Introduction
The brain is the most sensitive organ to ischemia
and hypoxia, and cerebral ischemia can lead to necrosis or
apoptosis of brain cells. Timely thrombolysis, rapid and effective
reconstruction of collateral circulation of microvessels and
recovery of blood reperfusion in ischemic regions and the penumbra
are the best treatment for cerebral ischemia (1). However, recanalization of blood flow
after ischemia can lead to ischemia/reperfusion injury. During
ischemic/reperfusion, there are a large number of inflammatory
factors synthesized and secreted in the ischemic area. The
activation and infiltration of inflammatory cells, and the
synthesis and secretion of adhesion molecules are part of a
positive feedback cascade that enhance and promote each other
(2). These responses signal through
a specific inflammatory signaling pathway, converting ischemic
brain tissues into inflammatory lesions (3,4).
Therefore, inflammation plays an important role in the mechanism of
cerebral ischemia/reperfusion injury.
Sphingomyelin (SM) is an important component of cell
membranes, mainly located in cell membranes, lipoproteins and other
lipid-rich tissue structures. SM is widely found in biological
tissues and is abundant in brain tissue (5). Previous studies have shown that
products and key enzymes in the metabolism of SM are involved in
many aspects of cerebral ischemia (6,7) and
play an important role in the development of cerebral ischemia
(8,9). Therefore, the regulation of the SM
metabolic pathway is expected to be a new target for the treatment
of ischemic cerebrovascular disease.
SM synthase (SMS) is the last enzyme in the SM
synthesis pathway. SMS has two isoenzymes, SMS1 and SMS2(10). SMS1 mainly exists in the Golgi
apparatus, and SMS2 mainly exists in the plasma membrane, with SMS2
being widely expressed in brain tissue (10).
Previous studies have found that SMS2 is an
important regulatory factor in inflammatory responses (11,12).
SMS2 deficiency inhibits the activation of NF-κB pathways in
macrophages induced by lipopolysaccharide (LPS) (13), which intimates that SMS2 may be
involved in the regulation of inflammatory responses. However, the
effect and mechanism of action of SMS2 on the inflammatory response
after cerebral ischemia/reperfusion injury are still unclear, and
remains to be further studied. Therefore, this present study used
wild-type and SMS2 knockout C57BL/6 mice as research subjects to
explore the role of SMS2 in cerebral ischemia/reperfusion injury in
mice, and to study its specific molecular mechanisms.
Materials and methods
Animals and experimental groups
Male wild-type (WT) C57BL/6 mice (8-12 weeks; 25-30
g; Cyagen Biosciences, Inc.) and SMS2 knockout (SMS2-/-)
mice (Cyagen Biosciences, Inc.) in the background of wild-type
C57BL/6 mice (8-12 weeks; 25-30 g) were acclimatized for 1 week at
room temperature (20-24˚C) with free access to water/food and
45-60% humidity in a 12 h light/dark cycle. A total of 26 WT mice
and 26 SMS2-/- were used in the present study. Mice were divided
into two subgroups: Sham group and middle cerebral artery occlusion
(MCAO) group. The present study was performed with the approval of
the Ethics Committee of The Third People's Hospital of Qingdao.
Cerebral ischemia/reperfusion
model
Longa's method (14)
was used to establish the cerebral ischemia/reperfusion mouse
model. Mice were injected intraperitoneally with 4% chloral hydrate
(Sinopharm Chemical Reagent Co., Ltd.) at a ratio of 350 mg/kg, for
anesthesia. Mice were fixed in a supine position and monitored to
ensure breathing was unobstructed. Mouse necks were sterilized with
an ethanol-soaked cotton ball and a 1-cm incision was made in the
neck. Under a stereo microscope, the submandibular glands were
dissected, the right common carotid artery, external and internal
carotid arteries were freed up, and the common carotid artery was
tied with a thread. At the bifurcation of the external carotid
artery and the common carotid artery, a loose knot was made with
silk; the distal end of the external carotid artery was ligated
with silk, and the external carotid artery was fused with an
electric coagulation pen. The internal carotid artery was fastened
with a silk thread, the external carotid artery was cut with
microscopic surgical scissors to insert a thread plug through the
small hole to the external carotid artery, and the line knot on the
internal carotid artery was loosened. The plug was slowly inserted
in the direction of the internal carotid artery until the
resistance was reached. To prevent bleeding, the external carotid
artery was tied up. After 2 h of ischemia, the plug was pulled out
and the arterial blood was allowed to circulate again for 24 h. The
surgical procedure in the sham group was the same as before,
however the line plug was not inserted. Post-surgery, animals were
maintained at 25-28˚C, with access to food and water ad
libitum.
Evaluation of neurological
deficits
The neurological deficit in the mice was evaluated
with reference to Longa's method (14): 0 points, no symptoms of neurological
deficits and normal activity; 1 point, contralateral forelimbs
cannot fully extend; 2 points, circling to the contralateral side
when crawling; 3 points, body dumping to the hemiparalysis side
when walking; 4 points, not autonomous walking and loss of
consciousness was 4 points; 5 points, death. If the score was 1-4,
the model was considered successful.
Determination of infarct volume
Magnetic resonance imaging (MRI) and
2,3,5-triphenyltetrazolium chloride (TTC) staining were used to
detect the infarct volume.
MRI
Mice were injected intraperitoneally with 4% chloral
hydrate at 350 mg/kg to be anesthetized. The Phillips
GyroscanIntera 1.5T MRI (Phillips Medical Systems B. V.) and the
rat coil (Shanghai Chenguang Medical Technology Co., Ltd.) were
used for fast spin echo T2-weighted (FSE T2W) imaging. The
parameters used were as follows: Time of Repetition was 1,600 msec;
Time of Echo was 80 msec; slice thickness was 1 mm; slice gap was
0; Field of View was 35x35 mm; and matrices were 256x256. ImageJ
software (version 1.41; National Institutes of Health) was used to
measure the area of cerebral infarction per layer. The percentage
cerebral infarction volume was calculated via the following
formula: Percentage cerebral infarction volume=(S1 + S2 + S3 + SN)
H/(S1 total + S2 total + S3 total + SN total), where S1-SN were the
infarct sizes of each layer, S1 total-SN total were the areas of
brain tissue in each layer, and H was slice thickness (15).
