Introduction
Chronic obstructive pulmonary disease (COPD) has
become a significant risk of public health, especially in adults
aged >70 years (1,2). In 2010, ~11.7% of people aged ≥30
years were diagnosed with COPD worldwide (3). According to the data from the
Institute for Health Metrics and Evaluation in 2017, the number of
deaths globally due to COPD is ~3 million every year; in 2030, it
is predicted that COPD will be the third most common disease of
death worldwide (4). Subjects with
COPD exhibit a number of symptoms, such as breathlessness and
persistent productive coughing (5).
Smoking is the leading risk factor for COPD, although according to
14 countries from the international Burden of Obstructive Lung
Disease study, 20% of non-smokers were diagnosed with COPD in
2008(6). However, the standard
treatment methods for COPD, such as cessation of smoking, may
result in anxiety and depression in smokers (7). Thus, research into the underlying
mechanism of how smoking influences COPD is essential for the
development of treatments for the disease.
Long non-coding RNAs (lncRNAs) are >200
nucleotides long and are important regulators of various biological
processes, including cell proliferation, apoptosis, and
immunization (8-10).
An increasing number of studies have demonstrated that lncRNAs are
aberrantly expressed in COPD and are involved in cell inflammatory
response and apoptosis (11-13).
The expression of sphingosine-1-phosphate receptor 1 is promoted by
the lncRNA long intergenic noncoding RNA antisense to S1PR1 to
increase angiogenic capacity by regulating the
sphingosine-1-phosphate signaling pathway in human umbilical vein
endothelial cells (14). The lncRNA
maternally expressed gene 3 (MEG3) has been identified as a tumor
inhibitor, which suppresses the apoptosis and migration of cancer
cells (15). Additionally, MEG3 is
significantly increased in lung tissues of subjects with COPD
compared with non-COPD lung tissues (16). In addition, a recent study has
indicated that knockdown of MEG3 inhibits human bronchial
epithelial (HBE) cell apoptosis and autophagy by regulating the
expression of p53(17). However,
the exact role and underlying mechanism of lncRNA MEG3 in COPD
requires further elucidation.
MicroRNAs (miRNAs/miRs) are non-coding RNA that
serve vital roles in silencing or cleaving target mRNA at the
post-transcriptional level (18).
miRNAs can bind to the 3'-UTR of the target gene mRNA by binding
the RNA-induced silencing complex (19-21).
In addition, miRNA activity can be impaired by lncRNA through
sequestration, upregulating target gene expression (22). A previous study has demonstrated
that miRNAs, such as miR-218, -203 and -146a, as well as miR-34
family (-34a/b/c) and let-7 family are potential biomarkers for the
early diagnosis and treatment of COPD (23). Recently, miR-149-3p was demonstrated
to be associated with smoking-induced COPD, and its downregulation
increased inflammation in subjects with COPD via the Toll-like
receptor-4/nuclear factor-κB (NF-κB) signaling pathway (24). The principal regulator of
inflammatory gene expression is NF-κB and its activity is
controlled by IκB proteins, whose stimulus-responsive degradation
and re-synthesis provide for transient or dynamic regulation. Thus,
NF-κB signaling serves a major role in mediating inflammatory and
innating immune responses (25). In
lung tissues from patients with mild emphysema, the expression of
miR-149-3p was decreased compared with tissues from patients with
moderate emphysema (26). Thus, it
was hypothesized that miR-149-3p may be involved in the NF-κB
signaling pathway by regulating MEG3 expression.
The aim of the present study was to verify the role
of lncRNA MEG3 in COPD, the underlying interaction between MEG3 and
miR-149-3p and the effect of this interaction on cell proliferation
and apoptosis in HEB cells.
Materials and methods
Preparation of cigarette smoke extract
(CSE)
CSE was collected as previously described (27). Briefly, the filters of two 3R4F
research cigarettes from the University of Kentucky were removed,
and the cigarettes were burned out in 5 min. All the smoke produced
by two cigarettes was transferred into a 50 ml tube with 20 ml
sterile saline solution (Sinopharm Group Co., Ltd.), and the
extract was filtered through a 0.22-µm polytetrafluoroethylene
filter (EMD Millipore).
Subjects and cell culture
A total of 66 subjects (16 women and 50 men; median
age 58 years (range, 40-80 years); 18 non-smokers without COPD, 24
smokers without COPD and 28 smokers with COPD) were recruited at
the Qinghai University Affiliated Hospital (Xining, China) between
October 2018 and May 2019. Table I
summarizes the patient characteristics and clinical features.
Healthy subjects [forced expiratory volume in one second as a
percentage of the predicted (FEV1%) ≥70% and forced expiratory
volume in one second/forced vital capacity (FEV1/FVC) ≥70%]
(5) without chronic bronchitis and
emphysema, and all patients with COPD were stable (FEV1/FVC<70%)
and had no exacerbations in the prior 6 months were enrolled.
Peripheral blood samples were collected from the subjects, and
maintained at -80˚C. The present study was approved by the Ethics
Committee of Qinghai University Affiliated Hospital, and all
subjects provided written informed consent prior to participation
in the study. The diagnosis of emphysema was made by the
pathologist based on histological examination. All blood samples
were maintained at -80˚C until processing of total RNA
isolation.
 | Table IClinical and demographic
characteristics of the study subjects (n=66). |
Table I
Clinical and demographic
characteristics of the study subjects (n=66).
| Variables | Non-smokers | Smokers without
COPD | Smokers with
COPD |
|---|
| Total subjects,
n | 20 | 22 | 24 |
| Age, years | 53.4±3.6 | 55.8±4.2 | 62.1±2.5 |
| Sex, n
male/female | 12/8 | 17/5 | 21/3 |
| BMI,
kg/m2 | 22.4±1.3 | 21.6±0.8 | 22.1±1.5 |
| FVC, %pred. | 91.3±6.4 | 80.5±10.2 | 64.2±11.0 |
| FEV1, %pred. | 80.0±7.5 | 66.3±8.1 | 40.8±7.8 |
| FEV1/FVC,
%pred. | 87.6±8.2 | 82.4±7.2 | 63.5±10.2 |
The HBE cell line (HBE135-E6E7) was purchased from
the American Type Culture Collection (cat. no. CRL-2741). HBE cells
were cultured in RPMI-1640 (HyClone; GE Healthcare life Sciences)
medium containing 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.) with penicillin (100 U/ml l/Streptomycin (100
µg/ml; Sigma-Aldrich; Merck KGaA). The cells were cultured at 37˚C
in a 5% CO2 incubator. Cells were pretreated at 37˚C
with 5% CSE for 24 h before transfection and subsequent
experiments.
Cell transfection
For transfection experiments, miR-149-3p mimics
(miR-149-3p), miR-negative control (miR-NC) mimics, miR-149-3p
inhibitor (anti-miR-149-3p), miR-NC inhibitor (anti-miR-NC), small
interfering (si)RNA targeting MEG3 (si-MEG3#1 and si-MEG3#2 were
used to screen the one with more significant suppressive effect on
MEG3) and si-NC were obtained from Guangzhou RiboBio Co., Ltd. The
MEG3 overexpression plasmid and empty vector was purchased from
Hanbio Biotechnology Co., Ltd. In brief, the plasmids, miRNA mimics
and miRNA inhibitors were transfected into 1x106
HBE135-E6E7 and pretreated at 37˚C with 5% CSE for 24 h using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific Inc.) according to the manufacturer's instructions.
