, x200). For immunofluorescence, the slides were
immunostained with primary EFG1 polyclonal antibodies (1:50; cat.
no. 14274-1-AP; Thermo Fisher Scientific, Inc.) overnight at 4˚C
and secondary antibodies (1:2,000; cat. no. A-11036; Thermo Fisher
Scientific, Inc.) for 1 h at RT. The nucleus was visualized with
DAPI staining for 1 min at RT. Images were taken under a
fluorescence microscope (Olympus Corporation; magnification,
x150).
Reverse transcription-PCR
Total RNA from the collected hepatic and renal
tissue samples were isolated using Total RNA Extraction reagent
(cat. no. R401; Vazyme Biotech Co., Ltd.). Following quantification
and unified concentrations, total RNA was reversed-transcribed into
complementary DNA (cDNA) using a reverse transcription kit (cat.
no. R212; Vazyme Biotech Co., Ltd.), according to the
manufacturer's protocol. The cDNA was amplified using the Green Taq
PCR Mix (cat. no. P131; Vazyme Biotech Co., Ltd.). Primers
targeting the GFM1 gene used for PCR analysis were as follows:
forward, 5'-CAAAAGGTATTGGCAGGTT-3' and reverse,
5'-CAGGGGCAGTAATGGTCT-3'. The thermocycling conditions were
performed at 95˚C for 3 min, followed by 25 cycles of 95˚C for 30
sec, 56˚C for 30 sec and 72˚C for 60 sec.
Western blot analysis
Western blot analysis was performed using whole cell
lysates from the liver and kidney of the index patient, as
previously described (11), using
the following specific antibodies: Anti-NADH:Ubiquinone
oxidoreductase subunit A13 (NDUFA13; cat. no. 10986-1-AP;
ProteinTech Group, Inc.), anti-succinate dehydrogenase complex iron
sulfur subunit B (SDHB; cat. no. ab178423; Abcam),
anti-ubiquinol-cytochrome c reductase core protein 2 (UQCRC2; cat.
no. ab203832; Abcam), anti-ATP synthase peripheral stalk subunit F6
(ATP5; cat. no. ab176569; Abcam) and anti-GAPDH (cat. no. ab181602;
Abcam). Differences in proteins levels were determined by visual
inspection.
Results
Clinical features and laboratory
findings
At 32 weeks' gestation, the index patient was
genetically diagnosed with a defect in the GFM1 gene using
amniocentesis. He was born to unrelated Chinese parents, with a
pregnancy complicated by a late 3rd trimester suspicion of fetal
growth restriction. The vaginal birth, following a previous
cesarean section, occurred at 40 weeks' gestation, without any
complications and no neonatal resuscitation was required. His
birthweight was 2.3 kg and he presented with an omphalocele and an
inguinal hernia. Soon after birth, he was admitted to the neonatal
unit of Women's Hospital, Zhejiang University School of Medicine
and received symptomatic treatment for metabolic acidosis,