TTC staining
Mice were sacrificed immediately after MRI scans,
and the brains were dissected immediately. The brain tissue of mice
was frozen for 20 min in a -20˚C refrigerator, and then cut into
2-mm-thick continuous slices. Slices were incubated at 37˚C for
15-30 min with 2% TTC (Sigma-Aldrich; Merck KGaA) in a darkroom.
They were then fixed with 4% paraformaldehyde (Sinopharm Chemical
Reagent Co., Ltd.) at room temperature for 15 min. ImageJ software
was used to assess the area of cerebral infarction per layer
(16). The cerebral infarction
volume was calculated by multiplying the infarct size of each brain
slice by the thickness (2 mm) using a Leica TCS SP5 fluorescent
microscope (Leica Microsystems, GmbH).
Reverse transcription-quantitative PCR
(RT-qPCR)
Each group of mice was anesthetized with 4% chloral
hydrate at 24 and 72 h after the surgery. The mice were decapitated
and the infarcted penumbra was harvested under aseptic conditions.
Brain tissue was rapidly homogenized in RNAiso Plus (Takara
Biotechnology Co., Ltd.) within a glass homogenizer (Shanghai
Broadcom Chemical Technology Co., Ltd.). Total RNA from brain
tissue was extracted according to the TRIzol RNA total extraction
method. RNA was dissolved in diethylpyrocarbonate-treated water
(Takara Biotechnology Co., Ltd.), and RNA concentration was
measured with a NanoDrop 2,000 Ultramicro Spectrophotometer (Thermo
Fisher Scientific, Inc.). The extracted RNA was reverse transcribed
into cDNA by using a PrimeScript™ RT master mix reverse
transcription kit (cat. no. RR036B; Takara Biotechnology Co.,
Ltd.). Reverse transcriptase parameters were as follows: 37˚C for
60 min, 85˚C for 5 sec. RNA samples (20 µl) were prepared with SYBR
Green according to the SYBR-Green qPCR Master Mix kit instructions
(cat. no. 638320; Takara Biotechnology Co., Ltd.) and amplified
using the Applied Biosciences 7500 fluorescence PCR system (Thermo
Fisher Scientific, Inc.). PCR parameters were as follows: 95˚C for
30 sec, 90˚C for 5 sec, 65˚C for 30 sec, for 40 cycles. β-actin was
used as the internal control and the relative expression level of
the target gene was calculated by 2-ΔΔCq method
(17). The primers used were as
follows: Galectin forward, 5'-TTTCAGGAGAGGGAATGATGTTG-3' and
reverse, 5'-CACAATGACTCTCCTGTTGTTCTCA-3'; arginase 1 (Arg1)
forward, 5'-CTCCAAGCCAAAGTCCTTAGAG-3' and reverse,
5'-GGAGCTGTCATTAGGGACATCA-3'; inducible nitric oxide synthase
(iNOS) forward, 5'-CTCTTCGACGACCCAGAAAAC-3' and reverse,
5'-CAAGGCCATGAAGTGAGGCTT-3'; IL-1β forward,
5'-GAAATGCCACCTTTTGACAGTG-3' and reverse,
5'-TGGATGCTCTCATCAGGACAG-3'; β-actin forward,
5'-TCACCCACACTGTGCCCATCTACG-3' and reverse,
5'-CAGCGGAACCGCTCATTGCCAATG-3'.
Western blotting
Brain tissue was submerged in RIPA lysate,
containing 1 mM PMSF (Beyotime Institute of Biotechnology) and
rapidly homogenized within a glass homogenizer, then placed on ice
for 10 min, and centrifuged at 13,800 x g for 10 min and the
supernatant was isolated, which contained the total tissue
protein.
Harvest buffer [10 mmol/l HEPES, pH 7.9; 50 mmol/l
NaCl; 1 mmol/l EDTA; 0.5% Triton X-100; 1 mmol/l dithiothreitol
(DTT), 0.5 mol/l sucrose, 1 mmol/l PMSF] and brain tissue were
homogenized within a glass homogenizer, and then placed on ice for
10 min, centrifuged at 13,800 x g for 10 min at room temperature
and the supernatant was isolated, which contained the cytoplasmic
proteins. Centrifugal sediment was washed with buffer A (10 mmol/l
HEPES, pH 7.9; 10 mmol/l KCl; 0.1 mmol/l EDTA; 0.1 mmol/l EGTA; 1
mmol/l DTT; 1 mmol/l PMSF). This solution was centrifuged (13,800 x
g for 10 min at room temperature) again and the supernatant was
removed. The supernatant was resuspended in buffer C (10 mmol/l
HEPES, pH 7.9; 500 mmol/l NaCl; 0.1 mmol/l EDTA; 0.1 mmol/l EGTA;
0.1% Igepal; 1 mmol/l DTT; 1 mmol/l PMSF), and then placed on ice
for 15 min. The solution was centrifuged at 13,800 x g for 10 min
at room temperature and the supernatant was isolated, which
contained the nuclear protein.
Buffer solution containing 20% SDS was added to the
supernatant until the final concentration of SDS was 1%, and the
mixture was boiled at 100˚C for 5 min. The concentration of protein
was determined by bicinchoninic acid protein concentration assay
kit (Beyotime Institute of Biotechnology). A mass of 75 µg of total
protein was loaded into each lane and separated by 15% SDS-PAGE (90
V for 0.5 h; 120 V for 1 h) and transferred (400 mA for 1.5 h) to
polyvinylidene fluoride film (GE Healthcare Life Sciences), fixed
with methanol for 1 min at room temperature, washed three times (5
min each) with TBST (10 mM Tris-HCl; 150 mM NaCl; 0.1% Tween-20, pH
7.6) and blocked with blocking buffer (5% skim milk in TBST) for 1
h at room temperature. Anti-galectin 3 (cat. no. ab76245; Abcam;
1:1,000), anti-NF-κB-p65 (cat. no. ab16502; Abcam; 1:1,000),
anti-lamin A (cat. no. ab8980; Abcam; 1:1,000), or anti-β-actin
antibody (cat. no. ab8227; Abcam; 1:2,000) diluted in 5% skim milk
was added and incubated at 4˚C overnight, followed by washing with
TBST three times (10 min each). Sheep anti-rabbit secondary
antibody (cat. no. ab205718; Abcam; 1:2,000) or sheep anti-mouse
secondary antibody (cat. no. ab6808; Abcam; 1:2,000) was added and
incubated for 1 h at room temperature, and then ECL solution
(Sigma-Aldrich; Merck KGaA) was added for detection. The expression
of the target protein was analyzed by ImageJ software (National
Institutes of Health), and the relative expression level of target
protein was characterized by the gray value of the target
protein/gray value of β-actin or lamin A protein.
