Introduction
HER2 is a member of the human EGFR family and a
well-established target for HER2-overexpressing tumour treatment
(1). E23 is a monoclonal antibody
derived from mice immunized with HER2-overexpressing cell
components and has been indicated to specifically bind to the
extracellular domain of HER2(2). In
addition, e23 was generated by fusing the variable region of the
light chain (VL) with that of the heavy chain (VH) to form a
single-chain variable fragment (scFv) named e23sFv (3). e23sFv has been indicated to exhibit
reduced molecular weight and more efficient penetration into solid
tumours compared with that of e23; therefore, it has been utilized
in immunoconjugates, such as immunotoxin or immune-proapoptotic
molecules, as the portion targeted to HER2 (3,4).
Despite its advantages in terms of size and trafficking, e23sFv
exhibits a 4-fold reduced binding affinity for HER2 compared with
that of e23 or the corresponding antigen-binding fragment (Fab)
(5). To achieve equivalent
effectiveness, a higher dose is required for antibodies with less
affinity; however, an increased dose of therapeutic antibodies can
result in more intensive immunogenic reactions and possibly
unexpected toxicities (6). To
ensure the safety and efficacy of e23sFv-derived
immuno-proapoptotic molecules in HER2-targeted tumour treatment, it
is necessary to enhance the affinity of e23sFv for the HER2
extracellular domain (ECD) (5).
In vitro antibody affinity maturation
enhances the antibody affinity using genetic engineering (7). Several approaches have been developed
to improve the antibody affinity, the majority of which have
focused on the mutagenesis of the complementarity-determining
region (CDR), because CDRs are directly involved in
antibody-antigen interactions (7).
The crystal structure of antibody-antigen complexes has revealed
that specific CDR residues of an antibody directly contact antigens
and thus determine the affinity and specificity of the antibody
(8). Affinity improvement in
vitro predominantly involves inducing random mutagenesis in
CDRs and screening the mutants for enhanced affinity and
site-directed mutagenesis to deliberately enhance the affinity
based on antibody conformation (9).
In addition to CDR manipulation, the pioneering work of Foote and
Winter (10) has suggested that
residues in the β-sheet structure of framework regions (FRs), which
support CDRs, serve critical roles in the adjustment of the loop
structures of CDRs. Although these residues, which are referred to
as ‘Vernier zone residues’, do not directly interact with the
antigen, careful selection of these residues may prove essential
for shaping the diversity of the structures in the primary
repertoire and affinity maturation (11).
In the present study, the affinity of a single
variable fragment, e23sFv, was more improved using FR engineering
compared with CDR mutagenesis. The e23sFv FR was substituted with
FRs from the two most homologous antibodies in the National Centre
for Biotechnology Information (NCBI) protein database and two
candidates named EX1 and EX2 were constructed. Another candidate
was constructed by e23sFv FR residue mutation based on the sequence
alignment with the variable region of the homologous antibodies.
All three recombinant scFvs retained the e23sFv CDRs. The affinity
assays demonstrated that EX1 exhibited the highest homology with
e23sFv, thereby significantly improving its affinity for HER2, and
was internalized into HER2-overexpressing cells more effectively
compared with the other candidates.
Materials and methods
Framework redesign of e23sFv-based
scFvs by mutagenesis and engraftment
Two strategies were used to reconstitute the FRs of
e23sFv, site-directed mutagenesis and CDR grafting, both of which
were based on the analysis of the e23sFv amino acid sequence
homology of proteins in the NCBI database (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Five VL
(L1-L5) and five VH (H1-H5) sequences with the highest similarity
to the VL and VH domains of e23sFv are presented in Fig. 1A, and in these candidates all FRs
were aligned and comparable to that of e23sFv. Detailed information
on the five VLs and five VHs selected is presented in Table I.
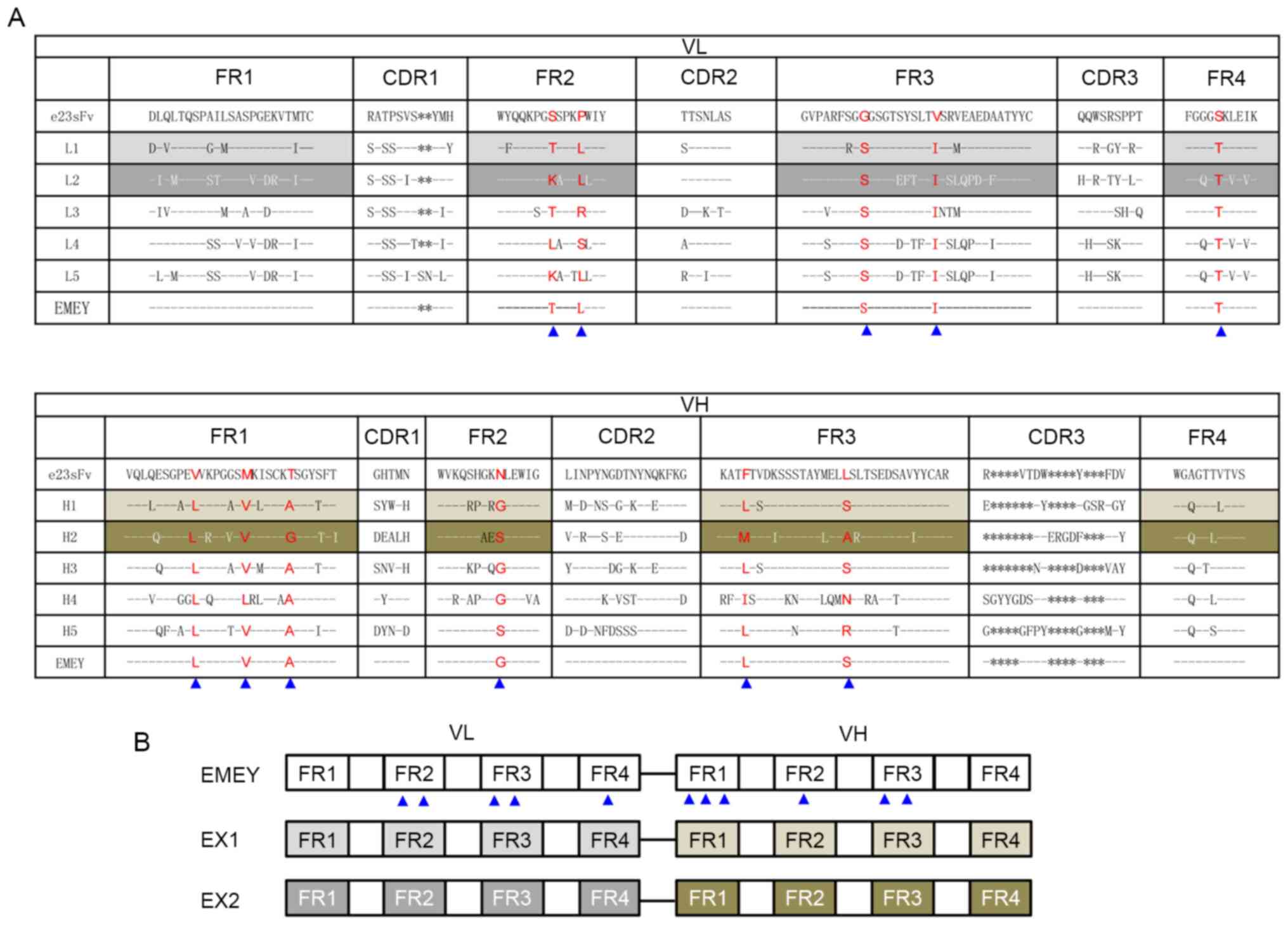 | Figure 1Design of FR-engineered e23sFv
derivatives. (A) Amino acid sequence alignment of the VL and VH
domains of mouse anti-HER2 single-chain variable fragment, e23sFv,
and their five most homologous counterparts identified in the
National Centre for Biotechnology Information protein database.