Cells were cultured at 37˚C with 5% CO2 for 48 h after
transfection. The sequences of miR-149-3p, miR-NC, anti-miR-149-3p,
anti-miR-NC, si-MEG3#1, si-MEG3#2 are: miR-149-3p,
5'-AGGGAGGGACGGGGGCUGUGC-3'; miR-NC, 5'-UUCUCCGAACGUGUCACGUTT-3';
anti-miR-149-3p, 5'-GCACAGCCCCCGUCCCUCCCU-3'; anti-miR-NC,
5'-CAGUACUUUUGUGUAGUACAA-3'; si-MEG3#1,
5'-AACAGCAAAUGGCACAGGAAGAGACGC-3'; and si-MEG3#2,
5'-AUUGGAGGUGAGGAAGGAAAGCAGC-3'.
Reverse transcription-quantitative PCR
(RT-qPCR)
TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific Inc.) was used to extract the total RNA from the
blood samples or HBE cells treated with various methods. The
quality and quantity of total RNA were measured using the NanoDrop
2000 (Thermo Fisher Scientific Inc.). RNA reverse transcription was
conducted using a GoScript Reverse Transcription System (Promega
Corporation), and qPCR was performed using the
SYBR®-Green I Supermix (Takara Biotechnology Co., Ltd.)
according to the manufacturer's instructions. The thermocycling
conditions were as follows: 95˚C pre-denaturation for 3 min,
followed by 40 cycles of 95˚C denaturation for 15 sec and 60˚C
annealing for 1 min. The relative expression of MEG3 or miR-149-3p
was calculated using glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) or U6 small nuclear RNA (snRNA), respectively, as an
internal control, and all data were calculated by the
2-ΔΔCq method (28) with
three replicates. The primers were synthesized by Sangon Biotech
Co, Ltd., and the primer sequences used were as follows: MEG3
forward, 5'-TCCATGCTGAGCTGCTGCCAAG-3' and reverse,
5'-AGTCGACAAAGACTGACACCC-3'; miR-149-3p forward,
5'-GAACCGGGATGGGAAGTGAC-3' and reverse, 5'-GCAAGCGGAACTTCTAGCCT-3';
GAPDH forward, 5'-GACTCCACTCACGGCAAATTCA-3' and reverse,
5'-TCGCTCCTGGAAGATGGTGAT-3'; and U6 forward,
5'-CTCGCTTCGGCAGCACA-3, and reverse 5'-AACGCTTCACGAATTTGCGT-3'.
Cell counting kit-8 (CCK-8) assay
HBE cell viability was assessed using the Cell
Counting Kit-8 (CCK-8; Beyotime Institute of Biotechnology)
according to the manufacturer's instructions. Following treatment
with CSE, cells were seeded into 96-well plates at a density of
~3,000 cells/well. Untreated cells were used as a negative control.
The cells were cultured at 37˚C with 5% CO2 for 48 h
prior to the addition of 10 µl CCK-8 reagent and incubated at 37˚C
for 2 h. The optical density was detected by a microplate reader
(Bio-Rad Laboratories Inc.) at 450 nm.
Enzyme-linked immunosorbent assay
(ELISA)
Interleukin 6 (IL-6) is the most commonly assayed
biomarkers to infer an underlying state of inflammation (29) and tumor necrosis factor-α (TNF-α)
plays an important pro-inflammatory role in COPD (30), thus IL-6 and TNF-α were selected to
detect the effect of MEG3 in inflammatory response. The expression
of interleukin-6 (IL-6) and TNF-α were detected using Human IL-6
and Human TNF-α ELISA Kits (cat. nos. ab46042 and ab181421,
respectively; both Abcam) as previously described (31). The detailed procedure of ELISA was
as follows: First, HBE cells induced by CSE and sterile saline
solution were added into a simpleStep ELISA plate that had been
coated with monoclonal antibody specific for IL-6 or TNF-α (from
ELISA kit). Following incubation at 37˚C for 30 min, the plates
were washed 3 times with PBS. Then, HBE cells were incubated with
Human TNF-α Detector Antibody or Human IL-6 Detector Antibody (from
the ELISA kits) at 37˚C for 1 h. Then 100 µl of the TMB substrate
was aded into each well and incubate for 30 min at room temperature
in the dark. Finally, the Stop Solution was added into each well
and the absorbance at 405 nm was measured by an ELISA instrument
(Thermo Fisher Scientific Inc.) after a 20-min incubation with the
chromogenic agent.
Apoptosis assay
To measure the apoptotic rates in HBE cells, the
Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI)
method was used, and the Annexin V-FITC/PI apoptosis detection kit
was purchased from Beijing Solarbio Science & Technology Co.,
Ltd.. According to the manufacturer's instructions, cells were
harvested following culture for 2 days at 37˚C with 5%
CO2 and resuspended in binding buffer. Subsequently, the
cell suspension was mixed with Annexin V-FITC (1:20) and PI (1:20),
followed by incubation at room temperature in the dark for 15 min.
Finally, the apoptotic cells were detected using a flow cytometer
(BD Accuri™ C6; BD Biosciences) and FlowJo 10.2 (BD Biosciences)
were used for analysis. The sum of apoptosis rate in right upper
quadrant and right lower quadrant were considered as the cell
apoptosis rate.
Western blotting
Total protein was extracted from HBE cells using
RIPA lysis buffer (Beyotime Institute of Biotechnology) for 30 min.
A sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) gel (10-15%) was used to separate proteins and each lane
had an equal amount of protein loaded in it (~30 µg). Subsequently,
proteins were transferred to polyvinylidene fluoride membranes (EMD
Millipore). 5% BSA was used to dilute primary antibodies. Following
blocking with 5% non-fat milk at room temperature for 1 h, the
membranes were incubated with primary antibodies against Bcl-2
(1:1,000; cat. no. ab32124; Abcam), caspase-3 (1:1,000; cat. no.
ab2302; Abcam), caspase-9 (1:1,000; cat. no. ab2324; Abcam), p65
(1:1,000; cat. no. ab16502; Abcam), phospho S536-p65 (1:1,000; cat.
no. ab86299; Abcam), IκBα (1:1,000; cat. no. ab95338; Abcam);
phospho-S36-IκBα (1:1,000; cat. no. ab133462; Abcam) or GAPDH
(1:1,000; cat. no. ab37168; Abcam), which HRP-conjugated secondary
antibody Goat Anti-Rabbit IgG H&L (1:20,000; cat. no. ab97051;
Abcam) was used to probe the proteins on the membranes at room
temperature for 1 h. Finally, protein bands were visualized using a
commercial enhanced chemiluminescence chromogenic substrate
(Beyotime Institute of Biotechnology), and the densitometry and
quantity of the protein was analyzed using Quantity One 1-D
Analysis software (v4.6.6; Bio-Rad Laboratories Inc.).