hypoglycemia, kaliopenia, hypocalcemia, hypernatremia, hepatic
impairment and anemia. He was treated with ursodeoxycholic,
levocarnitine, vitamins B1, -2, -6 and -12, coenzyme Q10 and
mecobalamine. He exhibited failure to thrive with poor weight gain
and his body weight was 2.9 kg at 2 months old. From 2 months of
age, he developed sporadic twitching in the corners of the mouth
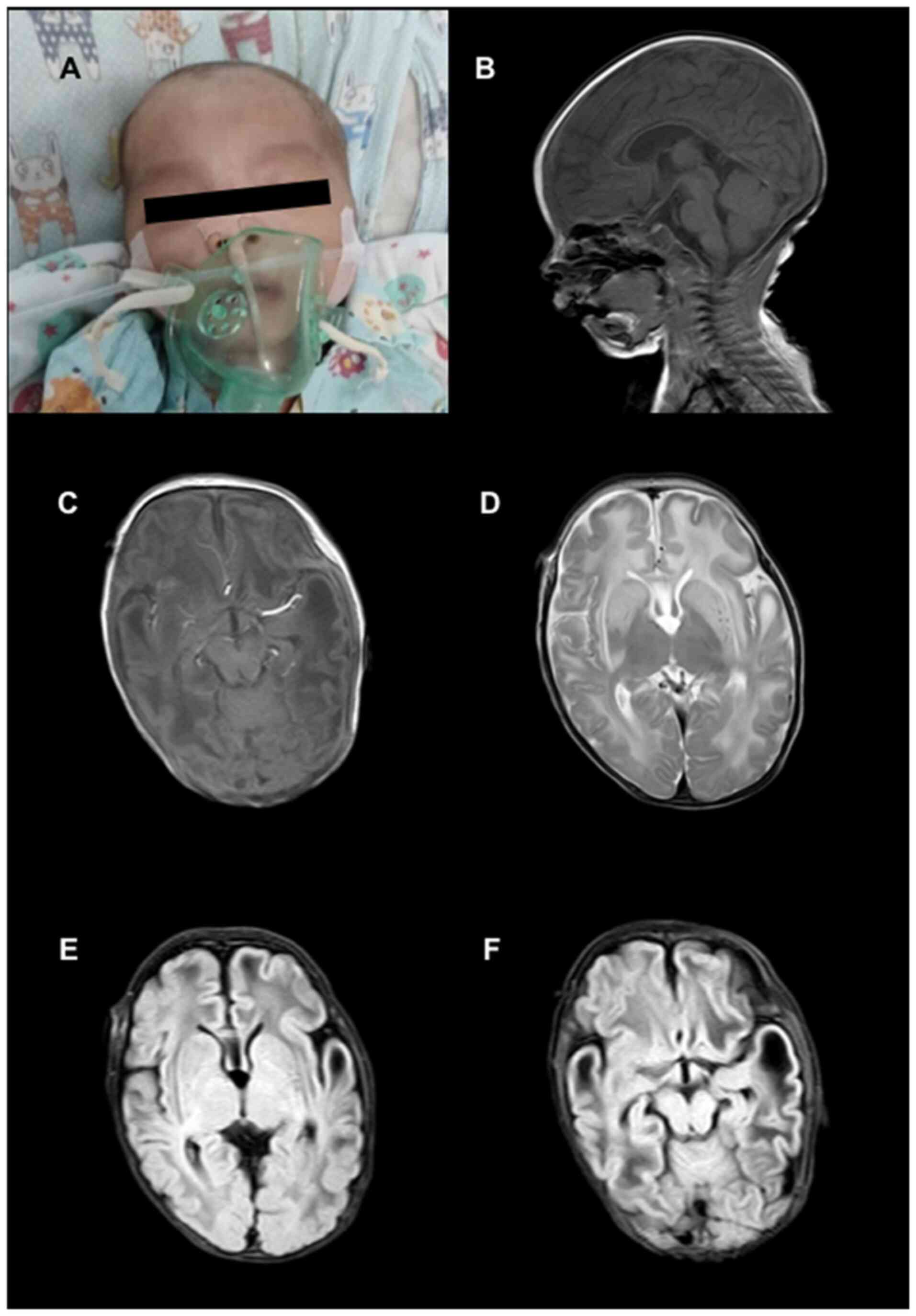
and eye blinking; however, there was no noticeable dysmorphic
features (Fig. 1A). An
electroencephalography identified multifocal epileptiform
discharges over the frontal and temporal lobes bilaterally;
however, he did not exhibit significant epileptic seizures. His
brain magnetic resonance imaging scan indicated symmetric swelling
in the frontal and temporal lobes, bilateral centra semiovalia,
gyri and white matter (Fig. 1B-F).
At 3 months of age, he developed severe metabolic acidosis [pH,
6.979 (normal range 7.350-7.450)]; PCO2, 11.9 mmHg
(normal range, 35.0-48.0 mmHg); PO2, 164 mmHg (normal
range, 83.0-108.0 mmHg); base excess -28.5 mM (normal range,
-3.0-3.0 mM); and lactic acid, 22.0 mM (normal range, 0.5-1.6 mM).
In the following days, several episodes of acute metabolic acidosis
were corrected by intravenous administration of bicarbonate.
Furthermore, he had severe liver impairment [albumin 28.5 g/l
(normal range, 32.0-52.0 g/l)]; total bile acids, 271 µM (normal
range, 0.0-12.0 µM); alanine aminotransferase, 220 U/l (normal
range, <50 U/l); aspartate amino transferase, 186 U/l (normal
range, 15-60 U/l) and high serum ammonia, 112 mM (normal range,
9-30 mM). However, the levels of blood urea nitrogen and creatinine
were not increased. In addition, a blood transfusion was
administered due to coagulation abnormalities: Prothrombin time,
49.8 sec (normal range, 9.0-14.0 sec); activated partial
thromboplastin time, 110 sec (normal range, 23-38 sec);
international normalized ratio, 4.15 (normal range, 0.8-1.2);
fibrinogen, 0.21 g/l (normal range, 1.8-4.0 g/l) and anemia
[hemoglobin 85 g/l (normal range, 110-155 g/l)]. Unfortunately, he
succumbed 4 days later due to multiple organ failure.
The female older sibling of the index patient was
born at 39 weeks' gestation by cesarean section due to fetal
distress. Her birth weight was 2.2 kg and no neonatal resuscitation
was required. After birth, she developed inconsolable crying and
opisthotonos posturing. At 2 days old, she received symptomatic
treatment in the neonatal unit of the hospital due to lethargy,
feeding difficulties and hypoglycemia; however, she did not receive
a precise diagnosis. Unfortunately, she succumbed 4 days later.
