Introduction
Colorectal carcinoma is the second most common type
of cancer. Although conventional treatments such as surgical
resection, chemotherapy and radiation therapy have decreased the
mortality rate of colon cancer, it has been projected that there
will be 148,950 estimated new cases and 53,200 estimated deaths in
the US in 2020(1). Accumulating
evidence has indicated that the existence of cancer stem cells
(CSCs) in human colorectal carcinoma is one of the major causes of
treatment failure (2,3). CSCs represent a subpopulation of cells
within cancer cells with an inherent capacity for unlimited
self-renewal and heterogeneous-lineage differentiation (4). They display distinct immunophenotypes
resistant to conventional chemotherapeutics and radiation therapy
and are responsible for tumorigenesis, relapse and metastasis
(5-9).
Hence, the elimination of CSCs in colon cancer may be a novel
approach for effective cancer management.
Reduced intratumoral oxygen tension (hypoxia) is a
feature that is common in a majority of solid tumors and occurs as
a result of the abnormal vasculature's limited capacity to deliver
oxygen, which cannot meet the demands of the rapidly proliferating
cancer cells (10). Evidence has
indicated that up to 60% of locally advanced solid tumors exhibit
substantial heterogeneity in the tissue oxygenation levels within
the tumor (11). In hypoxia, cancer
cells are able to adapt to their new microenvironment via multiple
mechanisms: Inhibition of apoptosis via Bcl-2 family proteins
(12), upregulation of MMPs
(13), and stimulation of
angiogenesis by vascular endothelial growth factor (14). Hypoxia also mediates resistance to
radiotherapy and chemotherapy, resulting in poor prognosis
(15). Several commercially
available drugs such as cisplatin, mitomycin C and sorafenib have
been associated with hypoxia-induced impairment of chemosensitivity
in several human cancer cell lines (16). Therefore, finding a chemotherapeutic
agent that is effective in a hypoxic environment is crucial in
cancer therapy.
Atovaquone (ATO) is an anti-parasitic drug approved
by the US Food and Drug Administration in the treatment of malaria
and pneumocystis pneumonia. ATO is structurally similar to coenzyme
Q10 and competitively inhibits its binding to the cytochrome bc1
complex, resulting in mitochondrial membrane potential (MMP)
collapse and parasite death (17).
ATO has attracted attention as a novel anticancer agent for
targeting various types of cancer cell (18-22).
Ashton et al (23) revealed
that ATO reduces the oxygen consumption rate by inhibiting
mitochondrial respiration complex III activity, reduces hypoxia in
both spheroids and xenografted tumors and causes tumor growth delay
in combination with radiation. However, studies on ATO targeting
CSCs are limited and the anti-cancer effects of ATO on hypoxic
colon CSCs have not been previously investigated. In the present
study, epithelial cell adhesion molecule (EpCAM) and CD44, which
are robust makers of human colon CSCs (2), were used to isolate
EpCAM+CD44+ cells from the HCT-116 colon
cancer cell line and the potential of ATO in eradicating colon CSCs
under hypoxic conditions was investigated. The present results
demonstrated that ATO inhibited cell growth and invasiveness,
induced apoptosis and caused S-phase arrest of
EpCAM+CD44+ HCT-116 cells under hypoxic
conditions.
Materials and methods
Cell lines and culture
The human HCT-116 colon cancer cell line was
purchased from the Cell Bank of the Chinese Academy of Sciences and
was cultured in high-glucose DMEM (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 10% FBS (Gibco; Thermo Fisher Scientific,
Inc.) at 37˚C with 5% CO2.
EpCAM+CD44+ HCT-116 cells were cultured in
serum-free DMEM/F12 (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 20 ng/ml epidermal growth factor (EGF), 20 ng/ml
basic fibroblast growth factor (bFGF; both from PeproTech, Inc.)
and 2% B27 (Gibco; Thermo Fisher Scientific, Inc.) at 37˚C with 5%
CO2. For hypoxic incubation, cells were cultured in a
hypoxic chamber at 37˚C in a humidified atmosphere of 5%
CO2, 1% O2 and 94% N2.
Magnetic-activated cell sorting and
FACS
EpCAM+CD44+ HCT-116 cells were
obtained by magnetic-activated cell sorting as previously described
(24). In brief, dissociated
HCT-116 colon cancer cells were labeled with biotin-conjugated
EpCAM antibodies (1:50; cat. no. 13-9326-82; eBioscience; Thermo
Fisher Scientific, Inc.). The cells were magnetically separated
using a CELLection Biotin Binder kit (Invitrogen; Thermo Fisher
Scientific, Inc.). The sorted EpCAM+ HCT-116 cells were
further labeled with biotin-conjugated CD44 antibody (1:50; cat.
no. 13-0441-82; eBioscience; Thermo Fisher Scientific, Inc.) and
then fractionated using the CELLection Biotin Binder kit. In the
meantime, 1x106 dissociated HCT-116 cells and
EpCAM+CD44+ HCT-116 cells in 0.1 ml PBS were
incubated with FITC-conjugated anti-EpCAM antibody (1:20; cat. no.
324203) and phycoerythrin-conjugated anti-CD44 antibody (1:20; cat.
no. 338807; both from BioLegend, Inc.) in the dark for 10 min at
4˚C. The cells were washed with PBS and then acquired and analyzed
using a Beckman Coulter FC500 Flow Cytometer with the CellQuest Pro
software (version 6.0; BD Biosciences) to determine the proportion
of EpCAM+CD44+ cells.
Tumorsphere-formation assay
In brief, a single-cell suspension of sorted
EpCAM+CD44+ HCT-116 cells was cultured in
serum-free DMEM/F12 supplemented with 20 ng/ml EGF, 20 ng/ml bFGF
and 2% B27. The cells were then seeded on uncoated 6-well culture
plates (Corning, Inc.) at a density of 1x104 cells/well.
Tumorsphere formation was observed for 4 days and representative
images of at least five random fields and were captured using an
inverted light microscope (Olympus Corp.) at a magnification of
x100.
To evaluate the effect of ATO on tumorsphere
formation, a single-cell suspension of
EpCAM+CD44+ HCT-116 cells was treated with 15
µM ATO for 3 days under hypoxic conditions, with 50 µM DDP and
0.05% DMSO as a positive and negative control, respectively. The
number of tumorspheres was counted under an inverted light
microscope (Olympus Corp.) at a magnification of x40.
Serum-induced differentiation
EpCAM+CD44+ HCT-116 cells were
resuspended and incubated in DMEM/F12 supplemented with 10% FBS at
37˚C with 5% CO2. Images of cells before and after 48 h
of serum induction were acquired using an inverted light microscope
(Olympus Corp.) at a magnification of x200.