L1-L5 represent VL homologous sequences and H1-H5 represent VH
homologous sequences. CDRs and FRs are indicated in columns. The
residues that are identical to those of e23sFv are indicated with
dashed lines, and missing residues in the CDRs are indicated with
asterisks. Non-identical FR residues in e23sFv and all their five
homologs in the VL or VH collection are in red. Introduced
site-directed mutations are indicated by blue triangles, above
which are the corresponding substituted residues. (B) The schematic
structure of three e23sFv derivatives. EMEY includes 11 mutated
residues in the FRs of e23sFv, as indicated by triangles. EX1 and
EX2 represent CDR grafts of e23sFv in the L1-H1 and L2-H2 FR
scaffolds, respectively. FR, framework region; VL, light-chain
variable region; VH, heavy-chain variable region; CDR,
complementarity-determining region. |
 | Table IDetailed information of selected
framework region scaffolds by homologous analysis. |
Table I
Detailed information of selected
framework region scaffolds by homologous analysis.
| Chain | Name | Origin | Entry ID | (Refs.) |
|---|
| L1 | Chain L, human
thrombopoietin neutralizing antibody TN1 Fab | Mus
musculus | pdb|2ZKH| | 16 |
| L2 | Chain B, crystal
structure of the Fab fragment of therapeutic antibody
daclizumab | Mus
musculus | pdb|3NFP| | 17 |
| L3 | Chain A, Fab fragment
of the engineered human monoclonal antibody A5B7 | Mus
musculus | pdb|1AD0| | 18 |
| L4 | Immunoglobulin H23
light chain kappa variable region | Humanized | gb|AAB38288.1| | 19 |
| L5 | Chain L, structure
of CD40L in complex with the Fab fragment of the humanized 5C8
antibody | Humanized | pdb|1I9R| | 20 |
| H1 | Chain B, crystal
structure of FabOX108 | Mus
musculus | pdb|3DGG| | 21 |
| H2 | Crystal structure
of sonic hedgehog bound to the 5E1 Fab fragment | Mus
musculus | pdb|3MXW| | 22 |
| H3 | Chain B, crystal
structure of IL-23 in complex with neutralizing Fab | Mus
musculus | pdb|3D85| | 23 |
| H4 | Immunoglobulin
heavy chain V region humanized bispecific antibody | Humanized | gb|AAB24133.1| | 24 |
| H5 | Immunoglobulin
heavy chain V region precursor | Homo
sapiens | pir|PN0444| | 25 |
For the site-directed mutations, five residues in
the VL and six residues in the VH sequences in the e23sFv FRs,
which differed among the five domains in the homologous FRs, were
replaced with the most frequently occurring amino acids in the
homologous proteins (Fig. 1A), with
the exception of the first S-T mutation in VL, which was not
substituted to ensure that the chemical characteristics of the
domains were retained. The resulting e23sFv derivative with 11
point mutations was genetically optimized with E.
coli-preferred codons, synthesized commercially (Beijing Augct
DNA-Syn Biotechnology Co., Ltd.) and designated EMEY (Fig. 1B).
For the CDR engraftment, FR scaffolds were selected
from the top two VL and VH homologous domains (Fig. 1A). The FRs of L1 (the VL of the TN1
Fab) and H1 (the VH of FabOX108) were substituted into e23sFv,
giving rise to a chimaera referred to as EX1 (Fig. 1B and Table I). Another chimaera, EX2, was
generated by substituting the FRs of L2 (the VL of daclizumab) and
H2 (the VH of 5E1 Fab) into e23sFv (Fig. 1B and Table I). The codons of EX1 and EX2 were
further optimized for prokaryotic expression and synthesized by
Beijing Augct DNA-Syn Biotechnology Co., Ltd.
Prokaryotic expression and
purification of the e23sFv derivatives
The genes encoding e23sFv-derived scFvs fused with
His-tag at the 3' end and with 5'- and 3'-flanking NcoI and
NotI restriction sites were cloned into the prokaryotic
expression vector pET28a (Takara Biotechnology Co., Ltd.). The
resulting recombinant plasmids were verified by agarose gel
electrophoresis using 1% gel and ethidium bromide visualization.
The recombinant plasmids were then transformed into E. coli
BL21 (DE3) (Takara Biotechnology Co., Ltd.), and after 3 h of
induction with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) at
37˚C, the bacteria were harvested and sonicated for 30 min at 2 sec
intervals on ice for use in SDS-PAGE analysis to demonstrate the
presence of the proteins of interest in inclusion bodies. For the
purification of inclusion bodies, the precipitates were dissolved
in 8 M urea buffer and subjected to Ni2+-NTA affinity
chromatography using Ni-NTA His•Bind Resins (Novagen;
MilliporeSigma) according to the manufacturer's instructions. After
extensive washing with 100 mM imidazole buffer, the bound proteins
were eluted at room temperature with 1,000 mM imidazole buffer and
refolded by linearly decreasing the urea gradient (7, 6, 4, 2, 1
and 0 M). The renatured proteins were finally dialyzed into PBS (pH
7.4) and quantified by BCA assay (Pierce; Thermo Fisher Scientific,
Inc.).