Immunofluorescence
Each group of mice was anesthetized with 4% chloral
hydrate at 24 and 72 h after surgery. The chest was cut to expose
the heart. The injection needle was then inserted into the left
ventricle and the right atrial appendage was cut. Rapid perfusion
with 100 ml normal saline 100 was performed, followed by perfusion
and fixation with 200 ml of 4% paraformaldehyde. The brain was
dissected immediately after perfusion. The brain tissue was fixed
in a 4% paraformaldehyde solution for 24 h at 4˚C, dehydrated by
sucrose gradient treatment, embedded in optimal cutting temperature
compound at 4˚C (Beijing Transgen Biotech Co., Ltd.), and serially
sectioned with a cryostat, to a thickness of 30 µm.
Brain tissue sections were removed and left at room
temperature for 20 min. After drying, the areas of
immunohistochemistry were marked. Rinsing was performed three times
(5 min each) with PBS solution (135 mM NaCl, 2.7 mM KCl, 1.5 mM
KH2PO4, 8 mM K2HPO4,
pH=7.2) at room temperature. After being blocked with 100% goat
serum (HyClone; GE Healthcare Life Sciences) at 37˚C for 1 h, the
goat serum was removed and sections were incubated with
anti-galectin 3 (cat. no. ab76245; Abcam; 1:1,000) and
anti-allograft inflammatory factor 1 (Iba 1) antibody (cat. no.
ab178847; Abcam; 1:100) at 4˚C overnight. Rinsing was performed
three times (5 min each) with PBS solution at room temperature in
the dark. Sections were then incubated with the goat anti-mouse
secondary antibody-Alexa Fluor Plus 488 (cat. no. A32723; 1:500;
Thermo Fisher Scientific, Inc.) or goat anti-mouse secondary
antibody-Alexa Fluor Plus 594 (cat. no. A32742; 1:500; Thermo
Fisher Scientific, Inc.) at 37˚C for 1 h, rinsed three times (5 min
each) with PBS at room temperature in dark, mounted and observed
with a fluorescence microscope (Carl Zeiss AG).
Statistical analysis
The data were analyzed with the SPSS 20.0 software
package (IBM Corp.) and the data are expressed as the mean ±
standard deviation. Student's t-tests were used to compare the
differences between two groups. One-way ANOVA with Duncan's
post-hoc test was used for comparing multiple groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
Lack of SMS2 attenuates cerebral
ischemia/reperfusion injury in mice
The neurological deficit score in different mice at
24 or 72 h after MCAO was evaluated by the Longa grading criteria
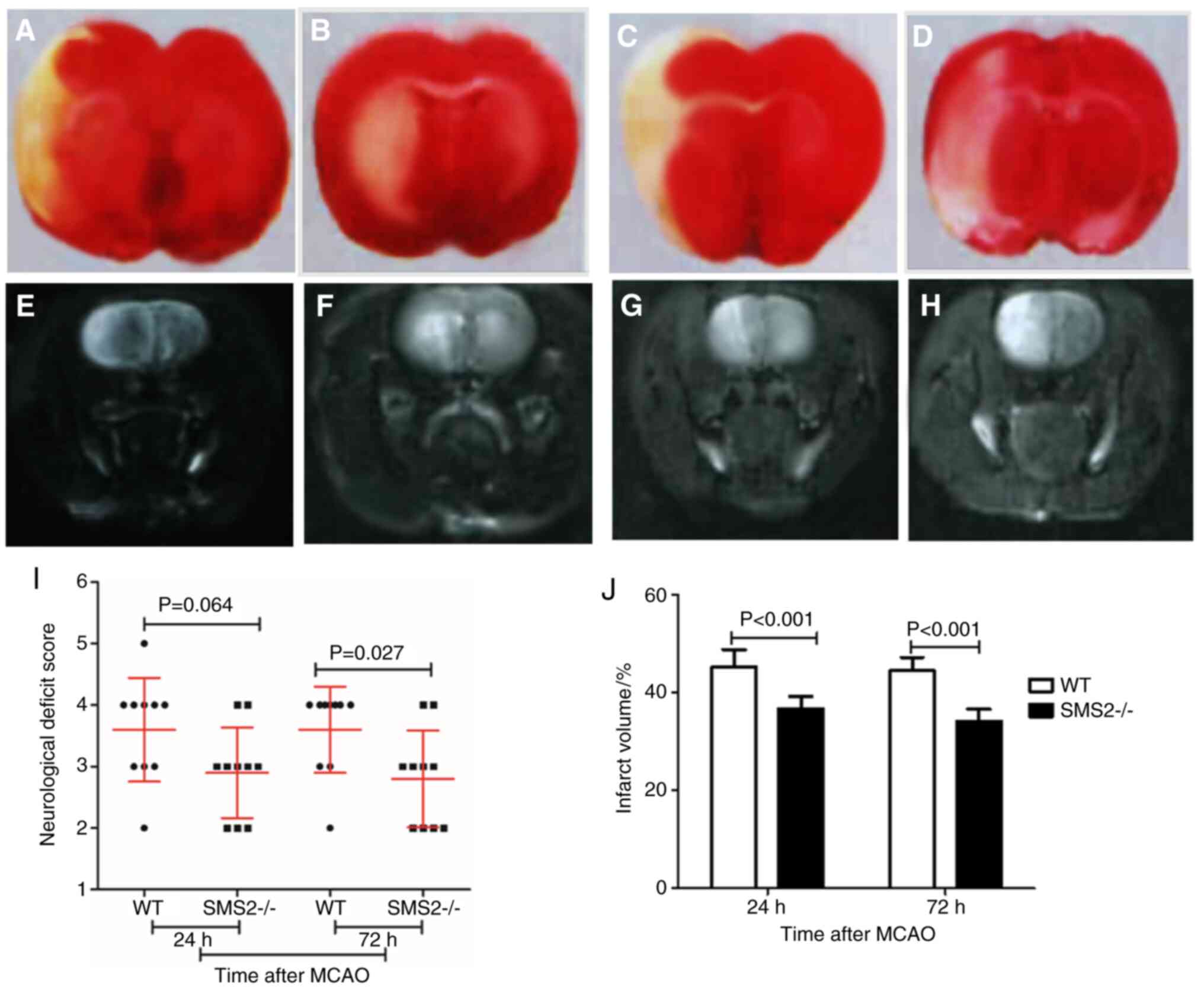
(14). As shown in Fig. 1I, there was no significant
difference in neurological deficit score between the two groups of
mice at 24 h after cerebral ischemia/reperfusion injury (P=0.064),
but the neurological deficit score was significantly lower in
SMS2-/- mice compared with WT mice at 72 h after
cerebral ischemia/reperfusion injury (P=0.027).