Dual luciferase reporter assay
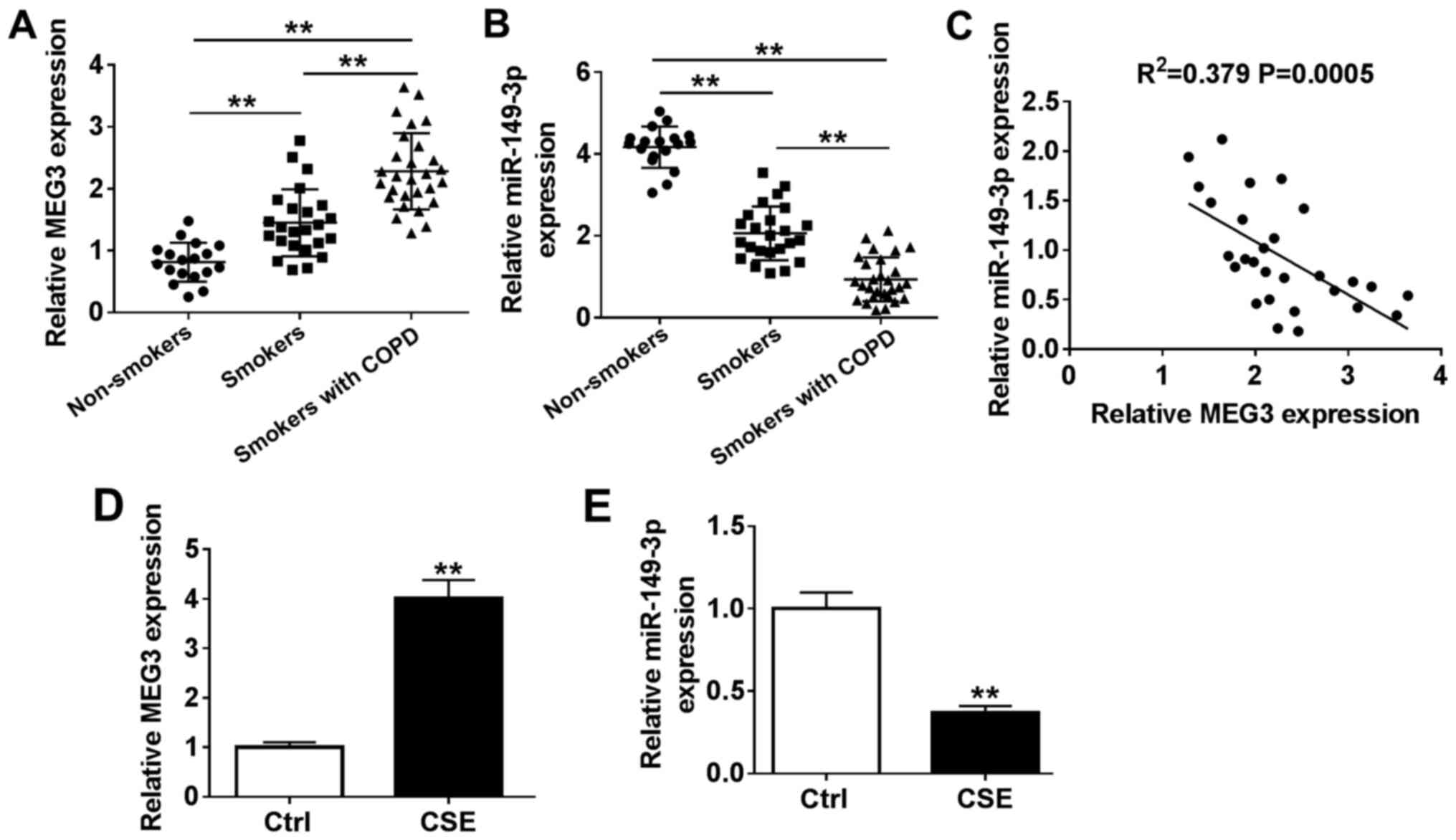
-0.616; P<0.001; Fig.
1C. In addition, the expression of MEG3 was significantly
upregulated in CSE-induced HBE cells compared with that in the
control cells, whereas miR-149-3p expression was reduced (Fig. 1D and E).
MEG3 inhibits cell proliferation,
induces apoptosis and regulates inflammatory cytokine expression in
CSE-induced HBE cells
To explore the role of MEG3 in COPD, HBE cells were
used for further investigation. RT-qPCR was performed in untreated
and CSE-induced HBE cells. The expression of MEG3 was elevated in
CSE-induced HBE cells compared with untreated HBE cells, but
reduced in CSE-induced HBE cells transfected with si-MEG3 compared
with HBE cells transfected with si-NC (Fig. 2A). The CCK-8 assay revealed that
cell proliferation in CSE-induced HBE cells was prompted by the
knockdown of MEG3 (Fig. 2B). In
addition, the levels of IL-6 and TNF-α were measured by ELISA, and
the results demonstrated that the levels of the two cytokines was
significantly reduced in CSE-induced HBE cells transfected with
si-MEG3 compared with CSE-induced HBE cells transfected with si-NC
(Fig. 2C and D). The apoptotic rate was markedly
decreased in CSE-induced HBE cells transfected with si-MEG3
compared with that in the CSE-treated si-NC group (Fig. 2E). Western blotting was used to
determine the protein expression levels of Bcl-2, cleaved-caspase-3
and cleaved-caspase-9; compared with the si-NC group, knockdown of
MEG3 enhanced the protein expression of Bcl-2 and inhibited that of
cleaved-caspase-3/pro-caspase-3 and cleaved-caspase-9/pro-caspase-9
in CSE-induced HBE cells (Fig. 2F).
In conclusion, MEG3 knockdown enhanced cell proliferation,
inhibited apoptosis and regulated inflammatory cytokine levels in
CSE-induced HBE cells.