Identification of the e23sFv
derivatives by SDS-PAGE and western blotting
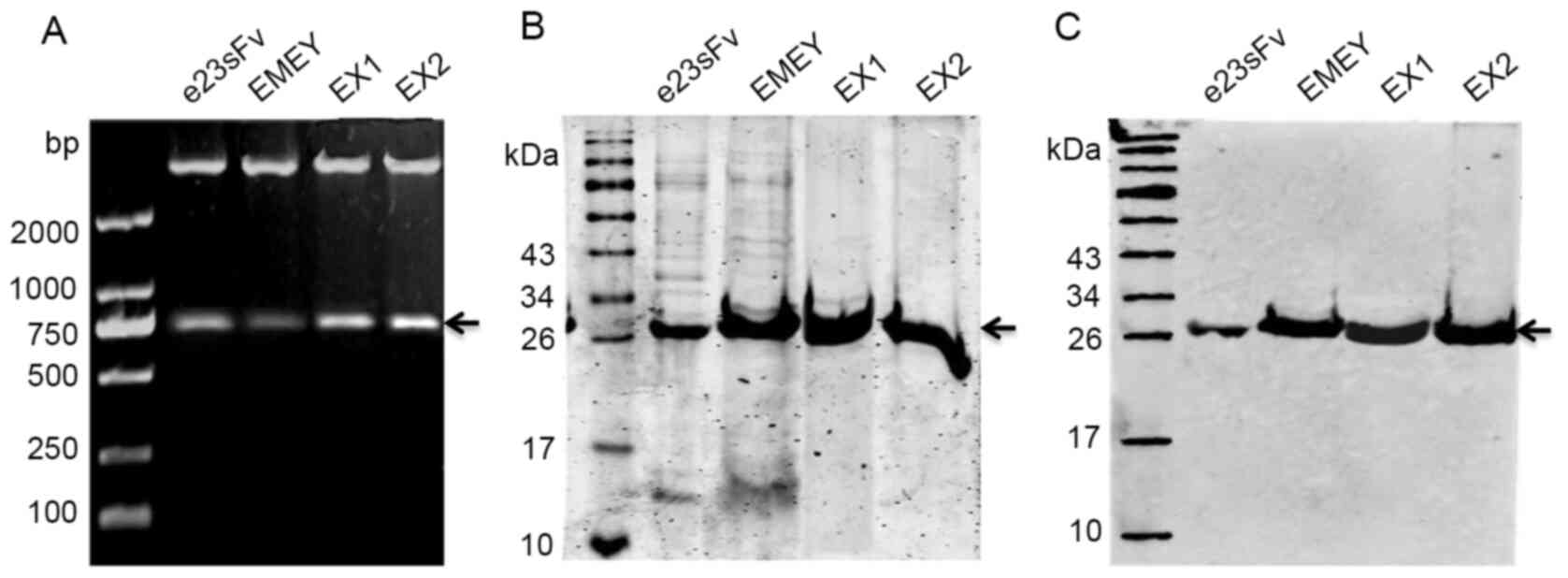
SDS-PAGE (12%) was performed using renatured e23sFv
proteins (30 µg per sample). The protein electrophoresis gel was
stained using Coomassie brilliant blue R-250 followed by greyscale
analysis of the purity of the scFvs with Image J (version 1.44;
National Institutes of Health). Specifically, the
background-corrected density of the protein band was divided by the
background-corrected density of the entire lane and multiplies by
100 to obtain the purity percentages. The e23sFv-based scFvs were
identified by immunoblotting using nitrocellulose membranes.
Membranes were blocked with 2% non-fat milk at room temperature for
1 h and was then incubated with anti-His antibody conjugated to
horseradish peroxidase (1:1,000 dilution; cat. no. 34460; Qiagen,
Inc.) at 4˚C overnight followed by chemiluminescent detection using
Pierce™ ECL Western Blotting Substrate (cat. no. 32209; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions.
FITC labelling of the e23sFv
derivatives
For the quantification of cellular binding and
visualization of the subsequent internalization by
HER2-overexpressing cells, the purified e23sFv-derived scFvs were
conjugated with FITC using Hook™ dye labelling kit (G-Biosciences).
The FITC-labelled scFvs were sterilized using 0.22 µm centrifugal
filters (EMD Millipore). The absorption of fluorescein at 495 nm
was detected by a UV-2450 spectrophotometer (Shimadzu Corporation),
and the molar ratio of FITC to scFv was ~2.5:1.
Binding affinity to recombinant HER2
as determined by ELISA
Recombinant human HER2 (Sino Biological; 500 ng per
well) was immobilized on 96-well plates at 4˚C for 16 h. The
HER2-coated plates were blocked with 1% BSA (Sigma-Aldrich; Merck
KGaA) at room temperature for 1 h and incubated with the
three-fold-serially diluted e23sFv derivatives from 3 µM for 4 h at
room temperature. ScFv15 served as the negative control. After
washing with PBS-Tween-20 (0.05%; PBS-T), the bound scFvs were
incubated with an HRP-conjugated anti-His antibody (1:2,000
dilution; cat. no. 34460; Qiagen, Inc.) for 1 h at room
temperature. Following extensive washing with PBS-T, the
microplates were incubated with a 2,2'-azino-bis
(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (7 mM)
substrate at 37˚C for 20 min, and the absorbance was measured at
409 nm using a Sunrise microplate reader (Tecan Group, Ltd.).
Measurement of the affinity constant
by surface plasmon resonance (SPR)
An SPR assay was used for the kinetic analysis of
the binding of the e23sFv-based scFvs to recombinant HER2 in
vitro. A ProteOn™ GLC sensor chip (Bio-Rad Laboratories, Inc.)
was immobilized with 5 µg/ml HER2 in 10 mM acetate (pH 5.5). Five
solutions in PBS-T with two-fold serial dilutions of the e23sFv
derivatives were injected and run through the sensor chip at a flow
rate of 50 µl/min. The analyte injection programme included a 180
sec association phase followed by a 600 sec dissociation phase. The
data were analysed in a 1:1 Langmuir binding model (12) using ProteOn Manager software
(Bio-Rad Laboratories, Inc.). The equilibrium constant
(KD) was calculated as the ratio of the dissociation
rate constant (Koff) to the association rate constant
(Kon).
Binding affinity for cellular HER2 as
determined by flow cytometry
HER2-positive cells (BT-474 and SKOV-3 cells) and
HER2-negative cells (MCF-7 cells) were purchased from American Type
Culture Collection and cultured in RPMI-1640 medium (cat. no.
10-040-CV; Corning, Inc.) with 10% FBS (cat. no. 10099; Gibco;
Thermo Fisher Scientific, Inc.) at 37˚C and 5% CO2.
After blocking in 5% BSA (cat. no. A1933; Sigma-Aldrich; Merck
KGaA) at 4˚C for 30 min, 1x106 HER2-positive cells
(BT-474 and SKOV-3 cells) and HER2-negative cells (MCF-7 cells)
were incubated with 125 nM FITC-conjugated e23sFv derivatives on
ice for 1 h. For the negative control, a non-specific scFv, scFv15
against hepatitis B virus surface antigen (HbsAg), which was
purified and labelled with FITC as previously described, was used
at 125 nM to exclude nonspecific binding (13). In addition, a FITC-labelled
anti-HER2 antibody (1:2,000; cat. no. 10004-R511-F; Sino
Biological) was used as a positive control. After antibody
incubation at 4˚C for 30 min, the cells were rinsed with PBS, and
FITC intensities were quantified using a FACSCalibur flow cytometer
(BD Biosciences) with CellQuest software (BD Biosciences).
Internalization as assessed by
fluorescence microscopy
HER2-positive cells (BT-474 and SKOV-3 cells) and
HER2-negative cells (MCF-7 cells) were cultured on coverslips
(1x106 cells/well in a six-well plate). After blocking
in 5% BSA (cat. no. A1933; Sigma Aldrich; Merck KGaA) at 4˚C for 30
min, cells were incubated with 500 nM FITC-labelled e23sFv
derivatives at 37˚C for 4 h. scFv15 (500 nM) was included as a
negative control, which was prepared and coupled with FITC as
previously described (13). After
incubation, the samples were fixed in 4% paraformaldehyde at 4˚C
for 30 min. Bright-field and fluorescence images of cells were
captured using a fluorescence microscope (IX-71; Olympus
Corporation).