Reverse transcription-quantitative PCR
(RT-qPCR)
To determine the expression of stemness-related
genes [OCT-4, SOX-2, Nanog homeobox (NANOG) and C-MYC], total RNA
of 1x106 HCT-116 and EpCAM+CD44+
HCT-116 cells was extracted using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). Complementary DNA was
synthesized by RT using a PrimeScript RT kit (Takara Bio, Inc.)
according to the manufacturer's instructions and quantified by
performing qPCR using the FastStart SYBR Green master mix (Roche
Diagnostics, GmbH) on a PikoReal 96 Real-Time PCR system (Thermo
Fisher Scientific, Inc.) with the following thermocycling
conditions: Initial denaturation for 5 min at 95˚C; and 40 cycles
of 95˚C for 5 sec and 60˚C for 30 sec. Likewise, in
EpCAM+CD44+ HCT-116 cells treated with 15 µM
ATO for 24 h under hypoxic conditions and in the respective
controls, the expression levels of apoptosis-associated genes
(Bcl-2 and Bax) and invasion-associated genes [MMP-2/-9 and tissue
inhibitor of MMPs 1 (TIMP-1)] were evaluated. GAPDH was detected as
an internal control. The average Cq values of the target genes were
normalized to those of the control genes (∆∆Cq) (25). Primers used for PCR are presented in
Table I. All experiments were
performed in triplicate.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Target gene | Sequence
(5'-3') | Product size
(bp) |
|---|
| GAPDH | F:
CAGGAGGCATTGCTGATGAT | 138 |
| | R:
GAAGGCTGGGGCTCATTT | |
| OCT4 | F:
GATGTGGTCCGAGTGTGGTTCTG | 195 |
| | R:
CGAGGAGTACAGTGCAGTGAAGTG | |
| NANOG | F:
ATGCCTGTGATTTGTGGGCC | 403 |
| | R:
GCCAGTTGTTTTTCTGCCAC | |
| c-MYC | F:
CACCAGCAGCGACTCTGAGGAG | 239 |
| | R:
ACTTGACCCTCTTGGCAGCAGG | |
| SOX2 | F:
TCCATGACCAGCTCGCAGA | 152 |
| | R:
GAGGAAGAGGTAACCACGGG | |
| MMP-2 | F:
GCCTCTCCTGACATTGACCTTGG | 112 |
| | R:
CACCACGGATCTGAGCGATGC | |
| MMP-9 | F:
GCACCACCACAACATCACCT | 284 |
| | R:
ACCACAACTCGTCATCGTCG | |
| TIMP-1 | F:
CCTGGCTTCTGGCATCCTGTTG | 162 |
| | R:
CGCTGGTATAAGGTGGTCTGGTTG | |
| BCL-2 | F:
GGGGAGGATTGTGGCCTTCTTT | 107 |
| | R:
TAATGTGCAGGTGCCGGTTCAG | |
| BAX | F:
TAACCAAGGTGCCGGAACTGA | 126 |
| | R:
GGGAGGAGTCTCACCCAACCA | |
MTS assay
EpCAM+CD44+ HCT-116 cells
(5x103/well) were seeded in a 96-well plate and cultured
under hypoxic conditions. Cells were incubated with ATO at
different concentrations (0, 5, 10, 15 and 20 µM) for 24 h.
Triplicate wells were set up for each concentration. Cell viability
was measured using an MTS kit (Promega Corp.) according to the
manufacturer's protocol. The absorbance was measured at 490 nm
using a Multiskan GO microplate reader (Thermo Fisher Scientific,
Inc.). All experiments were performed at least three times. The
cell viability was determined, and the inhibition ratio was
calculated using the following formula: Inhibition ratio (%) =
(1-optical density of the treatment group/optical density of the
solvent control) x100.
Detection of apoptosis by Annexin
V-FITC/PI staining
Flow cytometry was performed to analyze cell
apoptosis in hypoxia using an Annexin V/PI-FITC kit (Nanjing KeyGen
Biotech Co., Ltd.) according to the manufacturer's protocol. In
brief, the cells were seeded in a 6-well plate (1x106
cells/well) and cultured under hypoxic conditions. The cells were
then treated with ATO (5, 10 or 15 µM) for 24 h, with 50 µM DDP and
DMSO used as a positive and negative control, respectively. After
washing with PBS, the cells (5x105) were resuspended in
binding buffer (500 µl) and stained with Annexin V-FITC (5 µl) and
PI (5 µl) in the dark at room temperature for 5 min. The cells were
washed with PBS and analyzed within 1 h using a Beckman Coulter
FC500 Flow Cytometer with the CellQuest Pro software (version 6.0;
BD Biosciences). The experiment was performed for a total of three
times.
Detection of apoptosis by JC-1
assay
The MMP was detected by a JC-1 staining assay using
the JC-1 Apoptosis Detection kit (Nanjing KeyGen Biotech Co.,
Ltd.). In brief, the EpCAM+CD44+ HCT-116
cells were dissociated into single cells using TrypLE reagent and
then seeded in a 6-well plate (1x106/well). The cells
were incubated under hypoxic conditions and treated with 5, 10 or
15 µM ATO for 24 h, with 50 µM DDP and DMSO as a positive and
negative control, respectively. After washing with PBS, cells were
resuspended in 500 µl binding buffer, and 5x105 cells
were stained with JC-1 (5 µl) and incubated in the dark at room
temperature for 15 min at 37˚C with 5% CO2 under hypoxic
conditions. The cells were then resuspended in incubation buffer
(500 µl), washed with PBS and analyzed using a Beckman Coulter
FC500 Flow Cytometer with CellQuest Pro software (version 6.0; BD
Biosciences). The experiment was performed three times.
Cell-cycle analysis
In brief, 1x106
EpCAM+CD44+ HCT-116 cells treated with 15 µM
ATO were incubated under hypoxic conditions for 24 h. The cells
were dissociated into single cells using TrypLE reagent and fixed
with ice-cold 70% ethanol for 12 h at 4˚C. The cells were pelleted
at 500 x g for 5 min at 4˚C and washed by gently resuspending them
in 1 ml PBS. After carefully removing the supernatant, the cells
were treated with 50 µl RNaseA (100 µg/ml; Nanjing KeyGen Biotech
Co., Ltd.) and stained with 200 µl PI (50 µg/ml) at 37˚C in the
dark for 30 min. The stained cells were placed on ice in the dark,
washed with PBS and analyzed for PI fluorescence using a Beckman
Coulter FC500 flow cytometer with CellQuest Pro software (version
6.0; BD Biosciences). The experiment was performed three times.
Transwell invasion assay
An invasion assay was performed using 6.5-mm
Transwell plates with sterile 8.0-µm pore polycarbonate membrane
inserts (Corning, Inc.) and covered with a thin layer of BD
Matrigel (BD Biosciences). In brief, the
EpCAM+CD44+ HCT-116 cells (1x105
cells/well) were seeded in a Transwell plate and treated with 15 µM
ATO. As positive and negative controls, 50 µM DDP and DMSO were
respectively used. DMEM/F12 medium supplemented with 10% FBS was
loaded into the bottom chamber through the insert as a chemostatic
factor. After incubation for 24 h under hypoxic conditions, the
media in the upper and lower chambers were removed and the cells
that had invaded the membranes were fixed with 4% paraformaldehyde
(500 µl) for 20 min and stained with 0.01% crystal violet for 20
min at room temperature. The cells were counted on an inverted
light microscope (Olympus Corp.) and the mean value was determined
from counts of five random fields.