Postmortem histological analysis
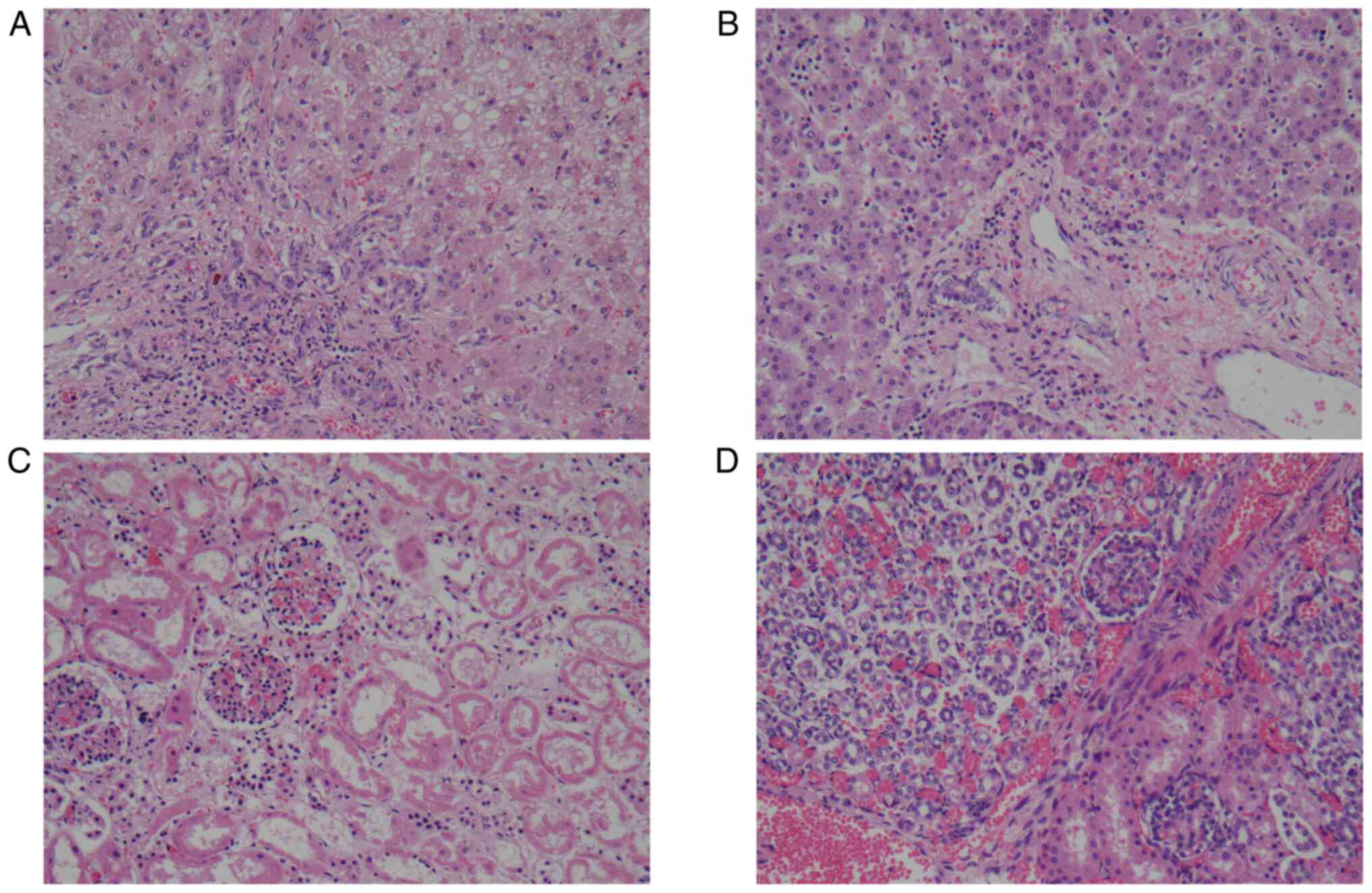
Postmortem histopathology of the index patient
revealed irregular hepatic plates, steatosis and cholestasis in the
liver (Fig. 2A) and glomerular and
tubular necrosis in the kidney (Fig.
2C); however, there was no cardiomyopathy (data not shown).