Molecular modelling of mutant
scFvs
The parental e23sFv and mutated variants were
generated using the web-based antibody modelling software BLASTP
(https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins),
which is based on a modified form of the algorithm used for antibody
modelling. We used the ‘dead-end elimination’ algorithm (14) for side chain building. We compared
both the heavy and light chains independently with the most
homologous antibodies based on the homologous protein structures
obtained from the Protein Data Bank (PDB) (https://www.rcsb.org/) using the Homology module in
Discovery Studio 4.5 software (BIOVIA; Dassault Systèmes). The
predicted structures of e23sFv and EX1 were subsequently docked to
the crystal structure of the human HER2 ECD using ZDOCK Server
(http://zdock.umassmed.edu.).
Statistical analysis
All assays were performed in triplicate on three
independent occasions unless otherwise stated. The data are
presented as the mean ± SD. ANOVA followed by Tukey's post hoc test
was used for the statistical analyses, which were performed with
SPSS v15.0 software (SPSS, Inc.). P<0.05 was considered to
indicate a statistically significant difference.
Results
Framework engineering of e23sFv and
prokaryotic expression of the e23sFv derivatives
To maintain the HER2-binding activity of e23sFv, all
CDRs were maintained intact and only the FRs in the VL and VH
domains were manipulated. For the purpose of FR manipulation,
homologous sequences of e23sFv were selected by browsing the NCBI
protein database (Fig. 1A and
Table I). Two FR engineering
methods were employed based on this homologous candidate list.
Firstly, site-directed mutagenesis was performed to
modulate the FRs of e23sFv with as few mutations as possible and
therefore only 11 amino acids, which were not identical to any of
the homologous repertoires identified in the NCBI database, were
replaced with the most frequently occurring residues (Fig. 1A). The mutated e23sFv derivatives
harboured an 11-residue substitution, which was named EMEY
(Fig. 1B). Secondly, CDR grafting
of e23sFv to FR scaffolds of the two most homologous mouse VL and
VH domains was performed to avoid the risk of structurally
interfering with antibody-antigen recognition. e23sFv was engrafted
with L1-H1 and L2-H2 as FR acceptors, and these constructs were
referred to as EX1 and EX2, respectively (Fig. 1B).
These three His-tagged-FR-engineered e23sFv-based
scFv genes were cloned into a pET28a prokaryotic expression plasmid
(Fig. 2A), followed by
transformation into E. coli BL21 (DE3). IPTG induction
resulted in a robust expression of the engineered scFvs in the
inclusion bodies of the bacteria, which were denatured and purified
by Ni2+-NTA affinity chromatography. The yield of the
refolded scFvs was 1 mg/l for e23sFv and 3-4 mg/l for the three
derivatives, as quantified by a BCA assay, suggesting improved
protein production presumably due to FR engineering. The purity of
both e23sFv and EMEY was approximately 93%, as quantified by
SDS-PAGE, slightly less than the 98% purity of EX1 and EX2, which
indicated a subtle difference in nickel-binding dynamics resulting
from the FR substitutions (Fig.
2B). All purified scFvs were confirmed by western blotting with
an anti-His antibody (Fig. 2C).
In vitro binding affinity of the
FR-engineered anti-HER2 scFvs
Since all the original CDRs of e23sFv were
maintained intact in the three FR-engineered scFvs, their
HER2-binding activities were theoretically expected to be retained.
To address this hypothesis, in vitro ELISA was performed to