Statistical analyses
Values are expressed as the mean ± standard
deviation of data from triplicate experiments. GraphPad Prism
(version 6.0; GraphPad Software Inc.) was employed for statistical
analysis. Student's t-test or one-way ANOVA followed by Dunnett's
test was used in the analysis when appropriate. P<0.05 was
considered to indicate statistical significance.
Results
Characteristics of isolated
EpCAM+CD44+ HCT-116 cells
CSCs have a crucial role in tumor initiation,
progression and metastasis (26).
To study the efficacy of ATO in colon CSCs, the
EpCAM+CD44+ cells were isolated from the
HCT-116 colon cancer cell line by magnetic-activated cell sorting
and the proportion of CSCs and non-CSCs was measured by flow
cytometric analysis using EpCAM+ and CD44+
monoclonal antibodies. A single cell suspension of the sorted
EpCAM+CD44+ HCT-116 cells cultured in
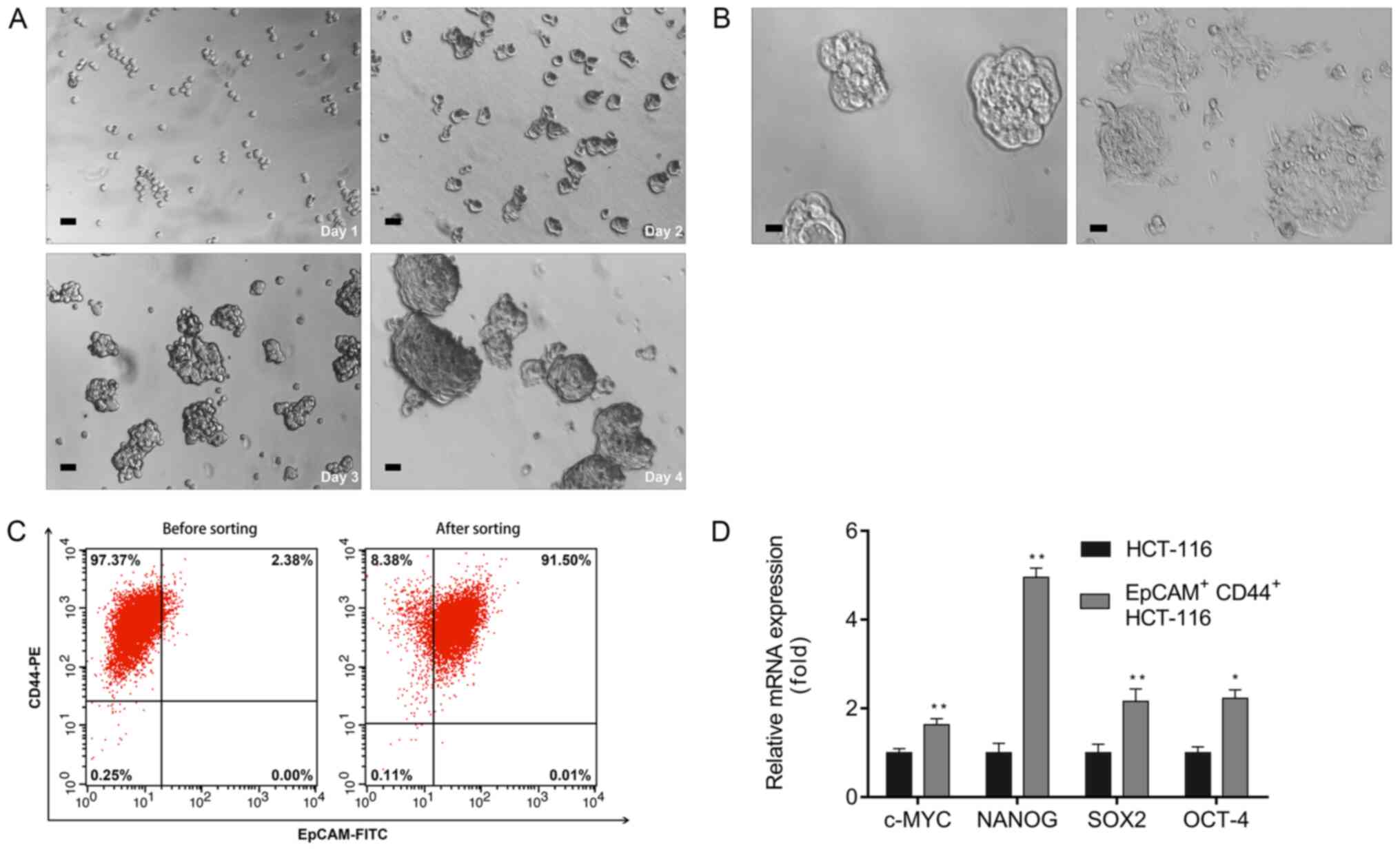
serum-free DMEM/F12 gradually proliferated and formed spheres on
day 4 (Fig. 1A) and were considered
to be colon CSCs. These spheroid cells switched to adherent
confluent cells after adding 10% FBS to the culture medium
(Fig. 1B). In addition, flow
cytometric analyses revealed that the percentage of
EpCAM+CD44+ cells in unsorted and sorted
HCT-116 cells was 2.38 and 91.50%, respectively (Fig. 1C). The RT-qPCR results indicated
upregulation of pluripotent-associated genes, including C-MYC,
NANOG, SOX-2 and OCT-4, in EpCAM+CD44+
HCT-116 cells relative to the HCT-116 cells (Fig. 1D).