TTC staining and MRI was used to detect the infarct
volume of MCAO mice after cerebral ischemia/reperfusion. After TTC
staining, the normal brain tissue was uniformly red and infarcted
brain tissue was white (Fig. 1A-D).
With MRI, infarcts showed high signal through FSE T2W imaging
(Fig. 1E-H). At 24 and 72 h after
cerebral ischemia/reperfusion, TTC staining showed a wide range of
white infarct areas in the infarct cortex and basal ganglia, which
was same as MRI in WT and SMS2-/2 mice. Infarct volumes of
SMS2-/- MCAO mice were significantly smaller than those
of WT MCAO mice (P<0.05; Fig.
1J).
Lack of SMS2 attenuates the expression
of inflammatory mediators
The inflammatory pathways that are mediated by the
Toll-like receptor (TLR) family play an important role in the
inflammatory response induced by cerebral ischemia (18). Galectin 3, an endogenous ligand for
TLR4(19), plays an important role
in the inflammatory response after organ ischemia (20,21).
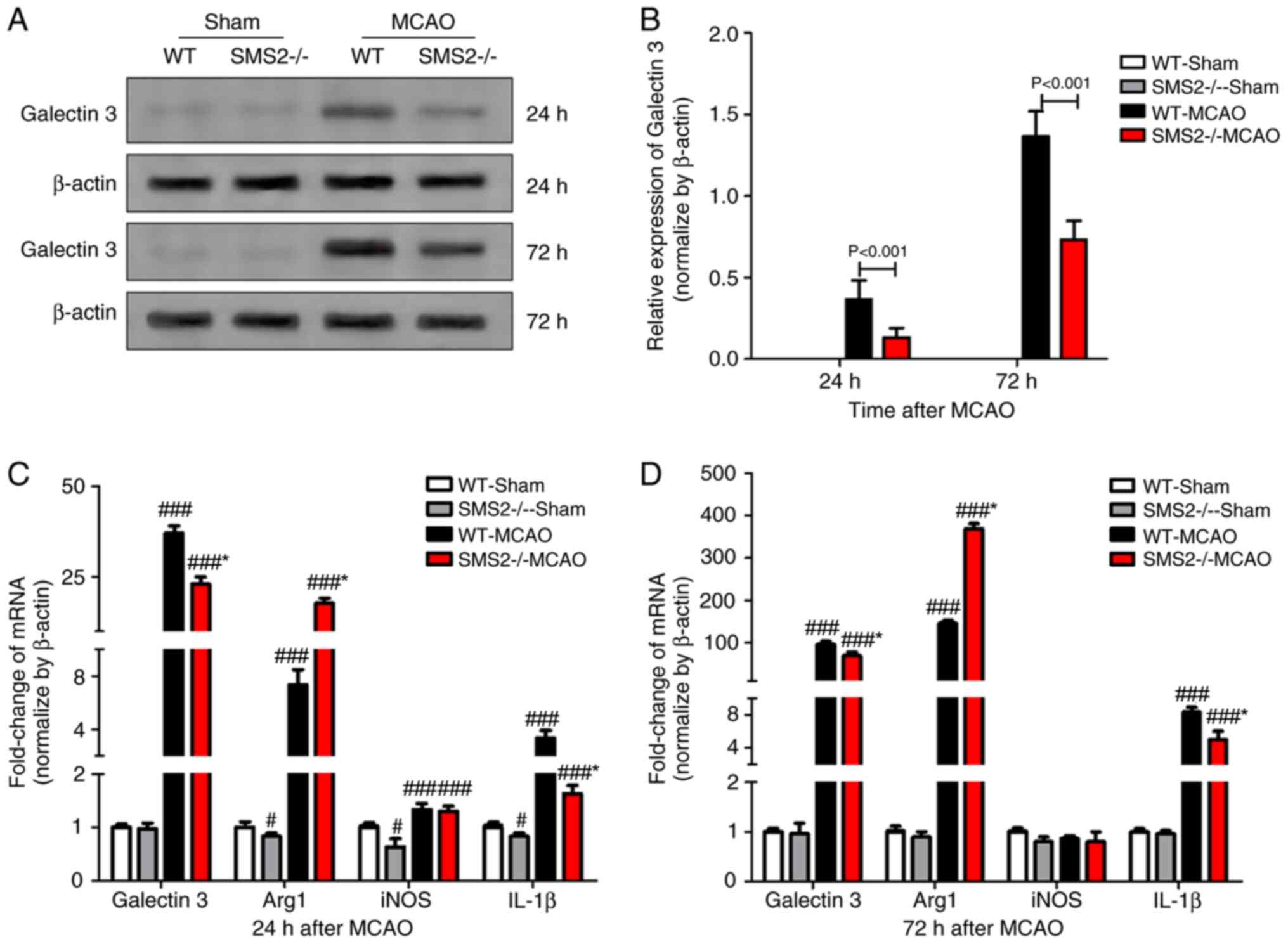
Western blotting was used to detect the expression of galectin 3
protein, and the results showed that galectin 3 protein expression
was increased at 24 and 72 h after cerebral ischemia/reperfusion in
mice compared to sham mice, with SMS2-/- group MCAO mice
expressing significantly lower levels of galectin 3 than those in
the WT group MCAO mice (P<0.001; Fig. 2A and B).