detect the binding capacities of the e23sFv derivatives to
recombinant HER2 that was immobilized on the microplates. As
illustrated in Fig. 3A, EMEY and
EX1 exhibited binding patterns similar to that of parental e23sFv,
but in contrast to e23sFv, they did not reach binding saturation at
the highest dose. By contrast, the binding curve of EX2 indicated
that the maximal binding level of EX2 was only 68.2% compared with
e23sFv, suggesting the considerably decreased ability of EX2 to
bind HER2.
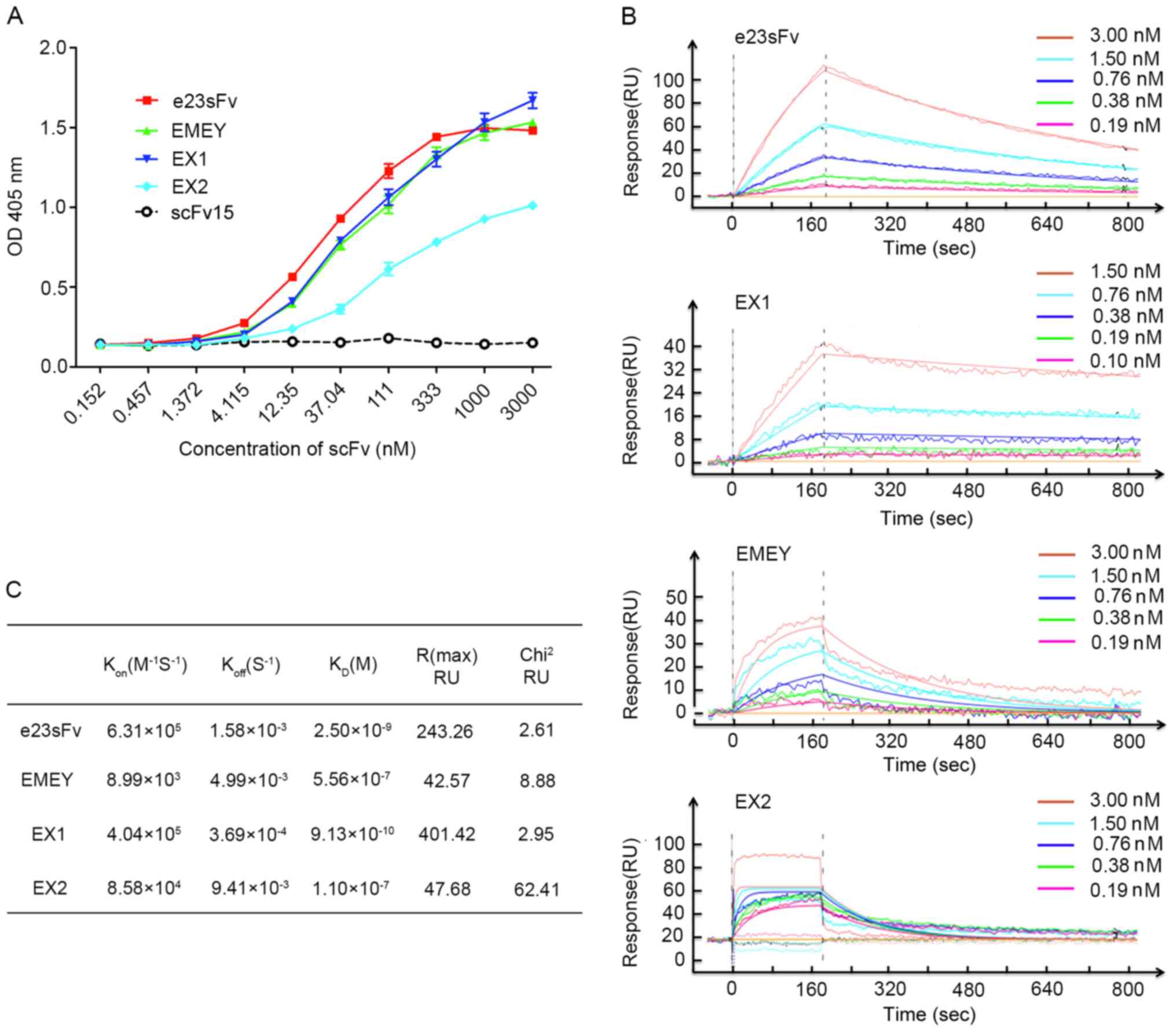 | Figure 3In vitro binding of the
e23sFv-derived scFvs to recombinant HER2. (A) Affinity measurement
by ELISA. HER2-coated microplates were incubated with the e23sFv
derivatives at various concentrations, and the bound scFvs were
detected using an anti-His antibody. scFv15 served as the negative
control. (B) One-shot kinetics of SPR. Five sensorgrams indicated
the response of HER2-immobilized sensor chips with five diluted
concentrations of the e23sFv derivatives. (C) Comparison of
Kon, Koff and KD of the e23sFv
derivatives calculated from SPR sensing. scFv, single-chain
variable fragment; SPR, surface plasmon resonance; RU, resonance
unit; Kon, association rate constant; Koff,
dissociation rate constant, KD, equilibrium constant;
Chi2, goodness-of-fit between the binding model and
theoretical affinity; OD, optical density. |
To further dissect the association and dissociation
properties of the e23sFv-derived scFvs with recombinant HER2, SPR
was performed to monitor the binding kinetics based on five
sensorgrams in which different concentrations of scFvs run through
a HER2-coated sensor chip. An increase in the SPR signal was
observed in the nanomolar range for EX1 and e23sFv, differing from
the micromolar range obtained for EMEY and EX2 (Fig. 3B). The KD of EX1 was ~0.9
nM, exhibiting nearly a 3-fold increased binding affinity compared
with that of e23sFv, with a KD of 2.5 nM (Fig. 3C). Given that the Kon of
EX1 was only ~0.6-fold higher compared with that of e23sFv, the
enhanced affinity of EX1 was mainly attributed to a ~4.3-fold lower
Koff. The binding capabilities of the other two scFvs,
EMEY and EX2, were diminished with a KD value of ~0.5
and ~0.1 µM, respectively (Fig.
3C). These results indicated that EX1, when bound to
recombinant HER2, exhibited a slightly faster on-rate and a slower
off-rate compared with those of parental e23sFv, resulting in a
stronger antibody-antigen interaction.
Cellular binding and internalization
of the FR-engineered anti-HER2 scFvs
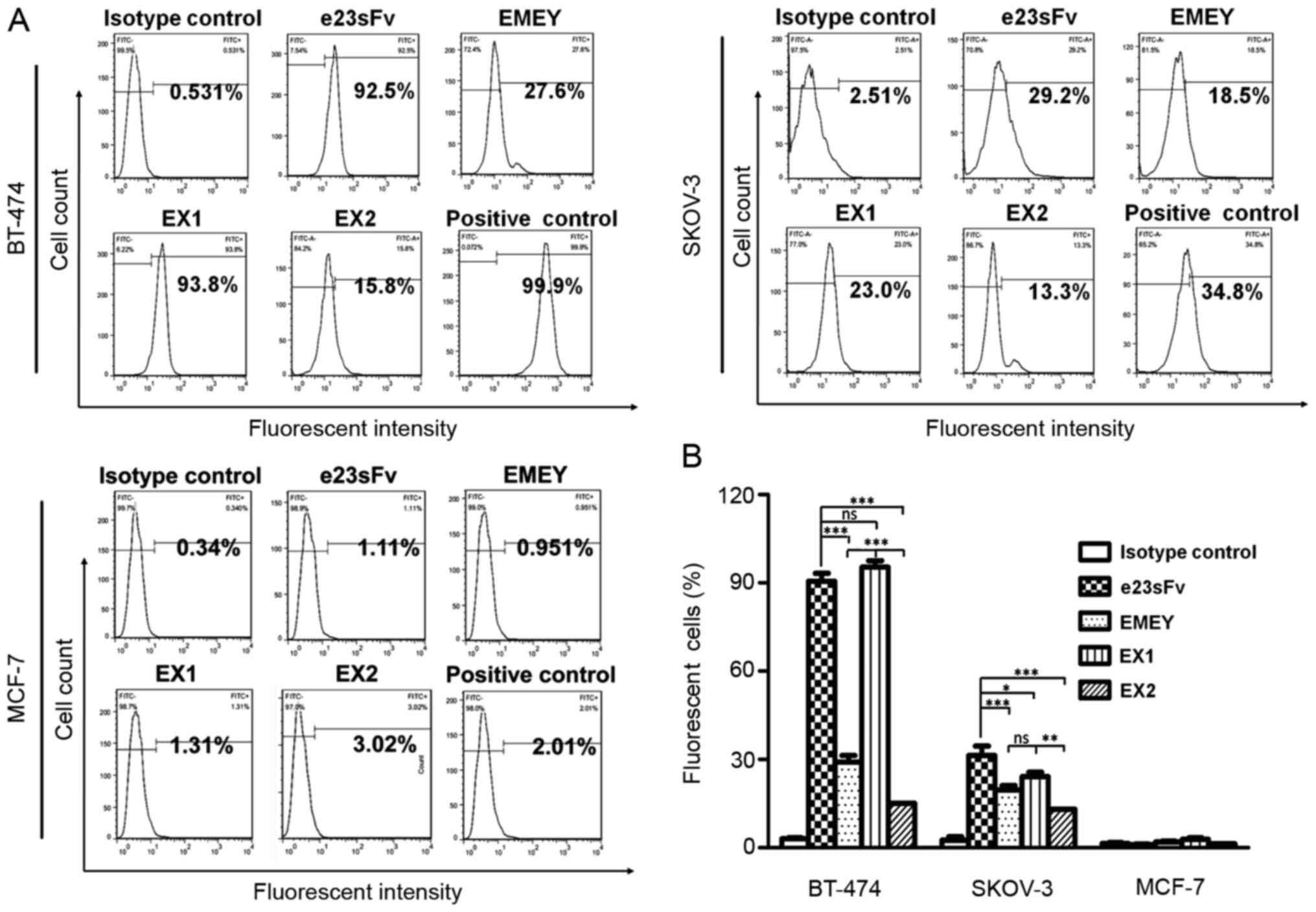
Subsequently, the potential of the e23sFv
derivatives to recognize endogenous HER2 was evaluated. Three cell
lines with differential HER2 expression levels were incubated with
FITC-labelled e23sFv derivatives, and the percentages of the bound
cells were quantified by flow cytometry. As demonstrated in
Fig. 4, in the BT-474 breast cancer
cells that express high levels of HER2 (~100% in the case of the
positive binding control), EX1 bound to >90% of the cell
population, comparable to the binding capacity of e23sFv, whereas
the cellular binding of EMEY and EX2 decreased to less than 30% of
the cell population. Similar results were observed in the SKOV-3
ovarian cancer cells with moderate HER2 expression, demonstrating
comparable binding activities between EX1 and e23sFv and impaired
binding of EMEY and EX2. Fluorescent HER2-negative MCF-7 breast
cancer cells were undetected, as was the non-specific
HbsAg-targeting scFv15, indicating the specificity of this
cell-based binding assay.