ATO inhibits the viability of
EpCAM+CD44+ HCT-116 cells
To determine the effects of ATO on cell viability,
the EpCAM+CD44+ HCT-116 cells were treated
with different concentrations (0-20 µM) of ATO under hypoxic
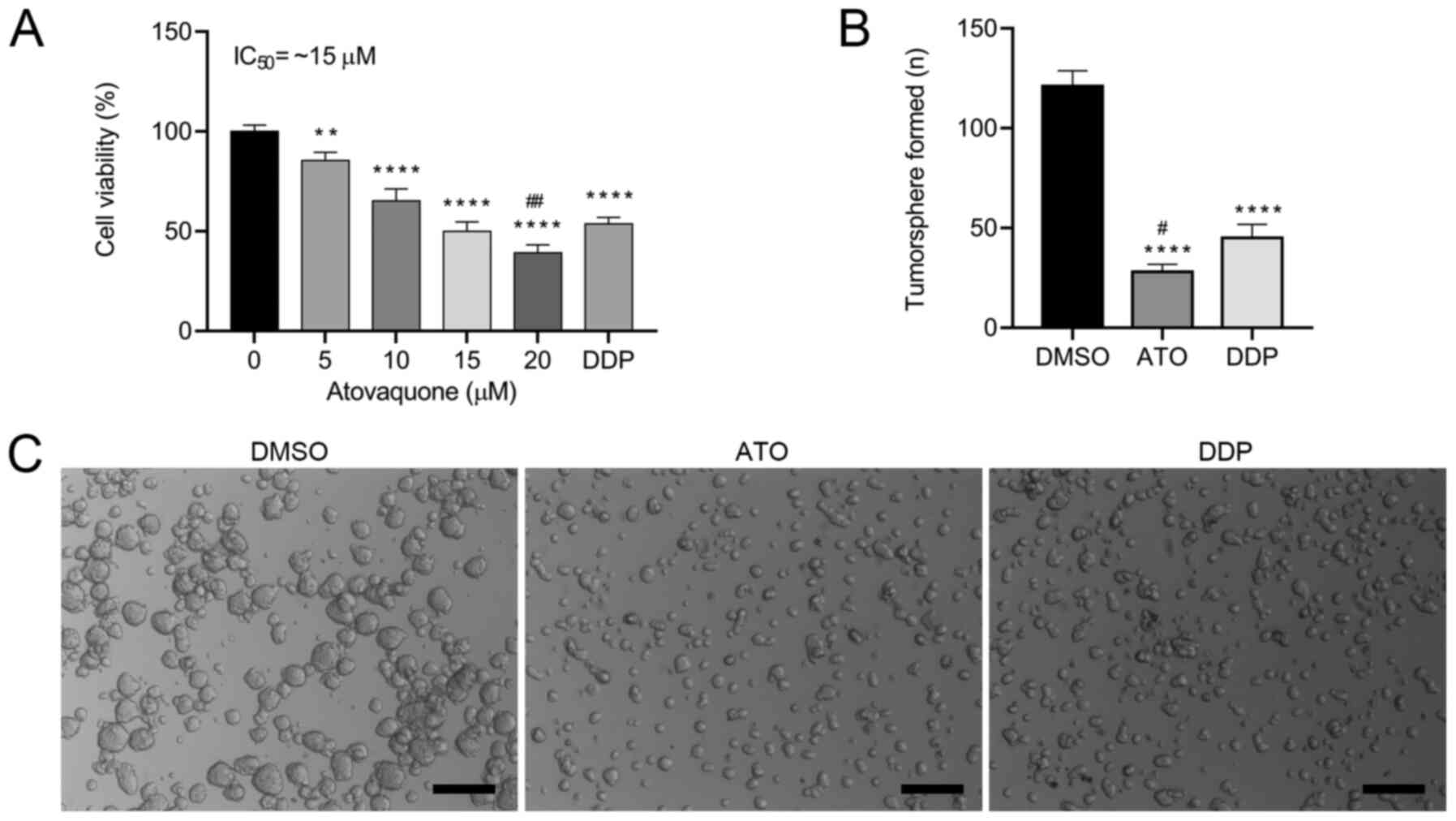
conditions for 24 h. The results indicated that ATO inhibited the
proliferation of EpCAM+CD44+ HCT-116 cells in
a concentration-dependent manner (Fig.
2A). The proliferation of EpCAM+CD44+
HCT-116 cells was significantly inhibited by 5 and 10 µM ATO and
the IC50 values was ~15 µM in hypoxia. When atovaquone
was administered with food at the standard regimen of 750 mg twice
daily, the average steady-state plasma concentration was ~57 µM
(27). Therefore, the obtained
IC50 of ATO is pharmacologically achievable and was used
for the subsequent experiments. To investigate whether ATO is
involved in the tumorigenic activity of the CSCs, a tumorsphere
formation assay was performed. ATO treatment induced the formation
of a lower number of tumorspheres (Fig.
2B) and smaller tumorspheres (Fig.
2C) in comparison with those of DMSO-treated cells. Likewise,
ATO treatment induced the formation of a lower number of
tumorspheres, but there were no significant differences in the size
of the tumorshperes in comparison with DDP-treated cells. These
results suggested that ATO suppressed the tumorsphere-formation
capacity of EpCAM+CD44+ HCT-116 cells.
ATO induced apoptosis of
EpCAM+CD44+ HCT-116 cells
An annexin V-FITC/PI double staining assay and JC-1
assay were performed to determine whether ATO is able to induce
apoptosis in EpCAM+CD44+ HCT-116 cells under
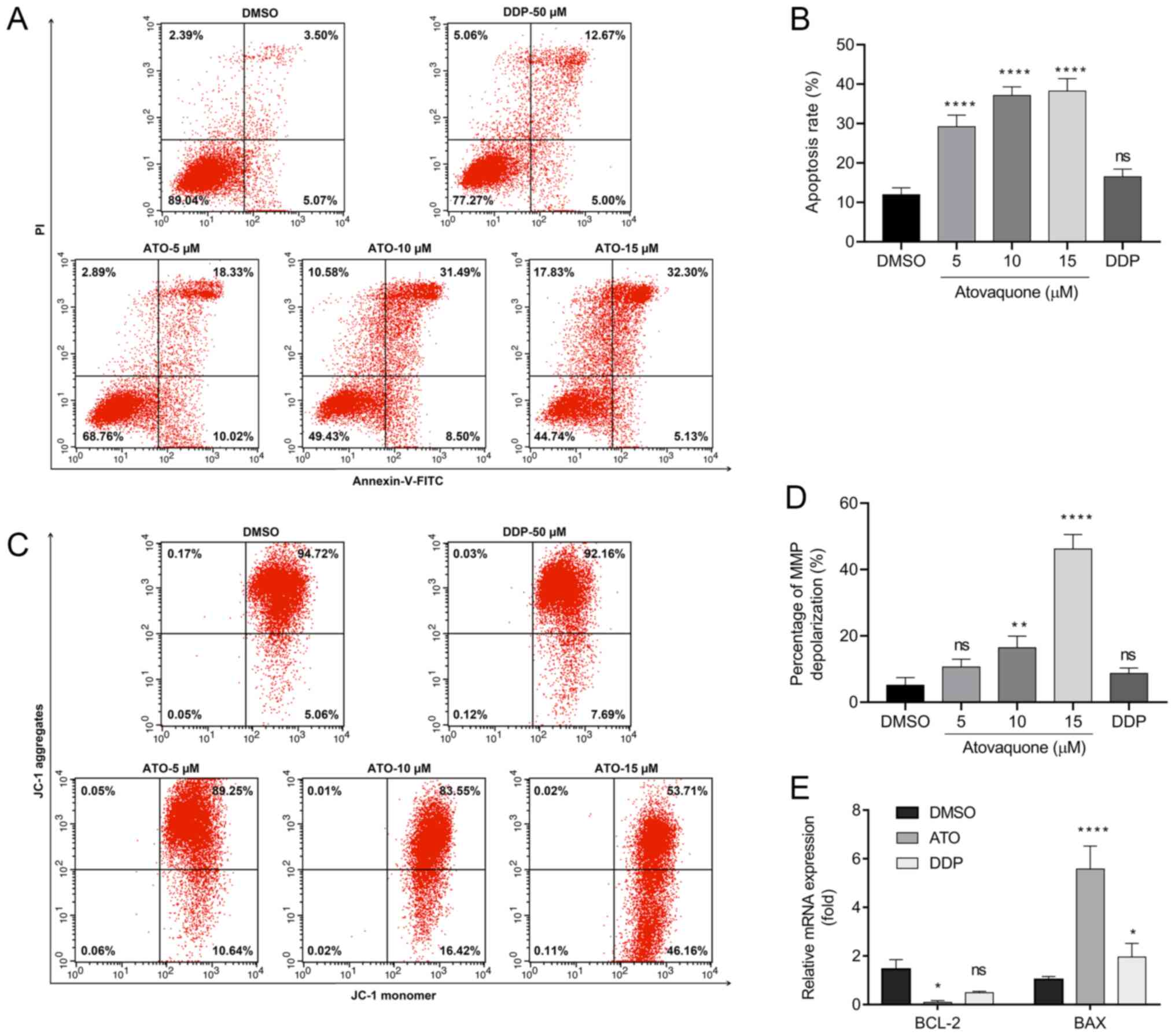
hypoxic conditions. The results suggested that treatment with 15 µM
ATO increased the percentage of Annexin V-positive cells to
38.22±5.18% compared with 8.48±2.75% in the DMSO group and
18.86±2.98% in the DDP group (Fig.