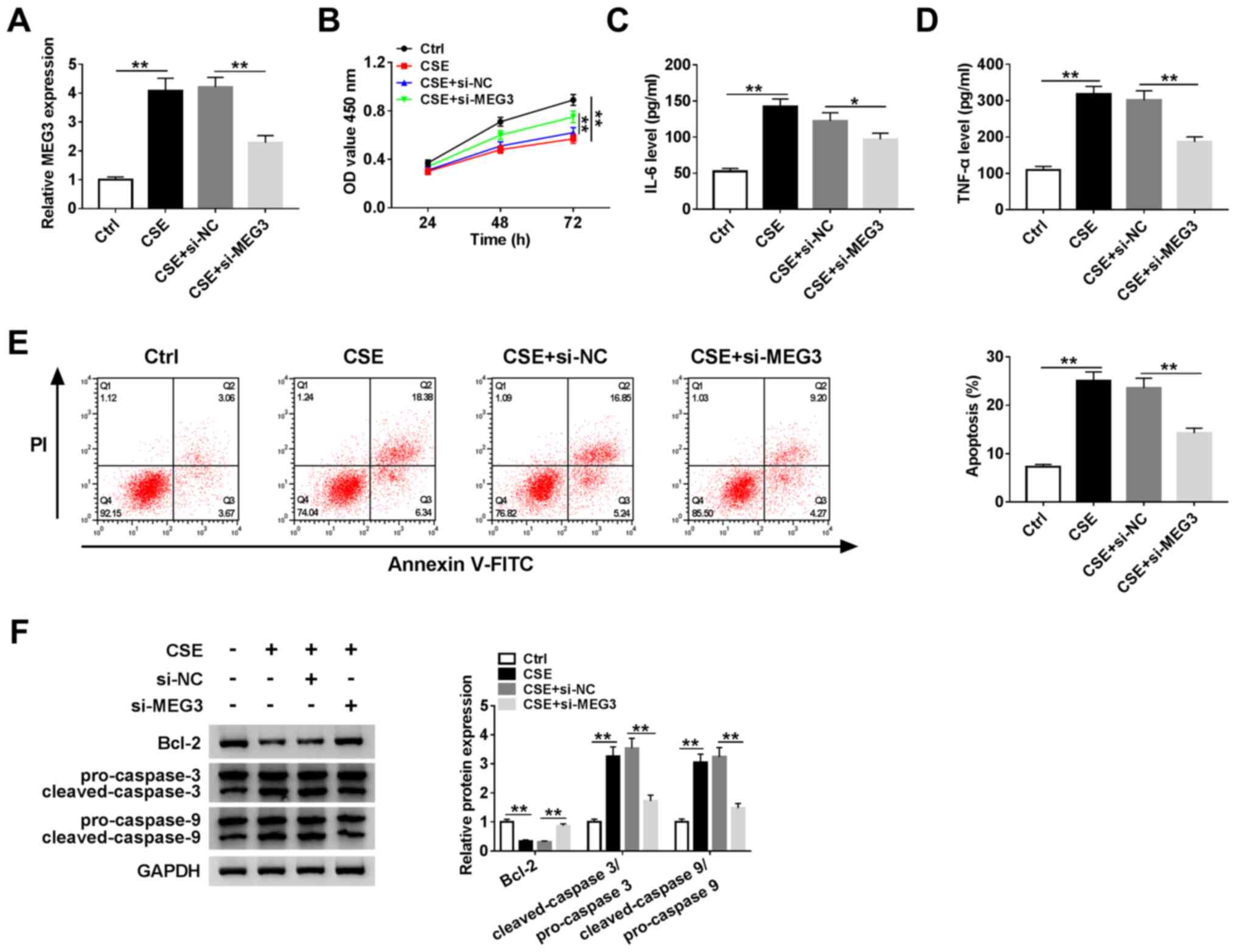 | Figure 2Effects of MEG3 knockdown on cell
proliferation, apoptosis and inflammatory cytokine expression in
CSE-induced HBE cells. Human bronchial epithelial cells were
treated with control (untreated HBE cells), CSE, CSE + si-NC or CSE
+ si-MEG3. (A) mRNA expression of MEG3 in treated HBE cells was
measured using reverse transcription-quantitative PCR. (B) Cell
proliferation was examined by the Cell Counting Kit-8 assay. (C and
D) The levels of IL-6 and TNF-α were detected using ELISA. (E)
Apoptosis was analyzed by flow cytometry. (F) Protein expression
levels of Bcl-2, caspase-3 and caspase-9 were determined by western
blotting. **P<0.01. MEG3, maternally expressed gene
3; miR, microRNA; CSE, cigarette smoke extract; OD, optical
density; PI, propidium iodide; NC, negative control; IL,
interleukin; TNF, tumor necrosis factor; si, small interfering
RNA. |
MEG3 targets miR-149-3p to
downregulate its expression
The DIANA online tool was used to predict the
binding site of MEG3 and miR-149-3p. It was identified that MEG3
may bind miR-149-3p. It was also observed that there was a
potential binding site for miR-149-3p in MEG3 and certain bases
were changed to obtain MEG3 mutant sequence (Fig. 3A). To verify this prediction,
MEG3-WT and MEG3-MUT vectors with luciferase reporter were
constructed and co-transfected into cells with miR-149-3p. The
transfection efficiency of miR-149-3p mimics was first verified
(Fig. S1A). The luciferase
activity of MEG3-WT in HEB cells significantly declined in the
presence of miR-149-3p mimics while HEB cells co-transfected with
MEG3-MUT and miR-149-3p did not exhibit a significant difference
(Fig. 3B). However, there were no
notable changes in the luciferase activity of MEG3-MUT in the
presence of miR-149-3p mimics (Fig.
3B). To further explore the relationship between MEG3 and
miR-149-3p, overexpression and knockdown MEG3 plasmids were
transfected into HBE cells, and RT-qPCR was used to verify the up-
and downregulation of MEG3 expression (Fig. 3C and D). Subsequently, the level of miR-149-3p
was measured in cells transfected with MEG3 or si-MEG3; the results
demonstrated that the expression of miR-149-3p was significantly
reduced in HBE cells overexpressing MEG3, but increased in cells
transfected with si-MEG3 compared with the respective negative
controls (Fig. 3E and F). These results suggested that miR-149-3p
was a target of MEG3 and was downregulated by MEG3.
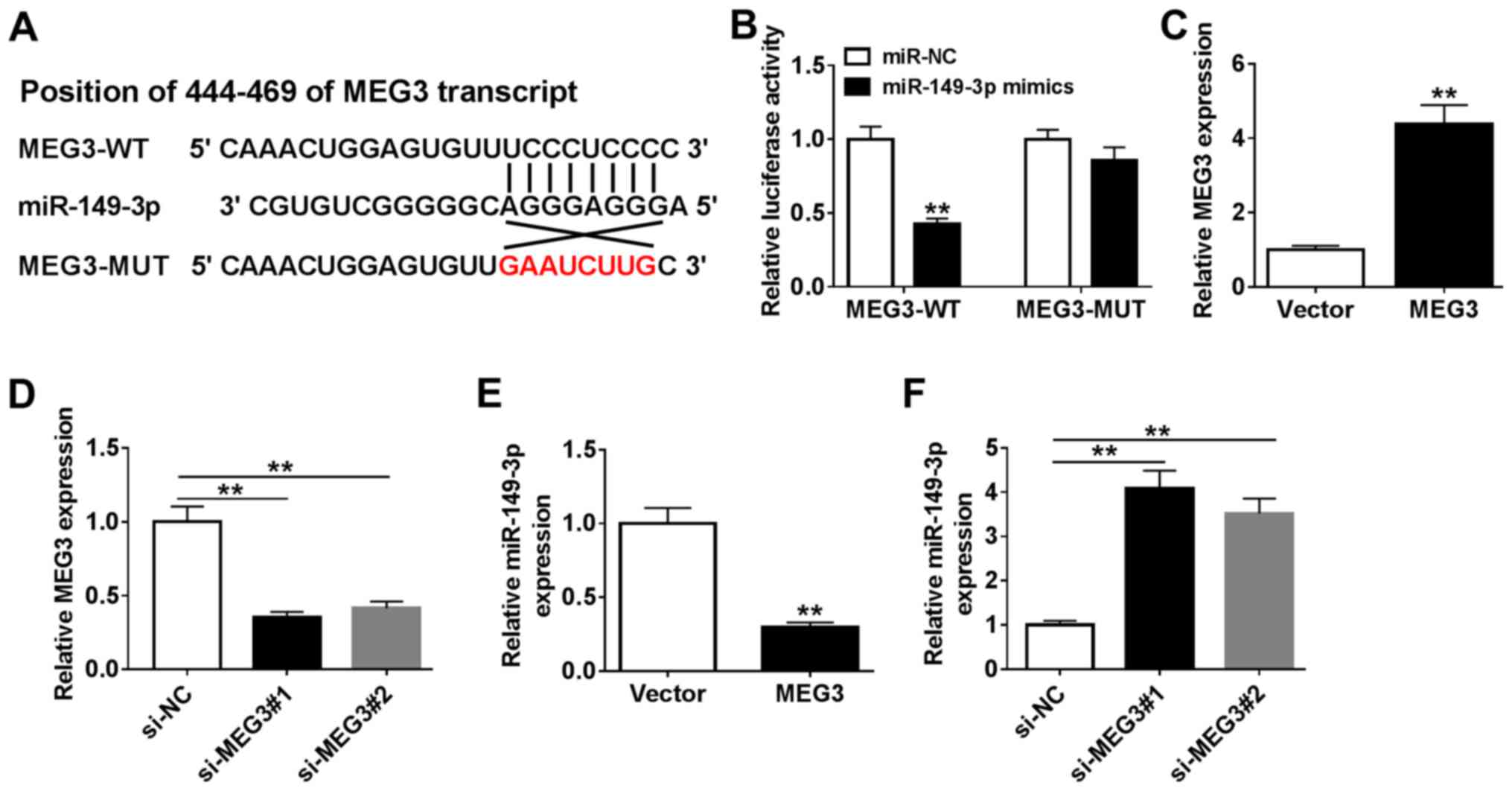 | Figure 3Relationship between MEG3 and
miR-149-3p. (A) The DIANNA online tool predicted the putative
binding sites of MEG3 and miR-149-3p and MUT sequence of MEG3 is
shown in red. (B) Luciferase activity was evaluated in HBE cells
co-transfected with MEG3-WT or MEG3-MUT and miR-NC or miR-149-3p
mimics. (C and E) The expression of MEG3 or miR-149-3p was examined
in HBE cells transfected with empty vector and MEG3. (D and F)
Levels of MEG3 or miR-149-3p were measured in HBE cells transfected
with si-NC, si-MEG3#1, and si-MEG3#2. **P<0.01. MEG3,
maternally expressed gene 3; miR, microRNA; CSE, cigarette smoke
extract; HBE, human bronchial epithelial; NC, negative control; si,
small interfering RNA; WT, wild-type; MUT, mutant. |
MEG3 enhances cell proliferation and
reduces the apoptotic rate in CSE-induced HBE cells by targeting
miR-149-3p
To verify the effects of MEG3 on regulating cell
proliferation and apoptosis, anti-miR-NC, miR-149-3p inhibitor or
