Introduction
Fabry disease (FD; OMIM 301500) is an X-linked
inherited error lysosomal storage disorder, caused by mutations in
the galactosidase α (GLA) gene that encodes the
α-galactosidase A enzyme (α-GAL A) (1-6).
Patients with α-GAL A deficiencies exhibit a harmful accumulation
of globotriaosylceramide (GL-3/Gb3) in capillary endothelial, renal
(podocytes, tubular, glomerular endothelial, mesangial and
interstitial cells), cardiac (cardiomyocytes and fibroblasts) and
nerve cells (7-10).
It is hypothesized that the accumulation of GL-3 causes a wide
spectrum of pathogenic symptoms that include progressive renal
insufficiency, cardiac involvement, and neuropathology (11-14).
End-stage renal disease and life-threatening cardiovascular or
cerebrovascular complications limit the life-expectancy of
untreated patients by 20 and 10 years, respectively, compared with
the general population (1). Almost
all FD-associated complications are non-specific and clinically
indistinguishable from similar abnormalities that occur in the
context of more common disorders, for example neuropathic pain,
sweating, gastrointestinal symptoms, and pulmonary symptoms
(15-21).
FD is treated via enzyme replacement therapy (ERT), which
substitutes the missing or altered, partially functional α-GAL A
(18). Additionally, DGJ
(1-deoxygalactonojirimycin), a pharmacological chaperone, is used
to treat amenable α-GAL A missense mutations with adverse side
effects (20). The aforementioned
therapeutic techniques do not reverse all symptoms experienced by
patients with FD (18,19,22).
Therefore, it is undetermined whether FD symptoms are solely
associated with the accumulation of GL-3 or whether other genetic
mechanisms may be involved.
The current study hypothesized that the causes of FD
are not limited to α-GAL A enzyme malfunction as a result of
pathogenic GLA genetic mutations. The GLA locus is
paired in a divergent configuration with heterogeneous nuclear
ribonucleoprotein (HNRNPH2) on chromosome X, sharing
regulatory sequences at the 5'-ends. The National Center for
Biotechnology Information (NCBI) database has previously suggested
that HNRNPH2 (Gene ID: 3188) is a likely cause of FD.
Previous studies have demonstrated that divergent genes are
governed by bidirectional promoters (BDP), which are characterized
by the absence of a TATA box, the presence of several transcription
factor motifs including nuclear respiratory factor 1 (NRF1), Yin
Yang 1 (YY1), GA binding protein transcription factor subunit α
(GABPA) and zinc finger protein 143 (ZNF143), an abundance of GC
and CpG islands (CGIs) (23-29).
Divergent mutations in the BDPs of paired genes are associated with
cancer (30-33),
cerebral cavernous malformations (34) and nicotine initiation or addiction
(35). Although heterozygous
females with FD usually present an attenuated form of the disease,
a recent study reported a severe case of FD observed in the
GLA of a heterozygous female, likely occurring from a
mutation that altered the CGI methylation status (36). The location of the aforementioned
mutation is in the discovered BDP reported in this study. The
current study therefore performed bioinformatics and additional
experiments to investigate the potential presence of BDP and its
association with GLA and HNRNPH2 expression.
Materials and methods
Genomics databases
Genomic features of the divergently paired
GLA and HNRNPH2 loci were searched in the following
genomic databases: Ensembl (https://uswest.ensembl.org/index.html), NCBI-Gene
(https://www.ncbi.nlm.nih.gov/gene/?term=), GeneCards
(https://www.genecards.org/), EMBL-EBI
(https://www.ebi.ac.uk/), UCSC genomic browser
(https://www.genome.ucsc.edu/), PrESSTo
(http://pressto.binf.ku.dk/about.php)
and the Eukaryotic promoter database, EPD (https://epd.epfl.ch//index.php). The precise genomic
map locations of the identified sequences were verified and
uploaded to the hg38 human genome sequence using the ‘get DNA’ and
‘BLAT’ tools of the UCSC Genome Browser database. EMBOSS Needle
(https://www.ebi.ac.uk/Tools/psa/emboss_needle/) was
used for the pairwise sequence alignment of nucleotide
sequences.
Regulatory sequences at the 5'-ends of
GLA and HNRNPH2
The 5'-end genomic regions of the divergently paired
GLA (ENSG00000102393) and HNRNPH2 (ENSG00000126945)
loci were searched for regulatory sequences. The motif tool of the
EPD database was used to search for TATA box motifs. Additionally,
the JASPAR database (http://jaspar.genereg.net/) of transcription factor
binding profiles (37,38) was used to determine the binding site
motifs of YY1, NRF1, E2F transcription factor 1 (E2F1) and GABPA
transcription factors in the BDP (23-29).