3A and B). Likewise, JC-1
staining revealed that the group treated with 15 µM ATO exhibited
loss of the MMP (48.96±4.42%) compared with the DMSO-treated
(5.25±1.63%) and DDP-treated groups (8.69±1.71%; Fig. 3C and D). Furthermore, cancer cells are able to
adapt to their surrounding hypoxic microenvironment by inhibition
of apoptosis via Bcl-2 family proteins. The RT-qPCR results
indicated suppression of Bcl-2 expression and upregulation of Bax
following treatment of EpCAM+CD44+ HCT-116
cells with ATO under hypoxia (Fig.
3E).
ATO induces cell-cycle arrest of
EpCAM+CD44+ HCT-116 cells
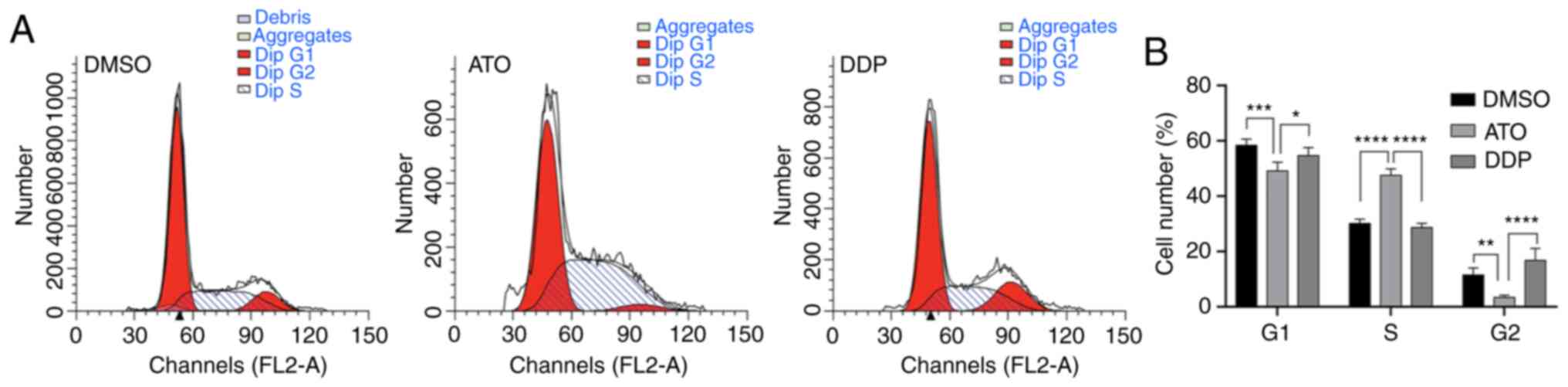
Flow cytometric analysis of the cell-cycle profile
of EpCAM+CD44+ HCT-116 cells after treatment
with 15 µM ATO revealed that the number of cells in S phase
significantly increased compared with that of the untreated cells
(P<0.01), while the number of cells in G1 and G2 phase decreased
accordingly (P<0.01). These results suggested that ATO treatment
of EpCAM+CD44+ HCT-116 cells under hypoxia
induced cell-cycle arrest in S-phase (Fig. 4A and B).