Moreover, the expression of inflammatory mediators
of cerebral ischemia/reperfusion in mice was detected by RT-qPCR
(Fig. 2C and D). At 24 and 72 h after cerebral
ischemia/reperfusion in mice, the expression of pro-inflammatory
cytokines galectin 3 and IL-1β was significantly increased
(P<0.001), and the SMS2-/- group MCAO mice expressed
significantly lower levels of both mRNAs than the WT group MCAO
mice at both time points (P<0.001). The expression of
anti-inflammatory factor Arg 1 was significantly increased after
cerebral ischemia/reperfusion (P<0.001), and the
SMS2-/- group MCAO mice expressed significantly higher
levels than the WT group MCAO mice at both time points
(P<0.001). While anti-inflammatory factor iNOS was also
significantly increased (P<0.001) at 24 h after
ischemia/reperfusion, there was no significant difference in iNOS
mRNA levels at 72 h in either the WT or SMS-/- groups
(P>0.05).
Lack of SMS2 inhibits the activation
of microglia
Microglia are activated soon after cerebral
ischemia/reperfusion injury and are the main cells that cause
serious inflammation (22,23). Iba 1 is a protein that is
specifically expressed on microglia (24,25).
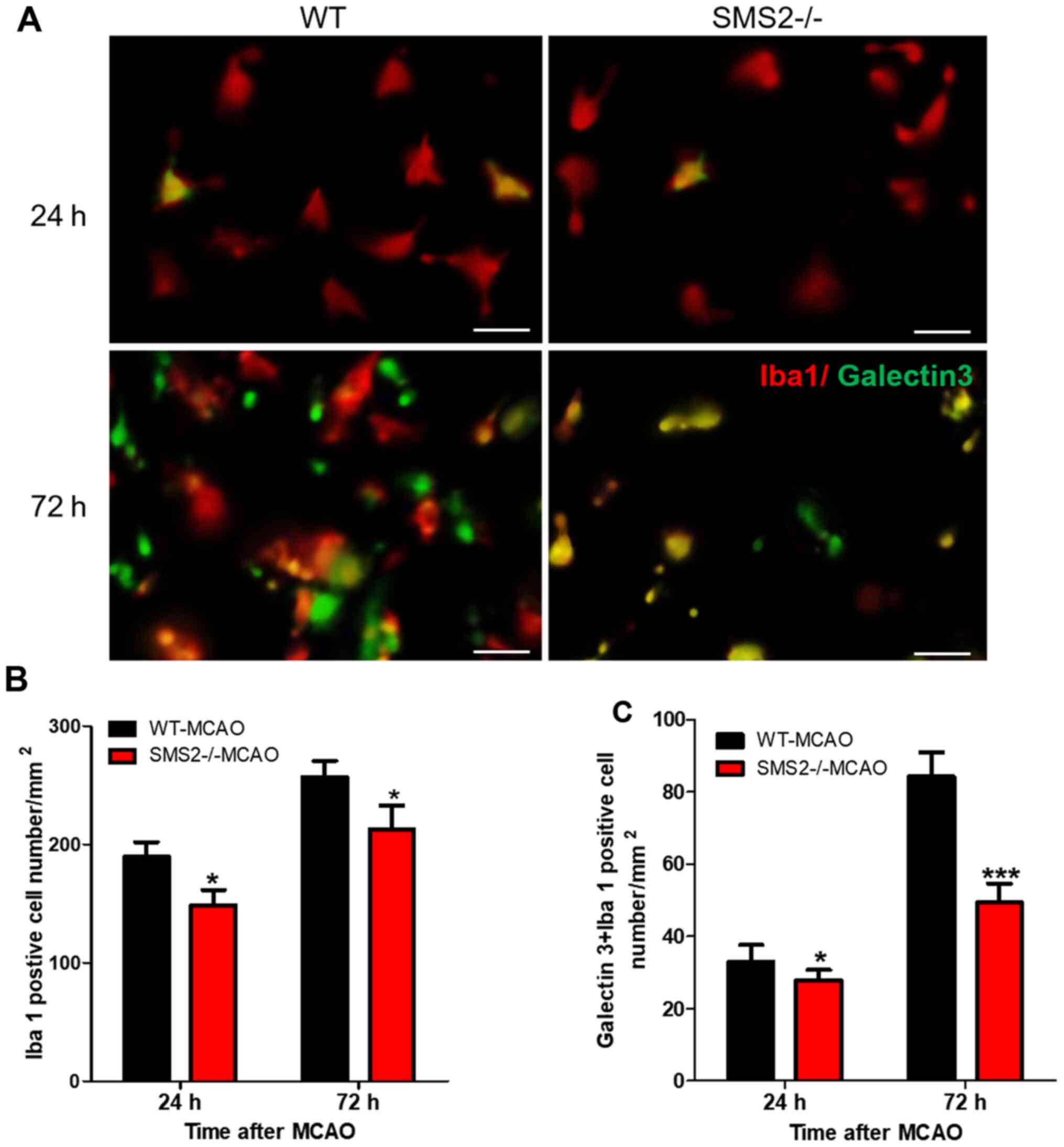
Therefore, immunofluorescence staining was used to count the number
of Iba 1-positive cells, which was used to characterize the
activation of microglia after cerebral ischemia/reperfusion. As
shown in Fig. 3, the number of Iba
1+ cells in SMS2-/- mice was significantly lower than
that in WT mice following MCAO at both 24 and 72 h (P<0.05).
Similarly, the number of galectin 3+/Iba 1+
cells significantly decreased at 24 and 72 h after cerebral
ischemia/reperfusion (P<0.05 and P<0.001 respectively).