CGI identification and plotting in the predicted BDP sequences were
achieved using the EMBOSS cpgplot tool (https://www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot/).
The parameters used to search for CGIs were as follows: Obs/Exp CpG
>0.6, G%+C% >50% and 100 nucleotides in length. The details
of CGI prediction are included in the EMBOSS cpgplot manual.
Human cell lines, RNA and DNA
293T (ATCC® CRL-1573™) cells were
purchased from The American Type Culture Collection and cultured in
Eagle's minimum Essential medium (cat. no. 30-2003) supplemented
with 10% heat-inactivated FBS (ATCC® 30-2020™), 100 U/ml
penicillin and 100 µg/ml streptomycin. Cells were cultured in a
humidified incubator in the presence of 5% CO2 at 37˚C.
A purification kit (cat. no. 48700; Norgen Biotek Corp.) was used
for the extraction of RNA and DNA from 293T cells. Human genomic
DNA for methylation analysis isolated from adult epidermal
keratinocytes (cat. no. 2119), renal glomerular endothelial cells
(cat. no. 4009) and renal epithelial cells (cat. no. 4129) were
purchased from ScienCell Research Laboratories, Inc.
Primer set design and quantitative
(q)PCR analysis
Primer sets for qPCR-intercalating dyes were
purchased from Integrated DNA Technologies, Inc. The PrimerQuest
Tool was used for the custom design of nuclear run-on (NRO) and
chromatin-immunoprecipitation (ChIP) assay primers. The methylation
primer sets were designed with the MethPrimer program (39) and used for methylation specific-PCR
(MSP) analysis. The real-time qPCR reactions were performed using
the iTaq universal SYBR-Green reaction mix on a Bio-Rad CFX96
Real-Time system. The thermocycling conditions were as follows:
Polymerase activation and DNA denaturation at 95˚C for 3 min
followed by 35 (MSP assay) and 45 cycles (NRO or ChIP assays) of
denaturation at 95˚C for 5 sec, and annealing and extension for 30
sec at 60˚C (NRO or ChIP assays) and 58˚C (MSP assay),
respectively. Replicate PCRs were run on the same sample where
target and reference are amplified in separate wells (40). At least three independent
experiments were performed for each sample (41).
ChIP analysis
ChIP was carried out using the EpiQuik™ Chromatin
Immunoprecipitation kit (Epigentek Group Inc.) in accordance with
the manufacturer's protocol. 293T cells were fixed using 1%
formaldehyde solution for 10 min at room temperature and sonicated
samples were subsequently immunoprecipitated at room temperature
for 90 min using NRF1 (ab175932) and YY1 (ab38422) antibodies
purchased from Abcam. Normal mouse IgG (1 mg/ml) (negative control)
and 1 mg/ml anti-RNA Polymerase II (positive control) were provided
as part of the kit; 1 µl of normal mouse IgG, 1 µl of anti-RNA
polymerase II, and 5 µg of anti-NRF1 and anti-YY1 were added to
each well. The wells were covered with Parafilm M and incubated at
room temperature for 120 min. Protein-DNA complexes were
de-crosslinked and the DNA was purified using the F-Spin column.
The primer sets were designed to produce amplicons that host the
NRF1 motifs (forward, 5'-AGCTGAGGAACCCAGAACTA-3' and reverse,
5'CAATCCATTGTCCAGTGCTCTA-3') and YY1 motifs (forward,
5'GTCATGAGCGTCCACCATTT-3' and reverse,
5'-CCTCTTTCGTTCTCTGCTTTCC-3'). The Fold enrichment method was used
to analyze ChIP-qPCR data. The 2-∆∆CT value was
calculated from (Ct IP)-(Ct mock). Normalization was completed
using the IgG Ct value. Duplicate PCRs were run on the same sample
and Ct average data were used (40). Four independent experiments were
performed for each sample (41).
Bisulfite DNA treatment and MSP
analysis
The methylamp DNA Modification kit (Epigentek Group
Inc.) was used to investigate the methylation status of predicted
BDP sequences in accordance with the manufacturer's protocol.