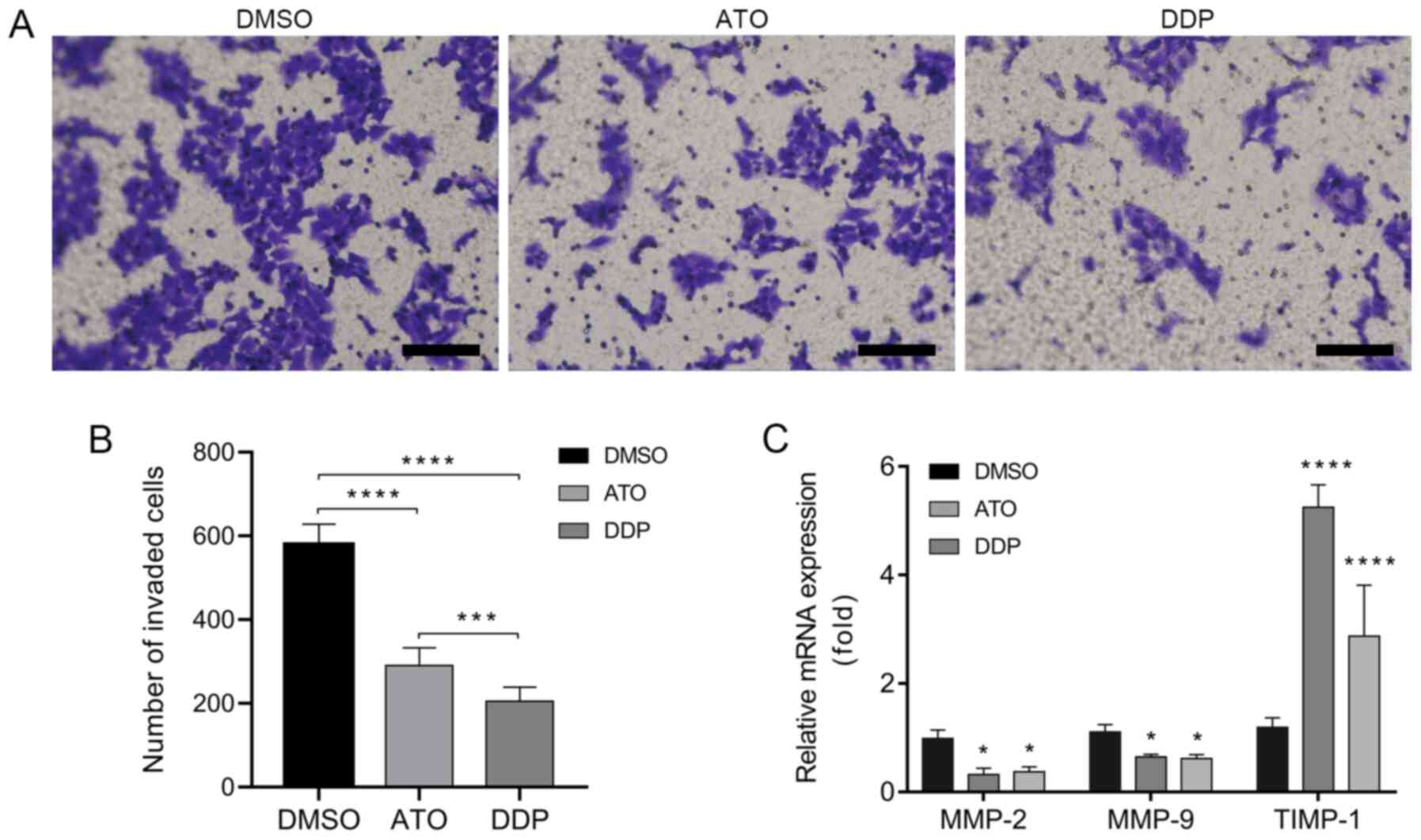
ATO inhibits the invasion of
EpCAM+CD44+ HCT-116 cells
A Transwell invasion assay was performed to
investigate the invasiveness of EpCAM+CD44+
HCT-116 cells following treatment with ATO under hypoxic conditions
for 24 h. The results indicated that treatment with ATO
significantly inhibited the invasion of
EpCAM+CD44+ HCT-116 cells as compared with
that in the DMSO group (Fig. 5A and
B). In addition, the RT-qPCR
results indicated downregulation of the invasion-associated genes
MMP-2 and MMP-9 and upregulation of TIMP-1 in
EpCAM+CD44+ HCT-116 cells after treatment
with ATO under hypoxic conditions for 24 h (Fig. 5C).
Discussion
Hypoxia is a major feature of the tumor
microenvironment and results from an imbalance between oxygen
supply and oxygen consumption of cancer cells (10). Tumor hypoxia was observed to
diminish the efficacy of several chemotherapeutic drugs (28). Recently, ATO has gained attention
due to its anticancer effects against various types of cancer,
including brain, breast and cervical cancer, as well as
hepatocellular carcinoma and retinoblastoma (18-22).
To the best of our knowledge, the present study was the first to
investigate the anticancer efficacy of ATO against colon CSCs in
vitro under hypoxic conditions. The results suggested that ATO
treatment inhibited the proliferation and invasion, induced
apoptosis and caused S-phase arrest of
EpCAM+CD44+ HCT-116 cells in
vitro.
EpCAM and CD44 were used as markers for the
isolation of colon CSCs from the colon cancer cell line HCT-116 by
magnetic-activated cell sorting; in the native cell population,
EpCAM+CD44+ HCT-116 cells accounted for 2.38%
of all HCT-116 cells, which is consistent with the result of
previous studies (29,30). Highly purified (>91%)
EpCAM+CD44+ HCT-116 cells were subsequently
obtained and the identity of CSCs was confirmed by performing a
tumorsphere formation assay and serum-differentiation assay, and by
evaluating the expression of stemness-related transcription
factors, including C-MYC, OCT-4, SOX-2 and NANOG. The expression of
these genes is associated with poor prognosis and malignant
progression (31). In the present
study, ATO inhibited the proliferation of
EpCAM+CD44+ HCT-116 cells in a
concentration-dependent manner with an IC50 of ~15 µM
under hypoxic conditions. However, cell inhibition showed a linear
trend with increasing ATO concentrations, while normally, a
sigmoidal dose-response relation would be expected. Several factors
may affect the accuracy of IC50 estimation (32), such as the total number of assay
concentrations, the number of concentrations beyond the lower and
upper bend point, dilution factor and the viability of the
responses at the same concentration. A linear trend obtained in the
present study may be partly due to the dilution factor and the
total number of drug concentrations. Five drug concentrations (0,
5, 10, 15 and 20 µM) were used and only one concentration was below
the IC50. Moreover, a difference of 5 µM between each
concentration point instead of various concentrations with a wide
range, for example, covering six logs of magnitude (0.001, 0.01
0.1, 1, 10, 100 and 1,000 µM) was used in the assay. A linear trend
was also observed in other studies, possibly due to similar reasons
(19,33). In the present study, the
IC50 (~15 µM) was lower than the average plasma
concentration (~57 µM) achieved in patients when ATO was
administered with food at a standard dose of 750 mg twice daily to
treat Pneumocystis carinii pneumonia (27) or when four 250/100 mg tablets of
ATO-proguanil were administered daily to treat malaria (34,35).
In an in vitro study, ATO treatment inhibited the viability
of the breast cancer MCF-7 cell line with an IC50 of ~10
µM and MCF-7-derived CSCs with an IC50 of ~1 µM,
examined by sulforhodamine B assay and mammosphere assay,
respectively (19). Similarly, ATO
at 10 µM markedly inhibited the proliferation in retinoblastoma
cells and renal cell carcinoma cell lines (22,33).
These results suggested that the effective concentration against
EpCAM+CD44+ HCT-116 cells and certain other
cancer cells is pharmacologically achievable (27).
ATO is a cytotoxic and apoptosis-inducing agent that
is potent against various cancer cell lines (36). In the present study, ATO treatment
induced apoptosis as detected by annexin V-FITC/PI double staining
assays and caused mitochondrial membrane potential depolarization,
as determined by a JC-1 staining assay. However, the trend of
different ATO concentrations to induce apoptosis in the former
assay appears to not be in accordance with the trend observed on
JC-1 staining. This may be caused by several factors, such as the
variation in trypsin digestion time and incubation time. In
apoptotic cells, phosphatidylserine translocates from the inner to
outer leaflet of the plasma membrane. Prolonged cell digestion time
or incubation time may increase the translocation of
phosphatidylserine, which can be identified and bound by Annexin V
and ultimately cause an increase in the number of apoptotic cells.