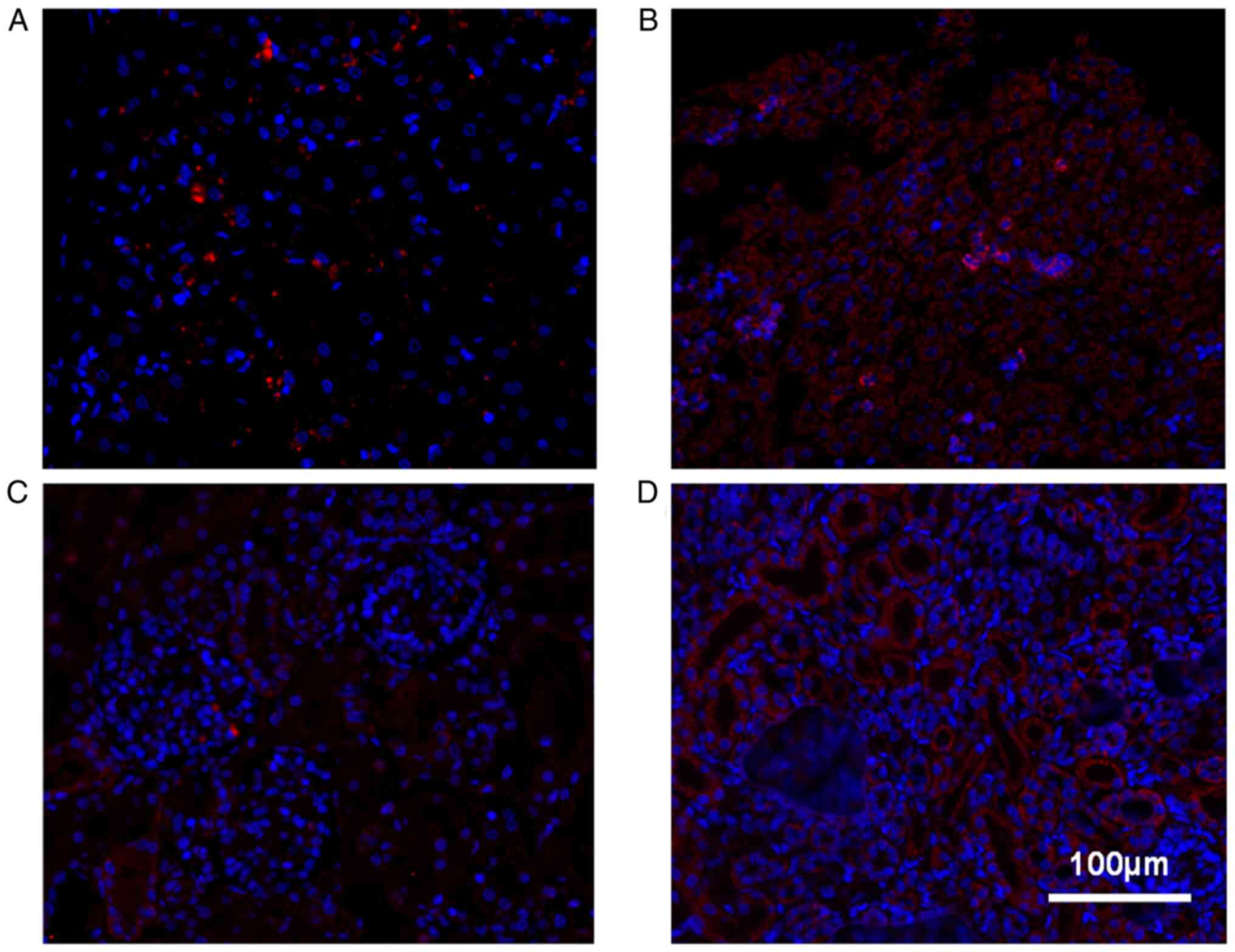
Furthermore, immunofluorescent staining with an EFG1 antibody
demonstrated markedly decreased protein expression levels of the
GFM1 gene in both the liver and renal tissues from the index
patient compared with the control (Fig.
3A-D).
Mutation identification and protein
prediction
The whole-exome sequencing and following variant
validation using Sanger sequencing, in both the amniotic fluid of
the index patient and the blood of his female older sibling,
demonstrated that these two patients carried 2 heterozygous
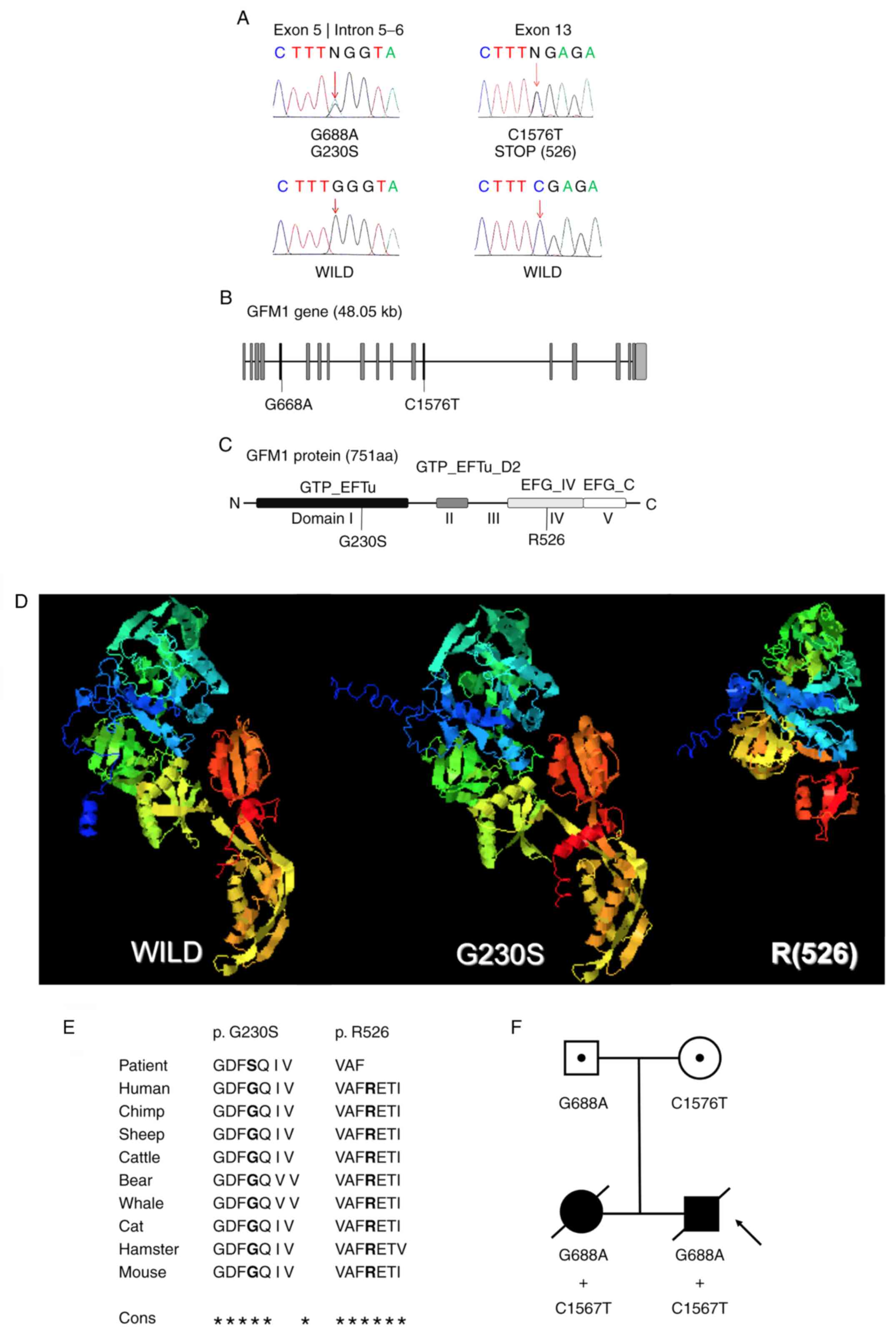
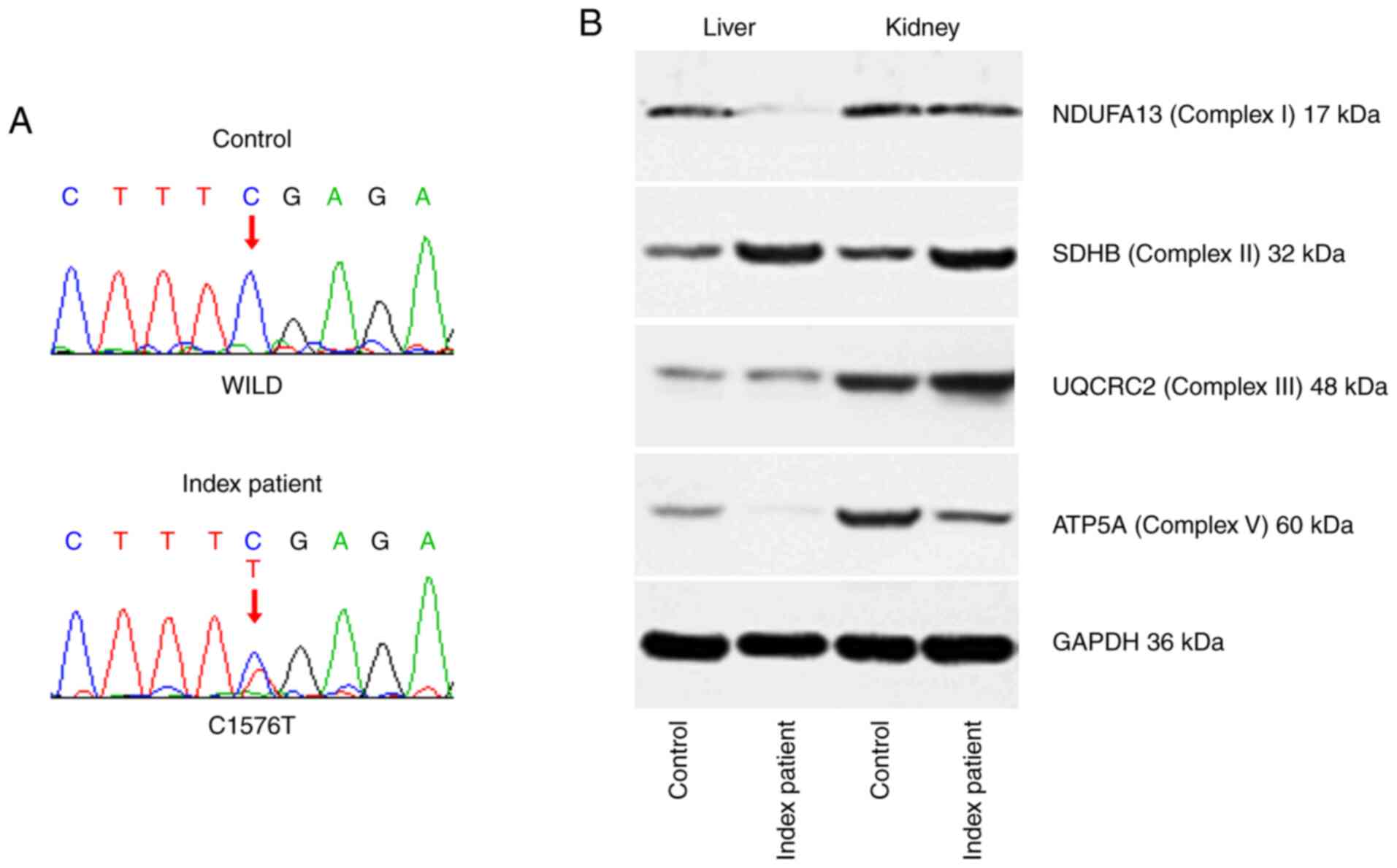
mutations in the GFM1 gene (NM_001308166.1): A G688A mutation in
the boundary between exon 5 and intron 5-6 (Fig. 4A and B), predicting a glycine to serine
substitution at position 230 (Fig.
4C); and a C1576T mutation in exon 13 (Fig. 4A and B), which resulted in a premature stop
codon at amino acid position 526 (Fig.
4C). These 2 aforementioned positions have been completely
conserved across multiple species (Fig.
4E). Molecular modeling indicated that the G230S substitution
changed the secondary and spatial structures in the α-helices and
β-sheets of the GTP-binding domain I and the R526 premature stop
codon resulted in the loss of the domains IV and V (Fig. 4D) in the EFG1 protein. To understand
the origin of the mutations in these patients, genome-wide DNA
sequencing from the peripheral blood of both their parents was