Lack of SMS2 inhibits the activation
of the NF-κB pathway
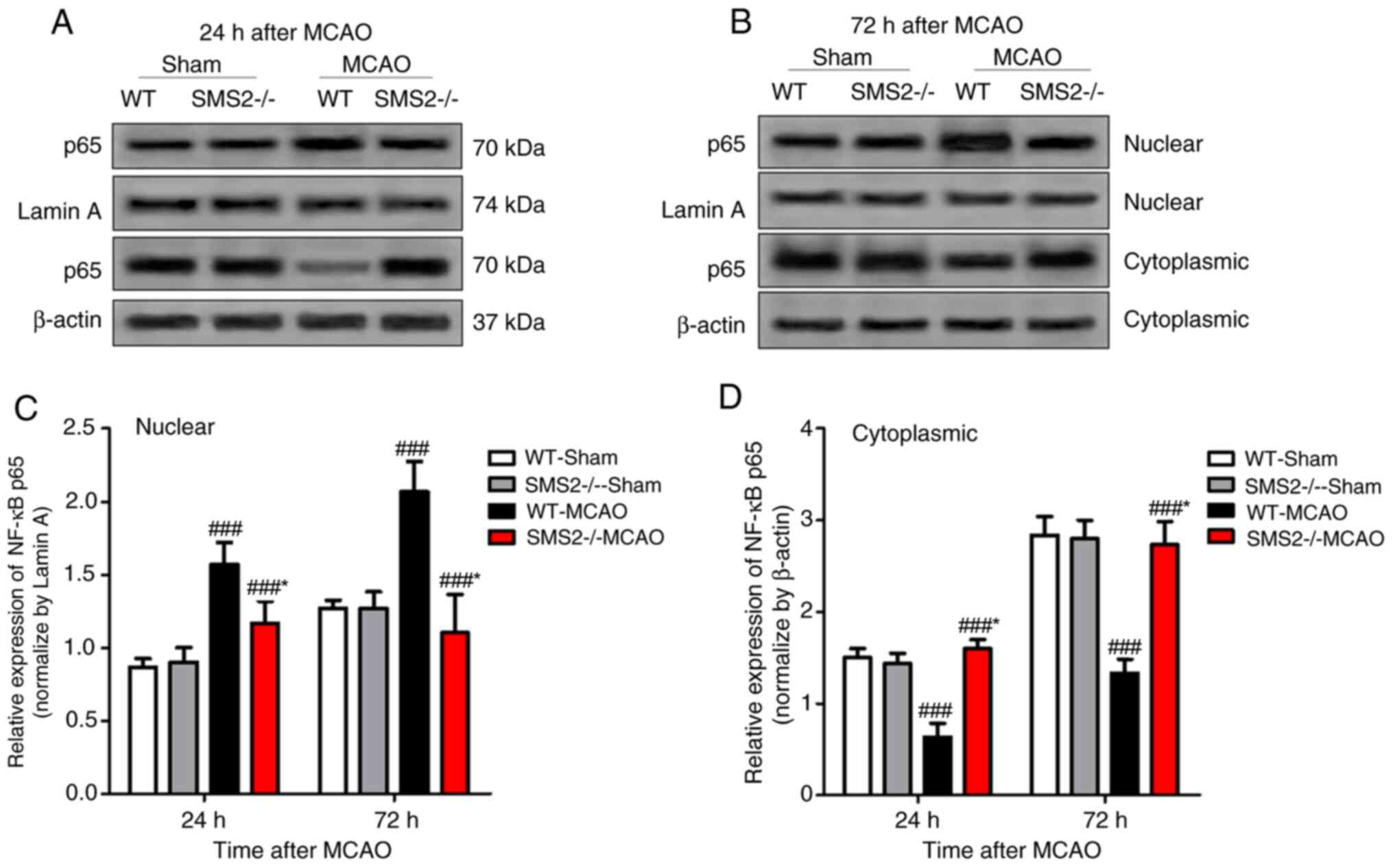
The expression of NF-κB p65 protein in nuclear and
cytoplasmic fractions was observed by western blotting. At 24 and
72 h after cerebral ischemia/reperfusion, the expression of NF-κB
p65 protein in the nucleus of SMS2-/- mice was
significantly lower than that in WT mice (P<0.001), but the
opposite was observed in the cytoplasm (P<0.001; Fig. 4). These data indicate that the
nuclear translocation of NF-κB p65 is significantly reduced in
SMS2-/- mice after ischemia/reperfusion, when compared
with WT MCAO mice.
Discussion
SMS2-/- mice were obtained by knocking
out intron 1 (1.1 kb), exon 2 (0.7 kb) and intron 2 (6.2 kb) in the
SMS2 gene of C57BL/6J mice. There was no difference in living
habits between C57BL/6J mice (26).
In this present study, WT and SMS2 knockout C57BL/6J were the
research subjects studied. This study found that SMS2-/-
mice not only had lower neurological deficit scores than WT mice
after ischemia/reperfusion, but also had a significantly lower
infarct volume than WT mice. This indicated that a lack of SMS2
attenuated cerebral ischemia/reperfusion injury in mice.
In addition, a previous study indicated (27) that SMS2 deficiency inhibits
LPS-induced inflammatory responses by impeding activation of the
NF-κB signaling pathway. Therefore, this present study examined the
expression of inflammatory mediators such as galectin 3, Arg 1,
iNOS and IL-1β in the infarcted brain after cerebral
ischemia/reperfusion in mice. The results showed that after
cerebral ischemia/reperfusion, the expression of both
pro-inflammatory and inhibitory factors increased, and the
expression of pro-inflammatory cytokines in the SMS2-/-
group MCAO mice was lower than that in the WT MCAO mice, but the
expression of anti-inflammatory cytokines in the SMS2-/-
group MCAO mice was higher than that in WT MCAO mice. This
suggested that SMS2 deficiency might reduce ischemia/reperfusion
injury in mice by reducing inflammatory responses after cerebral
ischemia/reperfusion.
Microglia are macrophages in the central nervous
system, and their activation is a hallmark of neuroinflammation.
Microglia in the normal mature brain are stationary. Resting
microglia become active after central nervous system damage. The
activated microglia mediate neurotoxic effects by releasing a
series of inflammatory cytokines, proteins and other biologically
active substances, and causing secondary brain injury (28,29).
Moon et al (30) used
immunohistochemistry to examine the temporal changes in the
activation of microglia after transient cerebral ischemic injury,
and they found that microglia were activated 3 h after cerebral
ischemia/reperfusion. Microglial activation state reached a peak 2
days after cerebral ischemia/reperfusion.