Cells may also undergo autophagy, which is another factor that
should be considered in the two assays. Autophagy can lead to
nonapoptotic cell death. Yu et al demonstrated that the
degradation of the reactive oxygen species scavenger, catalase,
results in the accumulation of reactive oxygen species in the cell,
leading to loss of membrane integrity and ultimately cell death
(37). The loss of membrane
integrity may cause an increase in the apoptotic rate in the
annexin V/PI double staining assays. In the JC-1 staining assay, 15
µM ATO treatment markedly increased the percentages of
mitochondrial membrane depolarization compared with the DMSO
control group and 5 and 10 µM ATO treatment groups. ATO-induced
mitochondrial membrane permeabilization is limited to only a subset
of mitochondria, autophagosomal elimination of the depolarized
mitochondria (known as mitophagy) will take place and promote cell
survival (38). On the other hand,
15 µM ATO treatment can lead to extensive mitochondrial membrane
permeabilization and finally results in apoptotic cell death, as
shown by increased percentages of mitochondrial membrane potential
depolarization. ATO treatment altered the expression of
apoptosis-related genes, as evidenced by downregulation of the mRNA
expression of Bcl-2 and upregulation of Bax. While the induction of
Bcl-2 leads to inhibition of apoptotic mechanisms and the
development of drug resistance (39), downregulation of Bcl-2 may be one of
the underlying mechanisms of apoptosis induced by ATO. The present
results suggested that cell proliferation was inhibited by inducing
cell-cycle arrest in S-phase. Similar results were also obtained in
a study on hepatocellular carcinoma, demonstrating that ATO
significantly inhibited the cell proliferation via inducing
cell-cycle arrest in S phase and the apoptotic pathway associated
with downregulation of cyclin A2 and cyclin-dependent kinase 2, and
upregulation of the expression of p53 and p21(21). mTOR is a central regulator of cell
growth, proliferation and differentiation. It also participates in
the regulation of tumor cell migration, invasion and cancer
metastasis (40). ATO induces
proapoptotic signaling and inhibits the mTOR pathway via
upregulation of activating transcription factor 4(41). In the present study, ATO suppressed
the invasiveness of EpCAM+CD44+ HCT-116 cells
and inhibited the activity of the zinc-dependent endopeptidases
MMP-2/-9 with an increased expression of TIMP-1. These proteins are
crucial in cell migration and invasion by controlling the
degradation of extracellular matrix. Previous studies suggested
that MMP-2/-9 expression is associated with the Akt/mTOR and
JAK2/STAT3 signaling pathways (42). Of note, ATO inhibits Akt/AMPK/mTOR
signaling and is a potent STAT3 inhibitor (43).
In summary, the present in vitro study
indicated that ATO inhibited the proliferation of
EpCAM+CD44+ HCT-116 cells via induction of
S-phase arrest, triggered apoptosis and inhibited cell invasion
under hypoxic conditions. However, the present study has several
limitations: Several factors, such as the differences in the
incubation time may affect the apoptotic rate in the Annexin V/PI
double staining assays and JC-1 staining assays; autophagy and
mitophagy may influence apoptosis and the interplay between
autophagy and apoptosis was not examined and the study focused on
the antitumor effects of ATO on colon cancer stem cells in
vitro. In future studies a xenograft model will be used to
determine the role of ATO in vivo. Nevertheless, the present
study laid a foundation for further investigation of the in-depth
mechanisms of the antitumor activity of ATO on colon CSCs and
provide a novel therapeutic regimen for colorectal carcinoma.
Acknowledgements
Not applicable.
Funding
The current study was supported by the Jilin
Province Science and Technology Support Program (grant no.
20200204036YY), the Education Department of Jilin Province (grant
no. JJKH20201122KJ) and the Jilin Province Health Technology
Innovation Project (grant no. 2017J062).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CF, WL and YW conceived and designed the
experiments. CF and XX performed the experiments. CF drafted the
manuscript and supervised the experiments. CF, XX and HX performed
data analysis and interpretation. WL and YW verified the results of
the experiments, helped with the statistical analysis and revised
the manuscript critically for intellectual content. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2020. CA Cancer J Clin. 70:7–30. 2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Dalerba P, Dylla SJ, Park IK, Liu R, Wang
X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, et al:
Phenotypic characterization of human colorectal cancer stem cells.
Proc Natl Acad Sci USA. 104:10158–10163. 2007.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ricci-Vitiani L, Lombardi DG, Pilozzi E,
Biffoni M, Todaro M, Peschle C and De Maria R: Identification and
expansion of human colon-cancer-initiating cells. Nature.
445:111–115. 2007.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Nguyen LV, Vanner R, Dirks P and Eaves CJ:
Cancer stem cells: An evolving concept. Nat Rev Cancer. 12:133–143.
2012.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Nandy SB and Lakshmanaswamy R: Cancer stem
cells and metastasis. Prog Mol Biol Transl Sci. 151:137–176.
2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Todaro M, Alea MP, Di Stefano AB,
Cammareri P, Vermeulen L, Iovino F, Tripodo C, Russo A, Gulotta G,
Medema JP and Stassi G: Colon cancer stem cells dictate tumor
growth and resist cell death by production of interleukin-4. Cell
Stem Cell. 1:389–402. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Was H, Czarnecka J, Kominek A, Barszcz K,
Bernas T, Piwocka K and Kaminska B: Some chemotherapeutics-treated
colon cancer cells display a specific phenotype being a combination
of stem-like and senescent cell features. Cancer Biol Ther.
19:63–75. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Szarynska M, Olejniczak A and Kmieć Z: The
role of cancer stem cells in pathogenesis of colorectal cancer.
Postepy Hig Med Dosw (Online). 70:1469–1482. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Dawood S, Austin L and Cristofanilli M:
Cancer stem cells: Implications for cancer therapy. Oncology
(Williston Park). 28:1101–1107, 1110. 2014.PubMed/NCBI
|
|
10
|
Muz B, de la Puente P, Azab F and Azab AK:
The role of hypoxia in cancer progression, angiogenesis,
metastasis, and resistance to therapy. Hypoxia (Auckl). 3:83–92.
2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Vaupel P, Mayer A and Höckel M: Tumor
hypoxia and malignant progression. Methods Enzymol. 381:335–354.
2004.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhou J, Schmid T, Schnitzer S and Brune B:
Tumor hypoxia and cancer progression. Cancer Lett. 237:10–21.
2006.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Stamenkovic I: Matrix metalloproteinases
in tumor invasion and metastasis. Semin Cancer Biol. 10:415–433.
2000.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Hicklin DJ and Ellis LM: Role of the
vascular endothelial growth factor pathway in tumor growth and
angiogenesis. J Clin Oncol. 23:1011–1027. 2005.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Graham K and Unger E: Overcoming tumor
hypoxia as a barrier to radiotherapy, chemotherapy and
immunotherapy in cancer treatment. Int J Nanomedicine.
13:6049–6058. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Strese S, Fryknas M, Larsson R and Gullbo
J: Effects of hypoxia on human cancer cell line chemosensitivity.