performed. The results identified that the father was heterozygous
for the G688A mutation and wild-type for the C1576T mutation, while
the mother was heterozygous for the C1576T mutation and wild-type
for the G688A mutation (Fig.
4F).
Effects of the C1576T mutation on GFM1
mRNA variants of the index patient
To examine if the mutant transcript was still
present, reverse transcription-PCR followed by sequencing was
performed in liver and kidney tissues, and the results identified
that the index patient carried the wild-type and mutant GFM1 mRNA
variants associated with the c.1576C>T change in a heterozygous
manner (Fig. 5A).
Effects of the GFM1 mutations on
protein expression in the OXPHOS complex of the index patient
To investigate the subsequent effects of the
heterozygous GFM1 mutations on the expression level changes in
representative proteins from the various OXPHOS complexes, western
blot analysis was performed using liver and kidney tissues from the
index patient, as presented in Fig.
5B. The protein expression levels of the nuclear-encoded OXPHOS
complex I protein NDUFA13 were decreased in the liver tissues;
however, they were unchanged in the kidney tissues. The protein
expression levels of SDHB and the core protein subunit, UQCRC2,
from the OXPHOS complex III, were not changed in either the liver
or the kidney, while ATP5A expression levels were decreased in both
the liver and kidney tissues.
Discussion
The association between GFM1 gene defects and the
recessively inherited mitochondrial disorder of oxidative
phosphorylation was first reported 15 years ago (3). Over the past decade, the number of
families identified to have mutations in this gene is only 25
(3-13).
These patients demonstrated diverse phenotypes; however, the most
prevalent clinical feature was liver disease, lactic acidosis,
encephalopathy, feeding problems and failure to thrive (11). In the present study, a family with
GFM1 mutations has been described; two siblings who were compound
heterozygous for two missense mutations: C1576T, which, to the best
of our knowledge; was reported for the first time; and G688A, which
expands the range of GFM1 variants to 28.
In the literature (3-13),
the majority of patients with GFM1 variants presented with fetal
intrauterine growth retardation or developmental delay after birth.
A variable neurological manifestation was identified from the
beginning stage of life, including hypotonia, dystonia and feeding
difficulties, as well as corresponding brain neuroimaging
abnormalities. Liver involvement was considered to be an important
diagnostic hallmark of GFM1-associated diseases; however, it has
not been observed in an increasing number of recently reported
patients with GFM1 mutations (4,5,8,12).
The index patient in the present study developed the most common
developmental, nervous and hepatic features, in concordance with
previously reported cases (3-7,9-13).