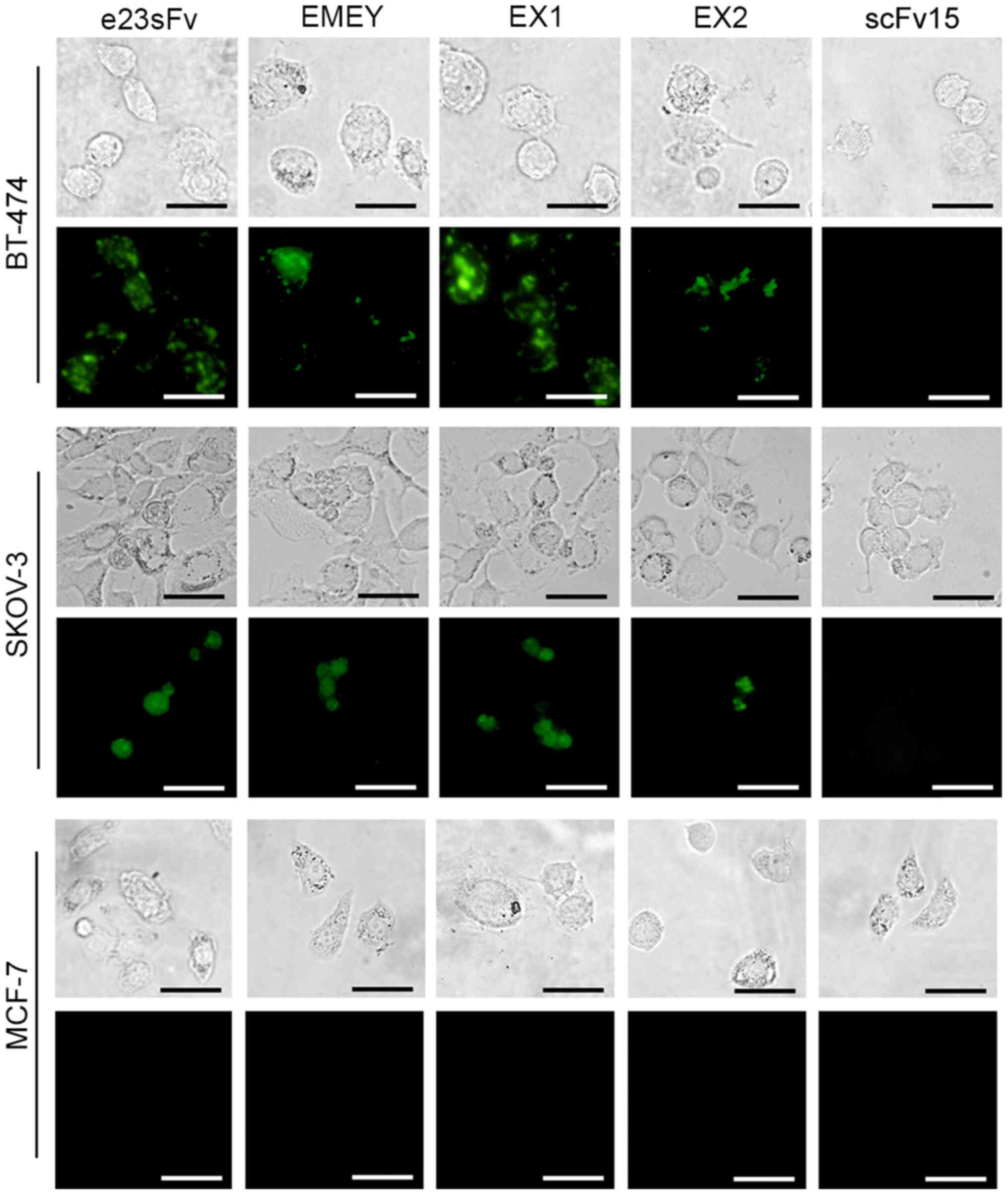
It was also determined whether interaction with cell
surface HER2 enabled the e23sFv derivatives to enter the cells.
Intracellular fluorescence of the FITC-labelled e23sFv derivatives
was monitored in HER2-positive cell lines 4 h post-incubation. In
contrast to the negative controls consisting of MCF-7 cells and
scFv15, a patchy distribution of EX1 fluorescence was observed in
both BT-474 and SKOV-3 cells, indicating endosomal localization
similar to the pattern of e23sFv (Fig.
5). However, the fluorescence of the other two variants was
faint compared with that of e23sFv and EX1, which indicated a
weaker HER2 binding and internalization.
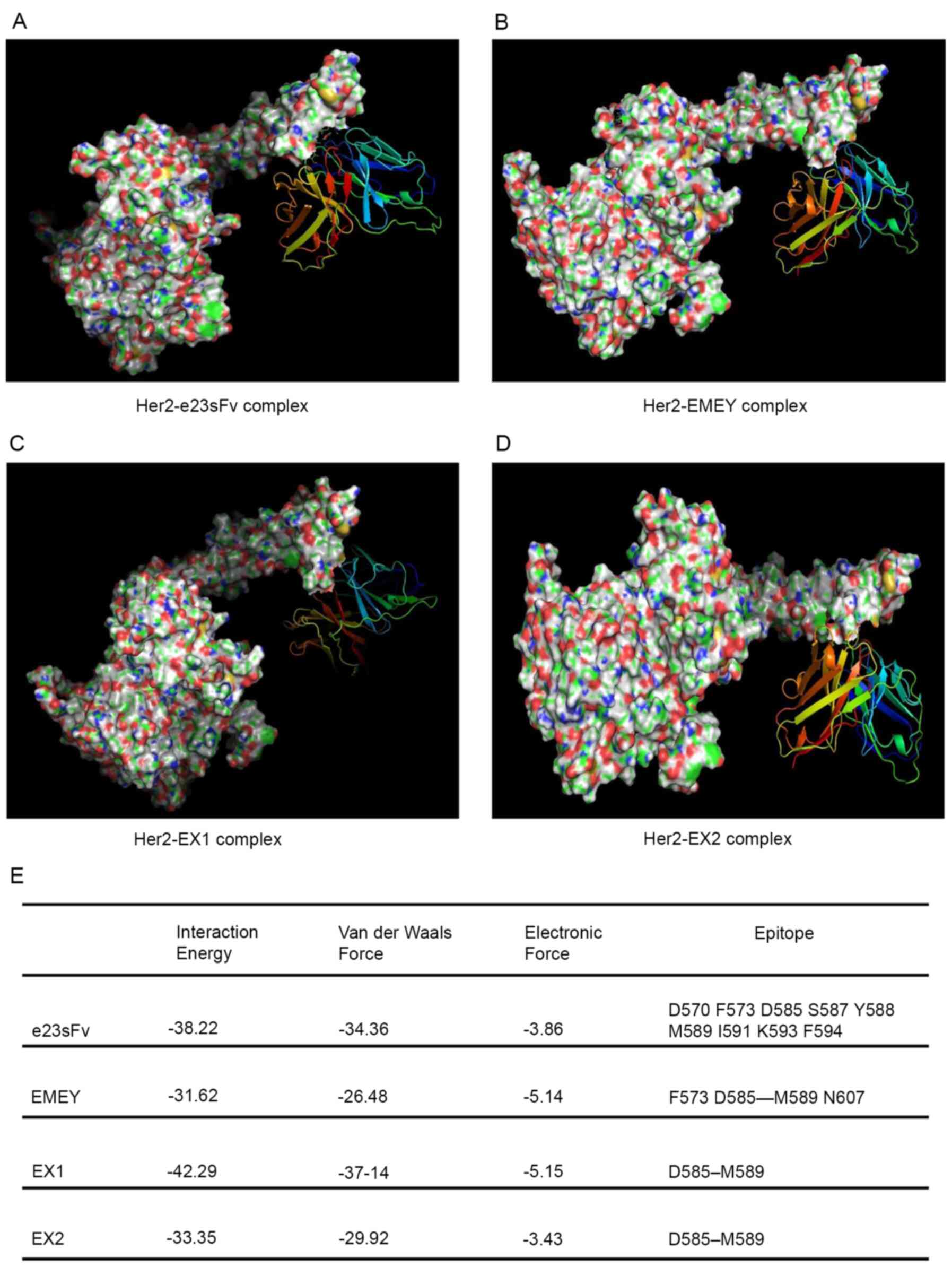
Docking mechanism of the enhanced
EX1-HER2 interaction
The in silico docking analysis revealed that
both e23sFv and the three mutants bound to domain IV of the HER2
ECD, and several residues were identified as putatively important
for these binding interfaces (Fig.