si-MEG3 were transfected into CSE-induced HBE cells. The
transfection efficiency of the miR-149-3p inhibitor was verified
and presented in Fig. S1B. RT-qPCR
was performed to detect the expression of miR-149-3p in CSE-treated
HBE cells. The results revealed that miR-149-3p expression was
significantly decreased in cells treated with the miR-149-3p
inhibitor compared with that in the miR-NC group; similarly, the
expression of miR-149-3p was upregulated in CSE-induced HBE cells
with MEG3 knockdown, but it was reduced when co-treated with
miR-149-3p inhibitor (Fig. 4A).
Next, the proliferation of HBE cells was analyzed using the CCK-8
assay. Compared with untreated CSE-induced HBE cells, miR-149-3p
inhibitor treated group reduced the proliferation of CSE-induced
HBE cells obviously (Fig. 4B). When
CSE-induced HBE cells were co-transfected with si-MEG3 and
miR-149-3p, cell proliferative ability was also decreased following
72-h culture compared with CSE-induced HBE cells co-transfected
with si-MEG3 and miR-NC (Fig. 4B).
Apoptosis of HBE cells was measured using flow cytometry, which
revealed that apoptosis was enhanced when miR-149-3p was inhibited
in GSE-induced HBE cells, and this apoptosis inducing effect was
reversed by co-transfected group (miR-149-3p inhibitor and si-MEG3)
(Fig. 4C and D). Western blotting was performed to
detect the expression of the apoptosis-related proteins Bcl-2,
cleaved-caspase-3 and cleaved-caspase-9. The results demonstrated
that the expression of Bcl-2 was downregulated, whereas
cleaved-caspase-3/pro-caspase-3 and cleaved-caspase-9/pro-caspase-9
were upregulated in CSE-induced cells transfected with si-MEG3 and
miR-149-3p inhibitor compared with CSE-induced cells transfected
with si-MEG3 and miR-NC (Fig. 4E).
An ELISA assay demonstrated that the levels of IL-6 and TNF-α,
which are important inflammatory cytokines, were increased by the
simultaneous knockdown of MEG3 and miR-149-3p compared with
CSE-induced cells transfected with si-MEG3 and miR-NC (Fig. 4F and G). Taken together, these results
demonstrated that miR-149-3p partly reversed the regulating effect
of MEG3 on cell proliferation, apoptosis and inflammatory cytokine
expression in CSE-induced HBE cells.
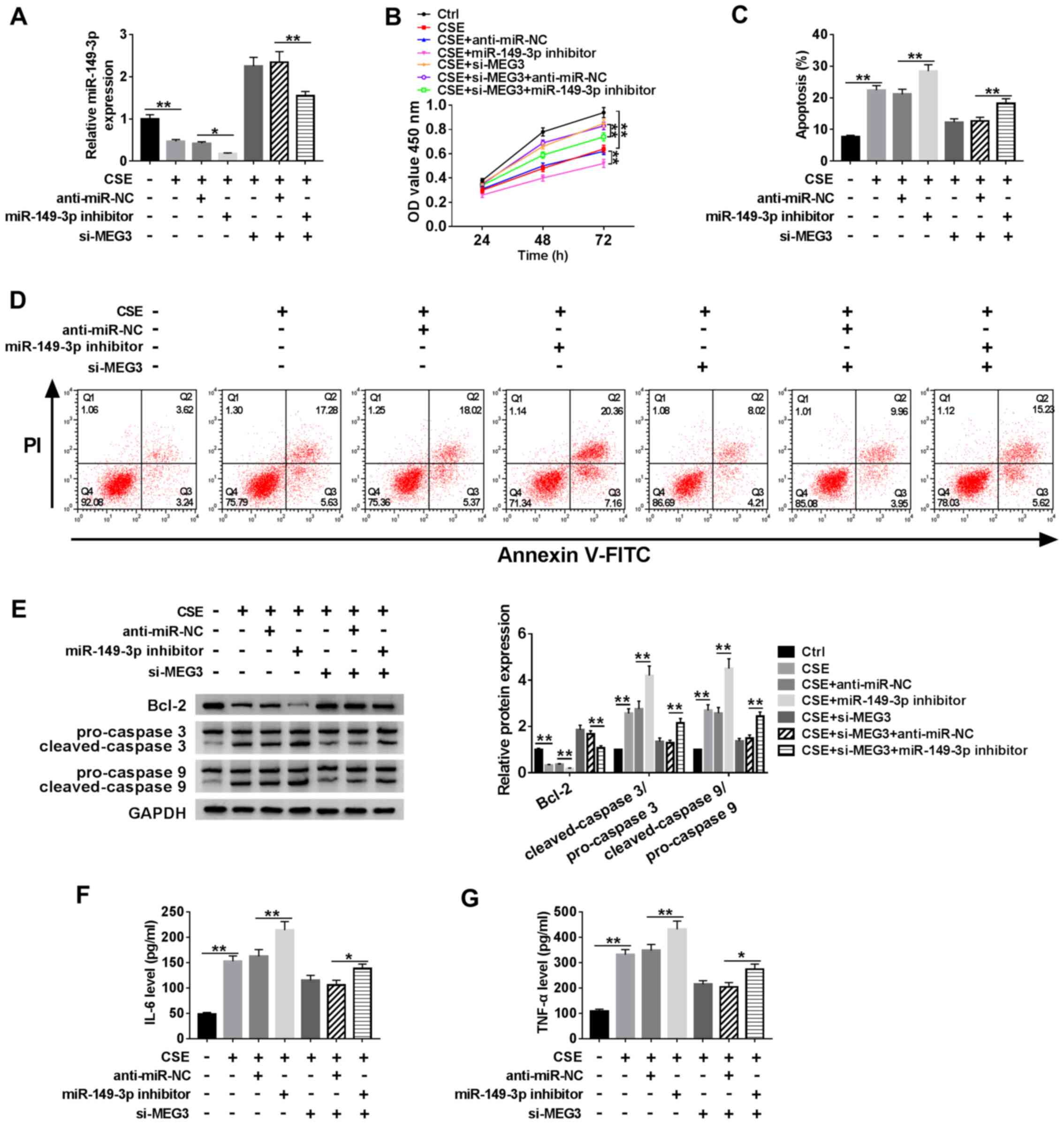 | Figure 4MEG3 regulatory effects on
miR-149-3p-mediated HBE cell proliferation, apoptosis and
inflammatory cytokine expression. HBE cells were treated with
control (untreated HBE cells), CSE, CSE + anti-miR-NC, CSE +
miR-149-3p inhibitor, CSE + si-MEG3, CSE + si-MEG3 + anti-miR-NC or
CSE + si-MEG3 + miR-149-3p inhibitor. (A) The expression of
miR-149-3p was determined using reverse-transcription quantitative
PCR in treated HBE cells. (B) Cell proliferation of treated HBE
cells was determined using the Cell Counting Kit-8 assay. (C and D)
Apoptosis analyzed by flow cytometry. (E) Protein levels of Bcl-2,
caspase-3 and caspase-9 were measured by western blotting. (F)
ELISA was used to measure the levels of IL-6 and (G) TNF-α.
**P<0.01. MEG3, maternally expressed gene 3; miR,
microRNA; CSE, cigarette smoke extract; OD, optical density; PI,
propidium iodide; NC, negative control; IL, interleukin; TNF, tumor
necrosis factor; si, small interfering RNA. |
MEG3 regulates the NF-κB signaling
pathway in CSE-induced HBE cells by targeting miR-149-3p
To elucidate the molecular mechanism of MEG3 and
miR-149-3p in the NF-κB signaling pathway, p-p65, t-p65, p-IκBα,