A previous study has shown that microglia can play a
protective role by regulating the expression of receptors, proteins
or cytokines after cerebral ischemia injury (31). Microglia can produce tumor necrosis
factor (TNF). Lambertsen et al (31) confirmed that TNF that was secreted
by microglia played a neuroprotective role in the acute phase of
focal cerebral ischemic injury through TNF-p55 receptor. This
present study found that the number of Iba 1+ and
galectin 3+/Iba 1+ cells in
SMS2-/- mice after cerebral ischemia/reperfusion was
significantly lower than that in WT mice (P<0.05). Iba 1 is a
specific surface antigen for microglia, and the decreased Iba
1+ cells in SMS2-/- mice after cerebral
ischemia/reperfusion indicate a decrease in microglial activation
in SMS2-/- mice after ischemia/reperfusion. The
decreased galectin 3+/Iba 1+ cells indicate a
decrease in the inflammatory response which is associated with
activated microglial galectin 3. After focal cerebral ischemia,
many pro-inflammatory cytokines, chemokines, and leukocyte adhesion
molecules are upregulated, and signal transduction by the cells was
necessary for the pathogenesis and pathological development
(32). TLRs, NF-κB,
Mitogen-activated protein kinases, and the JNK-STAT signaling
pathway are the most widely studied pathways which are involved in
the transduction of cerebral ischemia/reperfusion inflammation
(32). This present study has shown
that the expression of galectin 3 protein in SMS2-/-
mice was significantly lower than that of WT mice after
ischemia/reperfusion. Galectin 3 is an endogenous ligand of
TLR4(33). TLR4 binds to its ligand
to form the TLR/myeloid differentiation-2 (MD2) complex that
activates downstream inflammatory pathways (33). This study also found that the
expression of TLR4/MD2 on the surface of microglia was decreased in
SMS2-/- mice 72 h after cerebral ischemia/reperfusion
injury, but there was no significant difference in the expression
of TLR4. This suggests that SMS2 deficiency may inhibit subsequent
inflammatory responses by reducing TLR4/MD2 complex formation after
cerebral ischemia/reperfusion injury, rather than reducing TLR4
expression. The TLR family is a family of pathogen-associated
molecular pattern receptors that recognize and bind to conserved
sequences of pathogenic microorganisms (33). These are receptors that mediate
bacterial endotoxin LPS-induced inflammation. As this present study
progressed, TLRs were also found to be involved in cerebral
ischemia/reperfusion inflammatory induced injury (34). The brain is a sterile organ, and
inflammatory damage to the brain is mainly transduced through the
TLR pathway (34). DNA and protein
detection of TLRs was performed in normal mice and mice with
cerebral ischemia/reperfusion injury. The DNA and protein content
of TLR2 and TLR4 in mice with cerebral ischemia/reperfusion injury
were significantly higher than those in a normal group in a
previous study (35). TLRs mediate
NF-κB activation, and NF-κB upregulates the expression of
inflammatory factors, causing inflammatory damage.
NF-κB belongs to the Rel protein family and is an
important signal transduction molecule involved in inflammatory
reactions (36). During cerebral
ischemia, NF-κB is activated by inflammatory factors, cytokines,
increases in calcium concentration and other factors. After
activation, NF-κB can induce the expression of cytokines, adhesion
molecules and inflammatory enzymes, which forms a vicious circle of
inflammatory reactions, and cause brain edema and nerve cell
damage.
Howard et al (37) found that NF-κB was rapidly activated
at 15-30 min after hypoxia and reoxygenation in human brain
microvascular endothelial cells. In this present study, it was
found that the nuclear translocation of NF-κB p65 in
SMS2-/- mice was significantly less than that in WT mice
after ischemia/reperfusion. p65 is an important protein in the
NF-κB signaling pathway, and the entry of p65 from the cytoplasm to
the nucleus after phosphorylation is an important marker for
activation of NF-κB signaling (38). This indicated that the lack of SMS2
inhibited the activation of the NF-κB pathway following cerebral
ischemia/reperfusion in mice.
In conclusion, a shortage of SMS2 in mice may help
to reduce inflammation by inhibiting the activation of the NF-κB
signaling pathway, and further alleviate cerebral
ischemia/reperfusion injury in mice.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Author's contributions
YY, FH and QM conceived and designed the study;
collected data; drafted this study; and critically revised the
manuscript for the important intellectual content. GY analyzed and
interpreted the experimental data. QM read and approved the final
version of the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was performed with the approval of
the Ethics Committee of The Third People's Hospital of Qingdao.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lapi D and Colantuoni A: Remodeling of
cerebral microcirculation after ischemia-reperfusion. J Vasc Res.
52:22–31. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Jean WC, Spellman SR, Nussbaum ES and Low
WC: Reperfusion injury after focal cerebral ischemia: The role of
inflammation and the therapeutic horizon. Neurosurgery.
43:1382–1396. 1998.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wang YS, Li YX, Zhao P, Wang HB, Zhou R,
Hao YJ, Wang J, Wang SJ, Du J, Ma LJ, et al: Anti-inflammation
effects of oxysophoridine on cerebral ischemia-reperfusion injury
in mice. Inflammation. 38:2259–2268. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Xing B, Chen H, Zhang M, Zhao D, Jiang R,
Liu X and Zhang S: Ischemic post-conditioning protects brain and
reduces inflammation in a rat model of focal cerebral
ischemia/reperfusion. J Neurochem. 105:1737–1745. 2008.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kolesnick R and Golde DW: The
sphingomyelin pathway in tumor necrosis factor and interleukin-1
signaling. Cell. 77:325–328. 1994.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Mielke MM, Bandaru VV, Haughey NJ, Rabins
PV, Lyketsos CG and Carlson MC: Serum sphingomyelins and ceramides
are early predictors of memory impairment. Neurobiol Aging.
31:17–24. 2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Milhas D, Clarke CJ and Hannun YA:
Sphingomyelin metabolism at the plasma membrane: Implications for
bioactive sphingolipids. FEBS Lett. 584:1887–1894. 2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Khan M, Sekhon BK, Sekhon CS, Singh I and
Singh AK: Sphingolipids in rat model of transient focal cerebral
ischemia: Implication for stroke injury. J. Neurochem: Jun 28, 2008
(Epub ahead of print).
|
|
9
|
Tu R, Yang W and Hu Z: Inhibition of
sphingomyelin synthase 1 affects ceramide accumulation and hydrogen
peroxide-induced apoptosis in Neuro-2a cells. Neuroreport.
27:967–973. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Fikadu Geta T, Philipp T and Holthuis JCM:
The multigenic sphingomyelin synthase family. J Biol Chem.
281:29421–29425. 2006.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Qin R, Chen ML, Zhu K, Deng JB and Shi YY:
Sphingomyelin synthase 2 deficiency decreases atherosclerosis and
inhibits inflammation in mice. Sheng Li Xue Bao. 62:333–338.