6A-D). All the scFv variants formed distinct but overlapping
interfaces with domain IV of the HER2 ECD (Fig. 6E). The HER2 binding energy of EX1
was predicted to be slightly higher compared with that of e23sFv
while EMEY and EX2 showed decreased binding energy compared with
e23sFv (Fig. 6E).
Discussion
e23sFv is an scFv derived from the HER2-targeted
monoclonal antibody e23 by fusing its VL with its VF (3). e23sFv exhibits a decreased molecular
size compared with its IgG counterpart e23 and can therefore
penetrate solid tumours more effectively (3). However, e23sFv has been indicated to
exhibit a decreased affinity for HER2 by a factor of 4 compared
with that of e23 or the corresponding e23 Fab, according to the
results of a competitive binding assay (4). With a decreased affinity, the function
of e23sFv as the targeting moiety has been lessened (4). Therefore, improving the affinity of
e23sFv is of great importance to maintain the targeting function of
e23sFv. The decreased affinity of e23sFv compared with that of e23
has been primarily attributed to conformational alterations after
its reconstruction (4). Restoration
of the affinity of e23sFv may be effectively accomplished by
mutagenesis approaches to manipulate the interface of
antibody-antigen contact.
CDRs in the antibody variable region are well-known
as the antibody repertoire-determining regions and are responsible
for antigen-antibody interactions (7,8).
Hotspots of somatic mutations during the process of antibody
affinity maturation are predominantly located in CDRs, which
indicates that the diversity of the amino acids in CDRs mainly
accounts for antibody specificity and affinity (7,8). Based
on the critical role that CDRs serve in antigen binding, mutations
are frequently introduced into CDRs to obtain high affinity
variants in the practice of in vitro antibody affinity
improvement (15). However,
β-sheets in the FRs of the antibody variable region serve a
scaffold role for the loop structure of the CDRs, and specific
residues in the FRs supporting the CDR loops can affect loop
folding and fine-tune their binding to antigens (10). Furthermore, FR residues in the
periphery of CDRs have been indicated to be in direct contact with
antigens (16). Consequently, FR
engineering may serve as an alternative approach to improve
antibody affinity by modulating the antibody-antigen interface with
no risk of losing binding specificity. The present study used FR
grafting to improve the affinity of the scFv of e23sFv, which has
been indicated to bind HER2 with superior specificity in previous
studies (3). Firstly, NCBI was
searched for antibody variable region sequences homologous with the
VL and VH of e23sFv. Subsequently, the FRs in e23sFv were replaced
with the corresponding segments from the top two homologous
sequences to generate two engrafted scFvs, named EX1 and EX2.
The mutagenesis of the residues in the CDRs or FRs,
ranging from site-directed to undirected randomized mutations is
based on the conformational information of the antibody and
high-throughput screening procedures to obtain affinity-improved
antibodies (17). In the present
study, an affinity-improved antibody, EX1, was obtained by
transplanting the FRs of the homologous antibody heavy and light
chains into e23sFv. The affinity of EX1 was improved by a factor of
3 compared with that of e23sFv according to the SPR assay, and this
reconstruction was based on the absence of any conformational
information. Sequence alignment indicated that the residue
similarity of the Vernier zone of FRs in the e23sFv and the
candidates determined the extent of the affinity improvement. EMEY,
which exhibited a ~200-fold decreased affinity for HER2 compared
with that of e23sFv in the SPR assay, carried one mutation (G64S)
in the Vernier zone of VL and one mutation (F69L) in that of VH
compared with e23sFv. EX1, which exhibited a three-fold increased
affinity for HER2 compared with that of e23sFv, carried two
additional mutations (L2V and Y35F) in the VL Vernier zone and four
additional mutations (Q2K, S27T, F69L and V71S) in the VH Vernier
zone. For comparison with EMEY, EX1 was constructed by substituting
the FRs of e23sFv with counterparts from the top sequence based on
homology (18,19). The SPR binding assay demonstrated
that EX1 exhibited 3-fold increased affinity for recombinant human
HER2 compared with that of e23sFv, with an increased Kon
value and decreased Koff value. In the binding assay
performed with flow cytometry, EX1 exhibited higher affinity for
HER2-overexpressing cell lines compared with that of parental
e23sFv. The in silico docking analysis indicated that e23sFv
and the three mutants bound to domain IV of the HER2 ECD, with
several residues putatively important for establishing binding
interfaces. The binding energy of HER2 with EX1 was predicted to be
slightly higher compared with that of e23sFv. The docking analysis
also revealed that all the scFvs formed distinct but overlapping
interfaces with domain IV of the HER2 ECD, which indicated that the
manipulation of the FRs mildly influenced the binding specificity
of the scFvs.
A potent application of scFvs in tumour diagnosis
and treatment requires the administration of scFvs at a dose
sufficient for tumour targeting with minimal off-tumour effects
(5). The improved affinity of EX1
for efficient HER2 binding required a small dose, which indicated
that it poses a reduced risk of off-target effects and is less
likely to induce toxicity in organs, such as the liver and kidney.