and t-IκBα, which are involved in the NF-κB pathway, were selected
for evaluation using western blotting. CSE-induced HBE cells were
co-transfected with miR-149-3p inhibitor or/and si-MEG3 (Fig. 5). The ratio of p-p65/t-p65 and
p-IκBα/t-IκBα was increased when the expression of miR-149-3p was
inhibited by miR-149-3p inhibitor, compared with the negative
control group; however, it would be reversed when MEG3 was
downregulated with miR-149-3p inhibitor in CSE-induced HBE cells
(Fig. 5). These results suggested
that MEG3 regulates the NF-κB signaling pathway by targeting
miR-149-3p.
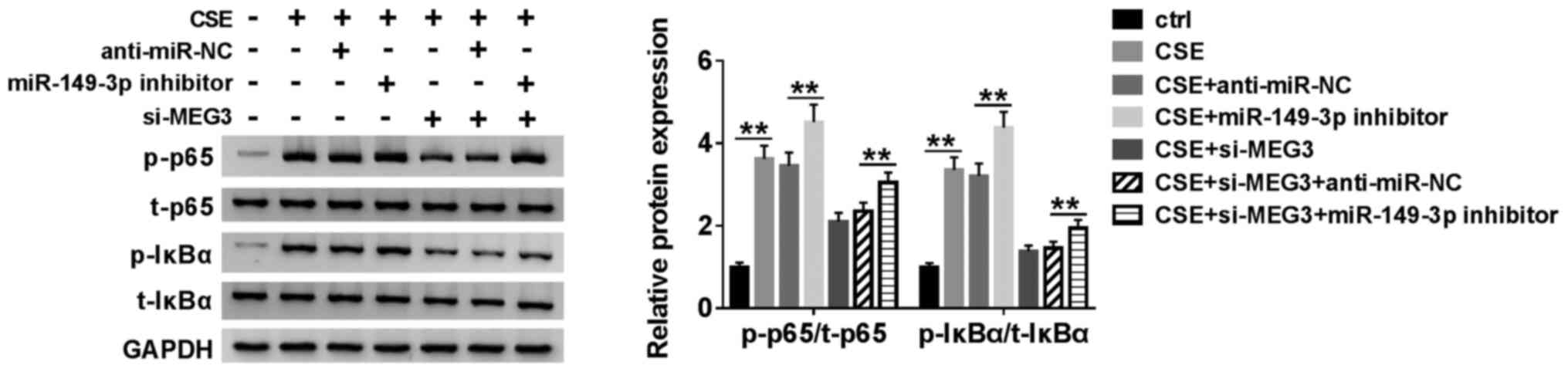 | Figure 5MEG3 regulates the NF-κB signaling
pathway by targeting miR-149-3p. HBE cells were treated with
control (untreated HBE cells), CSE, CSE + anti-miR-NC, CSE +
miR-149-3p inhibitor, CSE + si-MEG3, CSE + si-MEG3 + anti-miR-NC or
CSE + si-MEG3 + miR-149-3p inhibitor. The protein expression levels
of p-p65, t-p65, p-IκBα, p-IκBα in treated cells were determined by
western blotting. **P<0.01. MEG3, maternally
expressed gene 3; miR, microRNA; CSE, cigarette smoke extract; NC,
negative control; p, phosphorylated; t, total; si, small
interfering RNA. |
Discussion
The aim of the present study was to investigate the
potential functions and the underlying molecular mechanism of
lncRNA MEG3 in COPD. The results of the present study indicated
that MEG3 was highly expressed in blood samples from subjects with
COPD, and MEG3 knockdown promoted cell proliferation and inhibited
apoptosis in HBE cells. In addition, miR-149-3p mRNA expression was
downregulated in blood samples from subjects that smokers with COPD
compared with non-smokers or smokers without COPD and demonstrated
an inverse correlation with MEG3. The results of the present study
also revealed that miR-149-3p was a target of MEG3 and regulated
the expression of apoptotic proteins, such as bcl-2, cleaved
caspase3 and cleaved caspase 9, and inflammatory cytokines, such as
IL-6 and TNF-α. In summary, the present study demonstrated that the
effects of cell proliferation and apoptosis induced by MEG3 could
be restored by miR-149-3p mimics in CSE induced HBE cells, which
was involved in the NF-κB signaling pathway. The results of the
present study provide new evidence for the elucidation and better
understanding of the molecular mechanism of MEG3 in COPD.
lncRNAs have been reported to participate in various
biological and physiological processes, such as epigenetic
regulation, maintaining cell biological and morphological
characteristics and transcriptional regulation (32). lncRNAs, for example, TUG1 have been
demonstrated to regulate tumor growth and the development of lung
cancer (33). Silencing MEG3
suppresses HBE cell apoptosis and autophagy induced by fine
particulate matter (PM 2.5); however, the molecular basis of the
effects of lncRNA in subjects with COPD remains unclear (17). Compared with smokers and
non-smokers, MEG3 expression was significantly elevated in blood
samples from subjects with COPD in the present study, which was
consistent with the results of a previous study (16). In the present study, knockdown of
MEG3 triggered cell proliferation and inhibited apoptosis in
CSE-induced HBE cells. In addition, the rescue experiments of the
present study demonstrated that downregulation of miR-149-3p
rescued the effects of MEG3 in CSE-induced HBE cells. These results
indicated that MEG3 may serve a crucial role in COPD by binding
miR-149-3p.