BMC Cancer. 13(331)2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Srivastava IK, Rottenberg H and Vaidya AB:
Atovaquone, a broad spectrum antiparasitic drug, collapses
mitochondrial membrane potential in a malarial parasite. J Biol
Chem. 272:3961–3966. 1997.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Takabe H, Warnken ZN, Zhang Y, Davis DA,
Smyth HDC, Kuhn JG, Weitman S and Williams Iii RO: A Repurposed
drug for brain cancer: Enhanced atovaquone amorphous solid
dispersion by combining a spontaneously emulsifying component with
a polymer carrier. Pharmaceutics. 10(60)2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Fiorillo M, Lamb R, Tanowitz HB, Mutti L,
Krstic-Demonacos M, Cappello AR, Martinez-Outschoorn UE, Sotgia F
and Lisanti MP: Repurposing atovaquone: Targeting mitochondrial
complex III and OXPHOS to eradicate cancer stem cells. Oncotarget.
7:34084–34099. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Tian S, Chen H and Tan W: Targeting
mitochondrial respiration as a therapeutic strategy for cervical
cancer. Biochem Biophys Res Commun. 499:1019–1024. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Gao X, Liu X, Shan W, Liu Q, Wang C and
Zheng J, Yao H, Tang R and Zheng J: Anti-malarial atovaquone
exhibits anti-tumor effects by inducing DNA damage in
hepatocellular carcinoma. Am J Cancer Res. 8:1697–1711.
2018.PubMed/NCBI
|
|
22
|
Ke F, Yu J, Chen W, Si X, Li X, Yang F,
Liao Y and Zuo Z: The anti-malarial atovaquone selectively
increases chemosensitivity in retinoblastoma via mitochondrial
dysfunction-dependent oxidative damage and Akt/AMPK/mTOR
inhibition. Biochem Biophys Res Commun. 504:374–379.
2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ashton TM, Fokas E, Kunz-Schughart LA,
Folkes LK, Anbalagan S, Huether M, Kelly CJ, Pirovano G, Buffa FM,
Hammond EM, et al: The anti-malarial atovaquone increases
radiosensitivity by alleviating tumour hypoxia. Nat Commun.
7(12308)2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Fu C, Zhou N, Zhao Y, Duan J, Xu H and
Wang Y: Dendritic cells loaded with CD44 CT-26 colon cell lysate
evoke potent antitumor immune responses. Oncol Lett. 18:5897–5904.
2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Phi LTH, Sari IN, Yang YG, Lee SH, Jun N,
Kim KS, Lee YK and Kwon HY: Cancer stem cells (CSCs) in drug
resistance and their therapeutic implications in cancer treatment.
Stem Cells Int. 2018(5416923)2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Falloon J, Sargent S, Piscitelli SC,
Bechtel C, LaFon SW, Sadler B, Walker RE, Kovacs JA, Polis MA,
Davey RT Jr, et al: Atovaquone suspension in HIV-infected
volunteers: Pharmacokinetics, pharmacodynamics, and TMP-SMX
interaction study. Pharmacotherapy. 19:1050–1056. 1999.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Cosse JP and Michiels C: Tumour hypoxia
affects the responsiveness of cancer cells to chemotherapy and
promotes cancer progression. Anticancer Agents Med Chem. 8:790–797.
2008.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhang C, Tian Y, Song F, Fu C, Han B and
Wang Y: Salinomycin inhibits the growth of colorectal carcinoma by
targeting tumor stem cells. Oncol Rep. 34:2469–2476.
2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Xiong B, Ma L, Hu X, Zhang C and Cheng Y:
Characterization of side population cells isolated from the colon
cancer cell line SW480. Int J Oncol. 45:1175–1183. 2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Müller M, Hermann PC, Liebau S, Weidgang
C, Seufferlein T, Kleger A and Perkhofer L: The role of
pluripotency factors to drive stemness in gastrointestinal cancer.
Stem Cell Res. 16:349–357. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Larsson P, Engqvist H, Biermann J, Werner
Rönnerman E, Forssell-Aronsson E, Kovács A, Karlsson P, Helou K and
Parris TZ: Optimization of cell viability assays to improve
replicability and reproducibility of cancer drug sensitivity
screens. Sci Rep. 10(5798)2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Chen D, Sun X, Zhang X and Cao J:
Targeting mitochondria by anthelmintic drug atovaquone sensitizes
renal cell carcinoma to chemotherapy and immunotherapy. J Bioch Mol
Toxicol. 32(e22195)2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Baggish AL and Hill DR: Antiparasitic
agent atovaquone. Antimicrob Agents Chemother. 46:1163–1173.
2002.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Nixon GL, Moss DM, Shone AE, Lalloo DG,
Fisher N, O'Neill PM, Ward SA and Biagini GA: Antimalarial
pharmacology and therapeutics of atovaquone. J Antimicrob
Chemother. 68:977–985. 2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zhou J, Duan L, Chen H, Ren X, Zhang Z,
Zhou F, Liu J, Pei D and Ding K: Atovaquone derivatives as potent
cytotoxic and apoptosis inducing agents. Bioorg Med Chem Lett.
19:5091–5094. 2009.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Yu L, Wan F, Dutta S, Welsh S, Liu Z,
Freundt E, Baehrecke EH and Lenardo M: Autophagic programmed cell
death by selective catalase degradation. Proc Natl Acad Sci USA.
103:4952–4957. 2006.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Rambold AS and Lippincott-Schwartz J:
Mechanisms of mitochondria and autophagy crosstalk. Cell Cycle.
10:4032–4038. 2011.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Kinoshita M, Johnson DL, Shatney CH, Lee
YL and Mochizuki H: Cancer cells surviving hypoxia obtain hypoxia
resistance and maintain anti-apoptotic potential under
reoxygenation. Int J Cancer. 91:322–326. 2001.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Zhou H and Huang S: Role of mTOR signaling
in tumor cell motility, invasion and metastasis. Curr Protein Pept
Sci. 12:30–42. 2011.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Stevens AM, Xiang M, Heppler LN, Tošić I,
Jiang K, Munoz JO, Gaikwad AS, Horton TM, Long X, Narayanan P, et
al: Atovaquone is active against AML by upregulating the integrated
stress pathway and suppressing oxidative phosphorylation. Blood
Adv. 3:4215–4227. 2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Fang Z, Tang Y, Fang J, Zhou Z, Xing Z,
Guo Z, Guo X, Wang W, Jiao W, Xu Z and Liu Z: Simvastatin inhibits
renal cancer cell growth and metastasis via AKT/mTOR, ERK and
JAK2/STAT3 pathway. PLoS One. 8(e62823)2013.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Xiang M, Kim H, Ho VT, Walker SR,
Bar-Natan M, Anahtar M, Liu S, Toniolo PA, Kroll Y, Jones N, et al:
Gene expression-based discovery of atovaquone as a STAT3 inhibitor
and anticancer agent. Blood. 128:1845–1853. 2016.PubMed/NCBI View Article : Google Scholar
|