Moreover, a small dose may result in less intense immunogenic
reactions and lower costs for treatment, which are also important
issues to consider. Immunofluorescence experiments indicated that
EX1 can be internalized into cells after binding to HER2 on the
cell surface, which may suggest that proapoptotic molecules, such
as tBid, or caspase-6 among others, can be fused to the C-terminus
of EX1 as in our previous studies, and be internalized with scFv
into tumour cells (4). The
internalized molecules are presumed to enter endosomes with HER2
molecules and be carried and released to kill tumour cells, which
guarantees the success of subsequently applied antibody-based
therapeutics. The internalized proapoptotic molecules are
subsequently translocated to the cytoplasm where they execute
proapoptotic activity. The construction of an EX1-proapoptotic
fusion protein and analysis of its antitumour toxicity are ongoing
projects.
The approach of the present study for in
vitro antibody affinity maturation was based on protein
sequence alignment and domain grafting. Affinity-improved
candidates were achieved by retaining the CDRs of the parental scFv
and substituting the FRs with the counterparts from a homologous
antibody. This approach maintained the conformational structure of
the parental scFv and the maximal binding specificity to the
antigen. Moreover, FR grafting is more straightforward compared
with site-directed mutagenesis at specific residues because the
mutations are frequently based on the crystal structure of the
antigen-antibody complex (7).
However, several candidates in the present study were indicated to
exhibit a partially decreased affinity for HER2 compared with
e23sFv. One plausible explanation may be that the alterations in FR
core residues failed to fully contribute to antibody folding and
resulted in functional loss. Nonetheless, the methodology used in
the current study was indicated to be of satisfactory efficiency
and can be widely applied for antibody affinity improvement. In
conclusion, the FR grafting strategy was indicated to be more
effective and simple compared with site-directed mutagenesis to
improve e23sFv affinity. Moreover, it was indicated that the
affinity-improved candidate EX1 may be used for the diagnosis and
treatment of HER2-overexpressing tumours.
Acknowledgements
Teh authors would like to thank Dr Dandan Chai from
the Department of Immunology, Fourth Military Medical University
(Xi'an, China) for technical assistance.
Funding
The present study was funded by National Natural
Science Foundation of China (grant nos. 81630069, 81421003,
81172147, 81372459 and 81972871).
Availability of data and materials
The datasets used and/or analysed during the current
study are not publicly available because they are part of an
ongoing project but are available from the corresponding author on
reasonable request.
Authors' contributions
JLR, JZ and AGY designed the research. QOY and BY
performed all the experiments. JNF performed the molecular
modelling analysis. JZ and AGY wrote the manuscript. All author
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rimawi MF, Schiff R and Osborne CK:
Targeting HER2 for the Treatment of Breast Cancer. Annu Rev Med.
66:111–128. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kasprzyk PG, Song SU, Di Fiore PP and King
CR: Therapy of an animal model of human gastric cancer using a
combination of anti-erbB-2 monoclonal antibodies. Cancer Res.
52:2771–2776. 1992.PubMed/NCBI
|
|
3
|
Batra JK, Kasprzyk PG, Bird RE, Pastan I
and King CR: Recombinant anti-erbB2 immunotoxins containing
Pseudomonas exotoxin. Proc Natl Acad Sci USA. 89:5867–5871.
1992.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Jia LT, Zhang LH, Yu CJ, Zhao J, Xu YM,
Gui JH, Jin M, Ji ZL, Wen WH, Wang CJ, et al: Specific tumoricidal
activity of a secreted proapoptotic protein consisting of HER2
antibody and constitutively active caspase-3. Cancer Res.
63:3257–3262. 2003.PubMed/NCBI
|
|
5
|
Ou-Yang Q, Yan B, Li A, Hu ZS, Feng JN,
Lun XX, Zhang MM, Zhang MD, Wu KC, Xue FF, et al: Construction of
humanized anti-HER2 single-chain variable fragments (husFvs) and
achievement of potent tumor suppression with the reconstituted
husFv-Fdt-tBid immunoapoptotin. Biomaterials. 178:170–182.
2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Mazor R, Onda M and Pastan I:
Immunogenicity of therapeutic recombinant immunotoxins. Immunol
Rev. 270:152–164. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Sheedy C, Mackenzie CR and Hall JC:
Isolation and affinity maturation of hapten-specific antibodies.
Biotechnol Adv. 25:333–352. 2007.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Briney B, Sok D, Jardine JG, Kulp DW, Skog
P, Menis S, Jacak R, Kalyuzhniy O, de Val N, Sesterhenn F, et al:
Tailored immunogens direct affinity maturation toward HIV
neutralizing. Cell. 166:1459–1464.e11. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Prassler J, Steidl S and Urlinger S: In
vitro affinity maturation of HuCAL antibodies: Complementarity
determining region exchange and RapMAT technology. Immunotherapy.
1:571–583. 2009.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Foote J and Winter G: Antibody framework
residues affecting the conformation of the hypervariable loops. J
Mol Biol. 224:487–499. 1992.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Teplyakov A, Obmolova G, Malia TJ,
Raghunathan G, Martinez C, Fransson J, Edwards W, Connor J,
Husovsky M, Beck H, et al: Structural insights into humanization of
anti-tissue factor antibody 10H10. MAbs. 10:269–277.
2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhang L, Cai QY, Cai ZX, Fang Y, Zheng CS,
Wang LL, Lin S, Chen DX and Peng J: Interactions of bovine serum
albumin with anti-cancer compounds using a ProteOn XPR36 array
biosensor and molecular docking. Molecules. 21(1706)2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yan B, Ouyang Q, Zhao Z, Cao F, Wang T,
Jia X, Meng Y, Jiang S, Liu J, Chen R, et al: Potent killing of
HBV-related hepatocellular carcinoma by a chimeric protein of
anti-HBsAg single-chain antibody and truncated Bid. Biomaterials.
34:4880–4889. 2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
LuCore SD, Litman JM, Powers KT, Gao S,
Lynn AM, Tollefson WT, Fenn TD, Washington MT and Schnieders MJ:
Dead-end elimination with a polarizable force field repacks PCNA
structures. Biophys J. 109:816–826. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Tiller KE, Chowdhury R, Li T, Ludwig SD,
Sen S, Henry KA and Tessier PM: Facile affinity maturation of
antibody variable domains using natural diversity mutagenesis.
Front Immunol. 8(986)2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Chothia C and Lesk AM: Canonical
structures for the hypervariable regions of immunoglobulins. J Mol
Biol. 196:901–917. 1987.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Moreira GMSG, Fuhner V and Hust M: Epitope
mapping by phage display. Methods Mol Biol. 1701:497–518.
2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Tahara T, Kuwaki T, Matsumoto A, Morita H,
Watarai H, Inagaki Y, Ohashi H, Ogami K, Miyazaki H and Kato T:
Neutralization of biological activity and inhibition of receptor
binding by antibodies against human thrombopoietin. Stem Cells.
16:54–60. 1998.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wright GJ, Cherwinski H, Foster-Cuevas M,
Brooke G, Puklavec MJ, Bigler M, Song Y, Jenmalm M, Gorman D,
McClanahan T, et al: Characterization of the CD200 receptor family
in mice and humans and their interactions with CD200. J Immunol.
171:3034–3046. 2013.PubMed/NCBI View Article : Google Scholar
|