Research have indicated that miRNAs may be sponged
by lncRNAs, which have been identified as competing endogenous RNAs
(34). In the present study, a
relationship between MEG3 and miR-149-3p was identified. RT-qPCR
was conducted to detect the expression of miR-149-3p in peripheral
blood samples from non-smokers without COPD, smokers without COPD
and smokers with COPD and HEB cells treated with CSE or not.
Moreover, the low expression of miR-149-3p in smokers with COPD was
regulated by MEG3.
Previous studies have demonstrated that the NF-κB
signaling pathway serves a crucial role in the development of
lipopolysaccharide-induced acute lung injury (35) and in cell inflammatory response
(36,37). In addition, Bcl-2 has been
demonstrated to be an important biomarker for inflammatory function
in the NF-κB signaling pathway (38). In the present study, the protein
expression levels of Bcl-2 were measured in HBE cells in addition
to those of cleaved-caspase-3 and cleaved-caspase-9, which are
associated with cell apoptosis (39); the level of Bcl-2 was increased in
the presence of si-MEG3 in CSE-induced HBE cells compared with
CSE-induced HBE cells transfected with si-NC. However, an opposite
trend was observed for cleaved-caspase-3 and cleaved-caspase-9. In
addition, the inhibition of miR-149-3p repressed the level of Bcl-2
when cells were co-transfected with si-MEG3. All the aforementioned
results of the present study demonstrated that MEG3 regulated the
inflammatory response by targeting miR-149-3p.
The present study provided new evidence to elucidate
the function of lncRNA MEG3 in blood samples from subjects with
COPD. miR-149-3p mimics reversed the effects of MEG3 on cell
proliferation, apoptosis and inflammatory cytokine expression. In
addition, in the present study, MEG3 regulated the NF-κB signaling
pathway in HBE cells by targeting miR-149-3p. It has been reported
that miR-149-3p is involved in the Akt1 signaling pathway in
pancreatic cancer (40) and in the
p53-signaling pathway in non-small-cell lung cancer (41). Future studies should focus on
whether the regulatory effects of MEG3/miR-149-3p in COPD may be
mediated by other signaling pathways. Although the results of the
present study have revealed the potential role of MEG3 and
miR-149-3p in vitro, in vivo studies using animal
models are essential for gaining a deeper understanding of the
molecular mechanism of COPD. Smoking is the strongest, but not the
only risk factor for COPD, and further studies are required to
explore the regulatory mechanisms of other pathogenic factors of
COPD.
In conclusion, in the present study, upregulation of
MEG3 and downregulation of miR-149-3p expression was observed in
blood samples of smokers with COPD and in CSE-induced HBE cells. In
addition, inhibition of miR-149-3p reversed the MEG3
knockdown-mediated effects on cell proliferation and apoptosis in
CSE-induced HBE cells. MEG3 was demonstrated to regulate the NF-κB
signaling pathway in CSE-induced HBE cells by targeting miR-149-3p.
Thus, the results of the present study demonstrated a role of the
MEG3/miR-149-3p axis in CSE-induced HBE cells and provided new
insight into the molecular basis of COPD, which requires further
verification.
Supplementary Material
Transfection efficiency of (A)
miR-149-3p mimics and (B) miR-149-3p inhibitor was verified by
reverse-transcription quantitative PCR. **P<0.01.
miR, microRNA; NC, negative control.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SZ, ZL and JJ conceived and designed the study. ZL
and HG carried out the experiments. JS performed data mining,
acquisition and analysis of data. SZ, YK and JJ wrote and approved
the final manuscript. All authors have read and approved the
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Qinghai University Affiliated Hospital (Xining,
China), and all subjects provided written informed consent prior to
participation in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Centers for Disease Control and Prevention
(CDC). Chronic obstructive pulmonary disease among adults-United
States, 2011. MMWR Morb Mortal Wkly Rep. 61:938–943.
2012.PubMed/NCBI
|
|
2
|
Incalzi RA, Scarlata S, Pennazza G,
Santonico M and Pedone C: Chronic obstructive pulmonary disease in
the elderly. Eur J Intern Med. 25:320–328. 2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Adeloye D, Chua S, Lee C, Basquill C,
Papana A, Theodoratou E, Nair H, Gasevic D, Sridhar D, Campbell H,
et al: Global and regional estimates of COPD prevalence: Systematic
review and meta-analysis. J Glob Health. 5(020415)2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
GBD 2015 Chronic Respiratory Disease
Collaborators. Global, regional, and national deaths, prevalence,
disability-adjusted life years, and years lived with disability for
chronic obstructive pulmonary disease and asthma, 1990-2015: A
systematic analysis for the global burden of disease study 2015.
Lancet Respir Med. 5:691–706. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Vogelmeier CF, Criner GJ, Martinez FJ,
Anzueto A, Barnes PJ, Bourbeau J, Celli BR, Chen R, Decramer M,
Fabbri LM, et al: Global strategy for the diagnosis, management,
and prevention of chronic obstructive lung disease 2017 report GOLD
executive summary. Am J Respir Crit Care Med. 195:557–582.
2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lamprecht B, McBurnie MA, Vollmer WM,
Gudmundsson G, Welte T, Nizankowska-Mogilnicka E, Studnicka M,
Bateman E, Anto JM, Burney P, et al: COPD in never smokers: Results
from the population-based burden of obstructive lung disease study.
Chest. 139:752–763. 2011.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zarghami M, Taghizadeh F, Sharifpour A and
Alipour A: Efficacy of smoking cessation on stress, anxiety, and
depression in smokers with chronic obstructive pulmonary disease: A
randomized controlled clinical trial. Addict Health. 10:137–147.
2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bhan A, Soleimani M and Mandal SS: Long
noncoding RNA (lncRNA): Functions in health and disease. Gene
Regulation, Epigenetics and Hormone Signaling, 2017.
|
|
9
|
Hon KW, Abu N, Ab Mutalib NS and Jamal R:
MiRNAs and lncRNAs as predictive biomarkers of response to FOLFOX
therapy in colorectal cancer. Front Pharmacol.
9(846)2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Heward JA and Lindsay MA: Long non-coding
RNAs in the regulation of the immune response. Trends Immunol.
35:408–419. 2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zheng M, Hong W, Gao M, Yi E, Zhang J, Hao
B, Liang C, Li X, Li C, Ye X, et al: Long noncoding RNA COPDA1
promotes airway smooth muscle cell proliferation in chronic
obstructive pulmonary disease. Am J Respir Cell Mol Biol.