Notably, he also presented with structural alterations in the
kidneys, with functional compensation, which has not been mentioned
in previous case studies.
Apart from 2 cases (9,11), all
the patients with GFM1 defects did not survive beyond infancy. In
the present study, the index patient received a genetic diagnosis
prior to birth and high-quality medical care in a university
hospital and lived for >3 months, while his older sister only
lived for 1 week, even though both siblings carried the same GFM1
mutations. The different ages of death in the two siblings was
primarily due to intrafamilial variability, which was consistent
with previous reports (3,11). In addition, as the clinical and
laboratorial data of the female sibling were not adequate, we
hypothesized that the difference in severity between the two
siblings was also partly due to timeliness and the level of medical
care they received. Furthermore, preimplantation genetic diagnosis
was recommended to the parents in the present study for their next
pregnancy.
As a GTPase, the EFG1 protein catalyzes the delivery
of peptidyl-tRNA from the ribosomal A site to the P site, following
the formation of the peptide bond (6). EFG1 has been demonstrated to interact
with both 50S large ribosomal subunit, near the L7/L12 stalk and
the sarcin-ricin region of 23S rRNA (17). Only loss of both functional alleles
could cause the mitochondrial translation defect in patients
(13). EFG1 consists of 751 amino
acids and includes 5 Pfam domains (10). The predictive data in the present
study revealed that the two patients were heterozygous for a
missense allele, which produces a conformational change in the
GTP-binding domain I, and a non-sense allele, which has been
predicted to cause an elimination of part of domain IV and the
whole of domain V. The G688A mutation was reported by
Balasubramaniam et al in 2012(7); however, to the best of our knowledge,
the C1576T mutation was identified for the first time in the
present study (7). On one hand, the
G230 substitution is located in the GTP EFTu domain I, a GTP
binding domain, which is exposed to conformational changes mediated
by the hydrolysis of GTP to GDP (10); on the other hand, the R526 premature
stop codon is located in the EFG IV domain, which appears to be
essential for the extensive structural rearrangement for tRNA-mRNA
movement to occur, through extension similar to a lever arm
(18). Therefore, we hypothesized
that, if translated, it would generate a truncated polypeptide
without a functional C-terminal block, as domains IV and V form the
C-terminal block of EFG (18). In
the present study, the pathogenicity of the novel mutation is
theoretically supported by molecular modeling; the existence of the
mutant GFM1 transcript associated with the c.1576C>T change was
confirmed. The subsequent effects of the GFM1 protein on the
expression levels of representative proteins in various OXPHOS
complexes were further determined. The present study demonstrated
that the levels of NDUFA13 were decreased in the liver; however,
they were unchanged in the kidney. This observation was not
surprising given that the nuclear-encoded OXPHOS complex I protein
NDUFA13 is unstable during complex I assembly and is compromised in
mitochondrial translation deficiencies (12), possibly in a tissue-specific manner.
Consistent with previous observations (11), the expression level of the OXPHOS
complex II protein SDHB was not attenuated in the present study,
due to the exclusive nuclear genetic origin of the complex. The
levels of UQCRC2, the core protein subunit from the OXPHOS complex
III, were unaffected, as the only mtDNA polypeptide in complex III
is cytochrome b (1). However, the
ATP5A expression level was decreased in both the liver and kidney
tissues in the index patient, while levels were reported to be
stable in fibroblasts of a previous patient with a GFM1 mutation
(12). Unfortunately, further
research to clarify the activity of the OXPHOS complexes could not
be conducted, as the samples of the index patient were not
adequate.
In summary, the present study provided novel data
for the mutational spectrum of GFM1 and additional patient
phenotype information. Early genetic diagnosis and corresponding
treatment would lead to an improved prognosis of this recessively
inherited mitochondrial disorder.
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant no. 81873837 to FW), the
Zhejiang Traditional Chinese Medicine Foundation (grant no.
2015ZQ025 to FW) and the Zhejiang Provincial Natural Science
Foundation of China (grant no. LQ18H040005).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
FW made substantial contributions to the conception
and design of the study. CS performed the data acquisition,
interpretation and analysis of the data. CS and FW were involved in
data interpretation and drafting and revising the manuscript. FW
gave final approval to the version to be published. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Written informed consent and consent for publication
to use samples and clinical data were obtained from the parents and
the present study was formally approved by the Ethics Committee of
The Women's Hospital, Zhejiang University School of Medicine,
Zhejiang, China.