2010.PubMed/NCBI(In Chinese).
|
|
12
|
Hailemariam TK, Huan C, Liu J, Li Z, Roman
C, Kalbfeisch M, Bui HH, Peake DA, Kuo MS, Cao G, et al:
Sphingomyelin synthase 2 deficiency attenuates NFkappaB activation.
Arterioscler Thromb Vasc Biol. 28:1519–1526. 2008.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Gowda S, Yeang C, Wadgaonkar S, Anjum F,
Grinkina N, Cutaia M, Jiang XC and Wadgaonkar R: Sphingomyelin
synthase 2 (SMS2) deficiency attenuates LPS-induced lung injury. Am
J Physiol Lung Cell Mol Physiol. 300:L430–L440. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhang F, Guo RM, Yang M, Wen XH and Shen
J: A stable focal cerebral ischemia injury model in adult mice:
Assessment using 7T MR imaging. AJNR Am J Neuroradiol. 33:935–939.
2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Pérez JM and Pascau J: Image Processing
with ImageJ. Packt Publishing, 2016.
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Arumugam TV, Okun E, Tang SC, Thundyil J,
Taylor SM and Woodruff TM: Toll-like receptors in
ischemia-reperfusion injury. Shock. 32:4–16. 2009.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Rahimian R, Béland LC and Kriz J:
Galectin-3: Mediator of microglia responses in injured brain. Drug
Discov Today. 23:375–381. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Fernandes Bertocchi AP, Campanhole G, Wang
PH, Gonçalves GM, Damião MJ, Cenedeze MA, Beraldo FC, Teixeira VP,
Reis M, Mazzali M, et al: A role for galectin-3 in renal tissue
damage triggered by ischemia and reperfusion injury. Transpl Int.
21:999–1007. 2008.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Yan YP, Lang BT, Vemuganti R and Dempsey
RJ: Galectin-3 mediates post-ischemic tissue remodeling. Brain Res.
1288:116–124. 2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Song H, Liu S, Zhao Z, Sun W, Wei X, Ma X,
Zhao P and Gao D: Increased cycles of DC/CIK immunotherapy
decreases frequency of tregs in patients with resected NSCLC. Int
Immunopharmacol. 52:197–202. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Liu M, Eguchi N, Yamasaki Y, Urade Y,
Hattori N and Urabe T: Focal cerebral ischemia/reperfusion injury
in mice induces hematopoietic prostaglandin D synthase in microglia
and macrophages. Neuroscience. 145:520–529. 2007.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ohsawa K, Imai Y, Sasaki Y and Kohsaka S:
Microglia/macrophage-specific protein Iba1 binds to fimbrin and
enhances its actin-bundling activity. J Neurochem. 88:844–856.
2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Yoshinori I and Shinichi KJG:
Intracellular signaling in M-CSF-induced microglia activation: Role
of Iba1. Glia. 40:164–174. 2002.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wang M, Uchiumi O, Ogiso H, Shui Y, Zou J,
Hashizume C, Taniguchi M, Okazaki T and Kato N: Stressful learning
paradigm precludes manifestation of cognitive ability in
sphingomyelin synthase-2 knockout mice. Behav Brain Res. 319:25–30.
2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Luberto C, Yoo DS, Suidan HS, Bartoli GM
and Hannun YA: Differential effects of sphingomyelin hydrolysis and
resynthesis on the activation of NF-kappa B in normal and
SV40-transformed human fibroblasts. J Biol Chem. 275:14760–14766.
2000.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Liu GJ, Middleton RJ, Hatty CR, Kam WWY,
Chan R, Pham T, Harrison-Brown M, Dodson E, Veale K and Banati RB:
The 18 kDa translocator protein, microglia and neuroinflammation.
Brain Pathol. 24:631–653. 2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Vegeto E, Belcredito S, Ghisletti S, Meda
C, Etteri S and Maggi A: The endogenous estrogen status regulates
microglia reactivity in animal models of neuroinflammation.
Endocrinology. 147:2263–2272. 2006.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Moon JB, Lee CH, Park CW, Cho JH, Hwang
IK, Yoo KY, Choi JH, Shin HC and Won MH: Neuronal degeneration and
microglial activation in the ischemic dentate gyrus of the gerbil.
J Vet Med Sci. 71:1381–1386. 2009.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Lambertsen K, Clausen BH, Babcock AA,
Gregersen R, Fenger C, Nielsen HH, Haugaard LS, Wirenfeldt M,
Nielsen M, Dagnaes-Hansen F, et al: Microglia protect neurons
against ischemia by synthesis of tumor necrosis factor. J Neurosci.
29:1319–1330. 2009.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Wong CH and Crack PJ: Modulation of
neuro-inflammation and vascular response by oxidative stress
following cerebral ischemia-reperfusion injury. Curr Med Chem.
15:1–14. 2008.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Kim S, Kim SY, Pribis JP, Lotze M, Mollen
KP, Shapiro R, Loughran P, Scott MJ and Billiar TR: Signaling of
high mobility group box 1 (HMGB1) through toll-like receptor 4 in
macrophages requires CD14. Mol Med. 19:88–98. 2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Yang QW, Li JC, Lu FL, Wen AQ, Xiang J,
Zhang LL, Huang ZY and Wang JZ: Upregulated expression of toll-like
receptor 4 in monocytes correlates with severity of acute cerebral
infarction. J Cereb Blood Flow Metab. 28:1588–1596. 2008.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Ma Y, He M and Qiang L: Exercise therapy
downregulates the overexpression of TLR4, TLR2, MyD88 and NF-κB
after cerebral ischemia in rats. Int J Mol Sci. 14:3718–3733.
2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wullaert A, Bonnet MC and Pasparakis M:
NF-κB in the regulation of epithelial homeostasis and inflammation.
Cell Res. 21:146–158. 2011.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Howard EF, Chen Q, Cheng C, Carroll JE and
Hess D: NF-kappa B is activated and ICAM-1 gene expression is
upregulated during reoxygenation of human brain endothelial cells.
Neurosci Lett. 248:199–203. 1998.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Wagner SA, Satpathy S, Beli P and
Choudhary CJEJ: SPATA2 links CYLD to the TNF-α receptor signaling
complex and modulates the receptor signaling outcomes. EMBO J.
35:1868–1884. 2016.PubMed/NCBI View Article : Google Scholar
|