61:584–596. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Qian Y, Mao ZD, Shi YJ, Liu ZG, Cao Q and
Zhang Q: Comprehensive analysis of miRNA-mRNA-lncRNA networks in
non-smoking and smoking patients with chronic obstructive pulmonary
disease. Cell Physiol Biochem. 50:1140–1153. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ge J, Geng S and Jiang H: Long noncoding
RNAs antisense noncoding RNA in the INK4 locus (ANRIL) correlates
with lower acute exacerbation risk, decreased inflammatory
cytokines, and mild GOLD stage in patients with chronic obstructive
pulmonary disease. J Clin Lab Anal. 33(e22678)2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Josipovic I, Pflüger B, Fork C, Vasconez
AE, Oo JA, Hitzel J, Seredinski S, Gamen E, Heringdorf DMZ, Chen W,
et al: Long noncoding RNA LISPR1 is required for S1P signaling and
endothelial cell function. J Mol Cell Cardiol. 116:57–68.
2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Liu W, Luo M, Zou L, Liu X, Wang R, Tao H,
Wu D, Zhang W, Luo Q and Zhao Y: uNK cell-derived TGF-β1 regulates
the long noncoding RNA MEG3 to control vascular smooth muscle cell
migration and apoptosis in spiral artery remodeling. J Cell
Biochem. 120:15997–16007. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Tang W, Shen Z, Guo J and Sun S: Screening
of long non-coding RNA and TUG1 inhibits proliferation with TGF-β
induction in patients with COPD. Int J Chron Obstruct Pulmon Dis.
11:2951–2964. 2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Li X, Zheng M, Pu J, Zhou Y, Hong W, Fu X,
Peng Y, Zhou W, Pan H, Li B and Ran P: Identification of abnormally
expressed lncRNAs induced by PM2.5 in human bronchial epithelial
cells. Biosci Rep. 38(BSR20171577)2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Trionfini P and Benigni A: MicroRNAs as
master regulators of glomerular function in health and disease. J
Am Soc Nephrol. 28:1686–1696. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Bartel DP: Metazoan MicroRNAs. Cell.
173:20–51. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Li D, Li YP, Li YX, Zhu XH, Du XG, Zhou M,
Li WB and Deng HY: Effect of regulatory network of exosomes and
microRNAs on neurodegenerative diseases. Chin Med J (Engl).
131:2216–2225. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Karatas OF, Oner M, Abay A and Diyapoglu
A: MicroRNAs in human tongue squamous cell carcinoma: From
pathogenesis to therapeutic implications. Oral Oncol. 67:124–130.
2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Thomson DW and Dinger ME: Endogenous
microRNA sponges: Evidence and controversy. Nat Rev Genet.
17:272–283. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Huang X, Zhu Z, Guo X and Kong X: The
roles of microRNAs in the pathogenesis of chronic obstructive
pulmonary disease. Int Immunopharmacol. 67:335–347. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Shen W, Liu J, Zhao G, Fan M, Song G and
Zhang Y, Weng Z and Zhang Y: Repression of Toll-like receptor-4 by
microRNA-149-3p is associated with smoking-related COPD. Int J
Chron Obstruct Pulmon Dis. 12:705–715. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Adelaja A and Hoffmann A: Signaling
crosstalk mechanisms that may fine-tune pathogen-responsive NFκB.
Front Immunol. 10(433)2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Savarimuthu Francis SM, Davidson MR, Tan
ME, Wright CM, Clarke BE, Duhig EE, Bowman RV, Hayward NK, Fong KM
and Yang IA: MicroRNA-34c is associated with emphysema severity and
modulates SERPINE1 expression. BMC Genomics. 15(88)2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wang J, Li Q, Xie J and Xu Y: Cigarette
smoke inhibits BAFF expression and mucosal immunoglobulin A
responses in the lung during influenza virus infection. Respir Res.
16(37)2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Del Giudice M and Gangestad SW: Rethinking
IL-6 and CRP: Why they are more than inflammatory biomarkers, and
why it matters. Brain Behav Immun. 70:61–75. 2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Antoniu SA, Mihaltan F and Ulmeanu R:
Anti-TNF-alpha therapies in chronic obstructive pulmonary diseases.
Expert Opin Investig Drugs. 17:1203–1211. 2008.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Takahashi E, Kataoka K, Indalao IL, Konoha
K, Fujii K, Chida J, Mizuno D, Fujihashi K and Kido H: Oral
clarithromycin enhances airway immunoglobulin A (IgA) immunity
through induction of IgA class switching recombination and
B-cell-activating factor of the tumor necrosis factor family
molecule on mucosal dendritic cells in mice infected with influenza
A virus. J Virol. 86:10924–10934. 2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Lin C and Yang L: Long noncoding RNA in
cancer: Wiring signaling circuitry. Trends Cell Biol. 28:287–301.
2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Beermann J, Piccoli MT, Viereck J and Thum
T: Non-coding RNAs in development and disease: Background,
mechanisms, and therapeutic approaches. Physiol Rev. 96:1297–1325.
2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zhuan B, Lu Y, Chen Q, Zhao X, Li P, Yuan
Q and Yang Z: Overexpression of the long noncoding RNA TRHDE-AS1
inhibits the progression of lung cancer via the miRNA-103/KLF4
axis. J Cell Biochem. 120:17616–17624. 2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhu G, Xin X, Liu Y, Huang Y, Li K and Wu
C: Geraniin attenuates LPS-induced acute lung injury via inhibiting
NF-κB and activating Nrf2 signaling pathways. Oncotarget.
8:22835–22841. 2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Jin LY, Li CF, Zhu GF, Wu CT, Wang J and
Yan SF: Effect of siRNA against NF-κB on sepsis-induced acute lung
injury in a mouse model. Mol Med Rep. 10:631–637. 2014.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wu C, Zhao J, Zhu G, Huang Y and Jin L:
SiRNA directed against NF-κB inhibits mononuclear macrophage cells
releasing proinflammatory cytokines in vitro. Mol Med Rep.
16:9060–9066. 2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Badrichani AZ, Stroka DM, Bilbao G, Curiel
DT, Bach FH and Ferran C: Bcl-2 and Bcl-XL serve an
anti-inflammatory function in endothelial cells through inhibition
of NF-kappaB. J Clin Invest. 103:543–553. 1999.PubMed/NCBI View
Article : Google Scholar
|
|
39
|
Kopeina GS, Prokhorova EA, Lavrik IN and
Zhivotovsky B: Alterations in the nucleocytoplasmic transport in
apoptosis: Caspases lead the way. Cell Prolif.
51(e12467)2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Si L, Xu L, Yin L, Qi Y, Han X, Xu Y, Zhao
Y, Liu K and Peng J: Potent effects of dioscin against pancreatic
cancer via miR-149-3P-mediated inhibition of the Akt1 signalling
pathway. Br J Pharmacol. 174:553–568. 2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zou A, Liu X, Mai Z, Zhang J, Liu Z, Huang
Q, Wu A and Zhou C: LINC00472 acts as a tumor suppressor in NSCLC
through KLLN-mediated p53-signaling pathway via MicroRNA-149-3p and
MicroRNA-4270. Mol Ther Nucleic Acids. 17:563–577. 2019.PubMed/NCBI View Article : Google Scholar
|