Patient consent for publication
Written consent for publication was obtained from
all the parents.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wallace DC: A mitochondrial bioenergetic
etiology of disease. J Clin Invest. 123:1405–1412. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
Christian BE and Spremulli LL: Mechanism
of protein biosynthesis in mammalian mitochondria. Biochim Biophys
Acta. 1819:1035–1054. 2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Coenen MJ, Antonicka H, Ugalde C, Sasarman
F, Rossi R, Heister JG, Newbold RF, Trijbels FJ, van den Heuvel LP,
Shoubridge EA and Smeitink JA: Mutant mitochondrial elongation
factor G1 and combined oxidative phosphorylation deficiency. N Engl
J Med. 351:2080–2086. 2004.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Valente L, Tiranti V, Marsano RM, Malfatti
E, Fernandez-Vizarra E, Donnini C, Mereghetti P, De Gioia L,
Burlina A, Castellan C, et al: Infantile encephalopathy and
defective mitochondrial DNA translation in patients with mutations
of mitochondrial elongation factors EFG1 and EFTu. Am J Hum Genet.
80:44–58. 2007.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Smits P, Antonicka H, van Hasselt PM,
Weraarpachai W, Haller W, Schreurs M, Venselaar H, Rodenburg RJ,
Smeitink JA and van den Heuvel LP: Mutation in subdomain G' of
mitochondrial elongation factor G1 is associated with combined
OXPHOS deficiency in fibroblasts but not in muscle. Eur J Hum
Genet. 19:275–279. 2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Antonicka H, Sasarman F, Kennaway NG and
Shoubridge EA: The molecular basis for tissue specificity of the
oxidative phosphorylation deficiencies in patients with mutations
in the mitochondrial translation factor EFG1. Hum Mol Genet.
15:1835–1846. 2006.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Balasubramaniam S, Choy YS, Talib A,
Norsiah MD, van den Heuvel LP and Rodenburg RJ: Infantile
progressive hepatoencephalomyopathy with combined OXPHOS deficiency
due to mutations in the mitochondrial translation elongation factor
gene GFM1. JIMD Rep. 5:113–122. 2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Calvo SE, Compton AG, Hershman SG, Lim SC,
Lieber DS, Tucker EJ, Laskowski A, Garone C, Liu S, Jaffe DB, et
al: Molecular diagnosis of infantile mitochondrial disease with
targeted next-generation sequencing. Sci Transl Med.
4(118ra110)2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Brito S, Thompson K, Campistol J, Colomer
J, Hardy SA, He L, Fernández-Marmiesse A, Palacios L, Jou C,
Jiménez-Mallebrera C, et al: Corrigendum: Long-term survival in a
child with severe encephalopathy, multiple respiratory chain
deficiency and GFM1 mutations. Front Genet. 6(254)2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ravn K, Schonewolf-Greulich B, Hansen RM,
Bohr AH, Duno M, Wibrand F and Ostergaard E: Neonatal mitochondrial
hepatoencephalopathy caused by novel GFM1 mutations. Mol Genet
Metab Rep. 3:5–10. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Simon MT, Ng BG, Friederich MW, Wang RY,
Boyer M, Kircher M, Collard R, Buckingham KJ, Chang R, Shendure J,
et al: Activation of a cryptic splice site in the mitochondrial
elongation factor GFM1 causes combined OXPHOS deficiency.
Mitochondrion. 34:84–90. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Barcia G, Rio M, Assouline Z, Zangarelli
C, Gueguen N, Dumas VD, Marcorelles P, Schiff M, Slama A, Barth M,
et al: Clinical, neuroimaging and biochemical findings in patients
and patient fibroblasts expressing ten novel GFM1 mutations. Hum
Mutat. 41:397–402. 2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Bravo-Alonso I, Navarrete R, Vega AI,
Ruíz-Sala P, García Silva MT, Martín-Hernández E, Quijada-Fraile P,
Belanger-Quintana A, Stanescu S, Bueno M, et al: Genes and variants
underlying human congenital lactic acidosis-from genetics to
personalized treatment. J Clin Med. 8(1811)2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wang X, Liu A, Lu Y and Hu Q: Novel
compound heterozygous mutations in the SPTA1 gene, causing
hereditary spherocytosis in a neonate with Coombs-negative
hemolytic jaundice. Mol Med Rep. 19:2801–2807. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wang F, Pan J, Liu Y, Meng Q, Lv P, Qu F,
Ding GL, Klausen C, Leung PC, Chan HC, et al: Alternative splicing
of the androgen receptor in polycystic ovary syndrome. Proc Natl
Acad Sci USA. 112:4743–4748. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Notredame C, Higgins DG and Heringa J:
T-Coffee: A novel method for fast and accurate multiple sequence
alignment. J Mol Biol. 302:205–217. 2000.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Sergiev PV, Bogdanov AA and Dontsova OA:
How can elongation factors EF-G and EF-Tu discriminate the
functional state of the ribosome using the same binding site? FEBS
Lett. 579:5439–5442. 2005.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Salsi E, Farah E, Dann J and Ermolenko DN:
Following movement of domain IV of elongation factor G during
ribosomal translocation. Proc Natl Acad Sci USA. 111:15060–15065.
2014.PubMed/NCBI View Article : Google Scholar
|