Genomic DNA from adult epidermal keratinocytes (cat. no. 2119),
renal glomerular endothelial cells (cat. no. 4009), renal
epithelial cells (cat. no. 4129) and 293T cells was used for MSP
analysis. Purified genomic DNA (100 ng) was treated using the
Methylamp DNA Modification kit, after which the converted DNA was
cleaned, captured and eluted using R6 (Modified DNA Elution)
solution and an F-Spin column. Eluted DNA was analyzed using the
iTaq universal SYBR-Green reaction mix (Bio-Rad Laboratories,
Inc.). The methylated primer sets were designed using the
MethPrimer program (39) and used
for CGI methylation analysis in the BDP sequence. The MSP primers
for methylated and unmethylated regions of the 323 bp CGI-2 were as
follows: M pair (forward, 5'-TTTTTTTAAACGGTTATAGCGAGAC-3' and
reverse, 5'-CTTAATTTACCAAATAACCCGTA-3'), U pair (forward,
5'-TTTTTTAAATGGTTATAGTGAGATGG-3' and reverse,
5'-AATACAACACCTTAATAATCCCAAA-3'). The qPCR was performed as
aforementioned. The percentage of sample methylation was calculated
using the following equation: Percentage methylation =100/[1+2∆Ct
(meth-unmeth)]. ∆Ct (meth-unmeth) was calculated by subtracting the
Ct values of methylated CGI signals from the Ct values of the
unmethylated CGI signal (42,43).
Each sample was run in duplicate for qPCR analysis (40). Three independent experiments were
performed for each sample (41).
NRO analysis
NRO is the method of choice when measuring the
transcriptional activity of nuclear nascent mRNA transcripts
(44). NRO analysis was used to
investigate the expression and the quantification of GLA and
HNRNPH2 nascent RNA transcription. 293T cells were used for
the preparation of nuclei. The preparation of cell cultures, nuclei
collection NRO transcription, nuclear RNA extraction,
immunoprecipitation of bromouridylated NRO-RNAs, nascent nuclear
RNA extraction, cDNA preparation and NRO cDNA quantification was
performed using the bromouridine immunocapture nuclear run-on
RT-qPCR method (44). NRO
transcription was performed in fully resuspended nuclei by gentle
pipetting at 30˚C for 30 min using the transcription buffer
reaction cocktail master mix, which was composed of BrUTP, ATP,
GTP, CTP and UTP. A total of 4,000,000 nuclei were used per sample
for NRO transcription. Nuclear RNA was extracted using the
MEGAclear transcription clean-up kit (Thermo Fisher Scientific,
Inc.; cat no. AM1908) according to the manufacturer's protocol.
Immunoprecipitation of bromouridylated NRO-RNAs was performed using
2 µg/tube of mouse monoclonal IgG1 anti-BrdU antibody IIB5
(sc-32323) (Santa Cruz Biotechnology, Inc.). Nascent nuclear RNA
was extracted using the RNAzol method (GeneCopoeia, Inc., cat. no.
E01010A) and precipitated by adding an equal volume of isopropanol
and 20 µg RNase-free glycogen (1 µl of 20 mg/ml stock) to each
sample. The High-Capacity cDNA Reverse Transcription kit (Thermo
Fisher Scientific, Inc.; cat. no. 4368814) was used for the
quantitative conversion of the extracted NRO RNA to single-stranded
cDNA in a single 20 µl reaction by the thermal cycler with the
following thermocycling conditions: 10 min at 25˚C, 30 min at 37˚C,
5 min at 85˚C. Amplified DNA was prepared from NRO-cDNA using the
designed primer sets for HNRNPH2-E1 (forward,
5'-AGTAGTTCTGGTCGTCGT CTA-3' and reverse,
5'-ACACACCAACCTCTAACGATAC-3') and GLA-E1 (forward,
5'-AGGTTACCCGCGGAAATTTAT-3' and reverse, 5'-GAAACGAGGGCCAGGAAG-3').
Normalization was performed using hypoxanthine
phosphoribosyltransferase 1 (HPRT1) primer sets (forward,
5'-TGAGGATTT GGAAAGGGTGT-3' and reverse, 5'-GAGCACACAGAGGG
CTACAA-3'). The normalization and relative expression of target
sequences was determined using HPRT1 as a reference gene.
Duplicate PCRs were run on the same sample and Ct average data were
used to calculate the 2-ΔΔCT value (40). Three independent experiments were
performed for each sample (41).
Statistical analysis
Microsoft Excel 2010 (Microsoft Corporation) and
GraphPad Prism 7.01 (GraphPad Software, Inc.) were used to analyze
the data of various parameters. Two-sample comparison was
determined by performing a Student's t-test. One-way ANOVA with
Tukey's multiple comparisons test/post hoc test was used to
evaluate four independent groups simultaneously and to test
statistical differences between every possible pair of all groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
GLA and HNRNPH2 loci
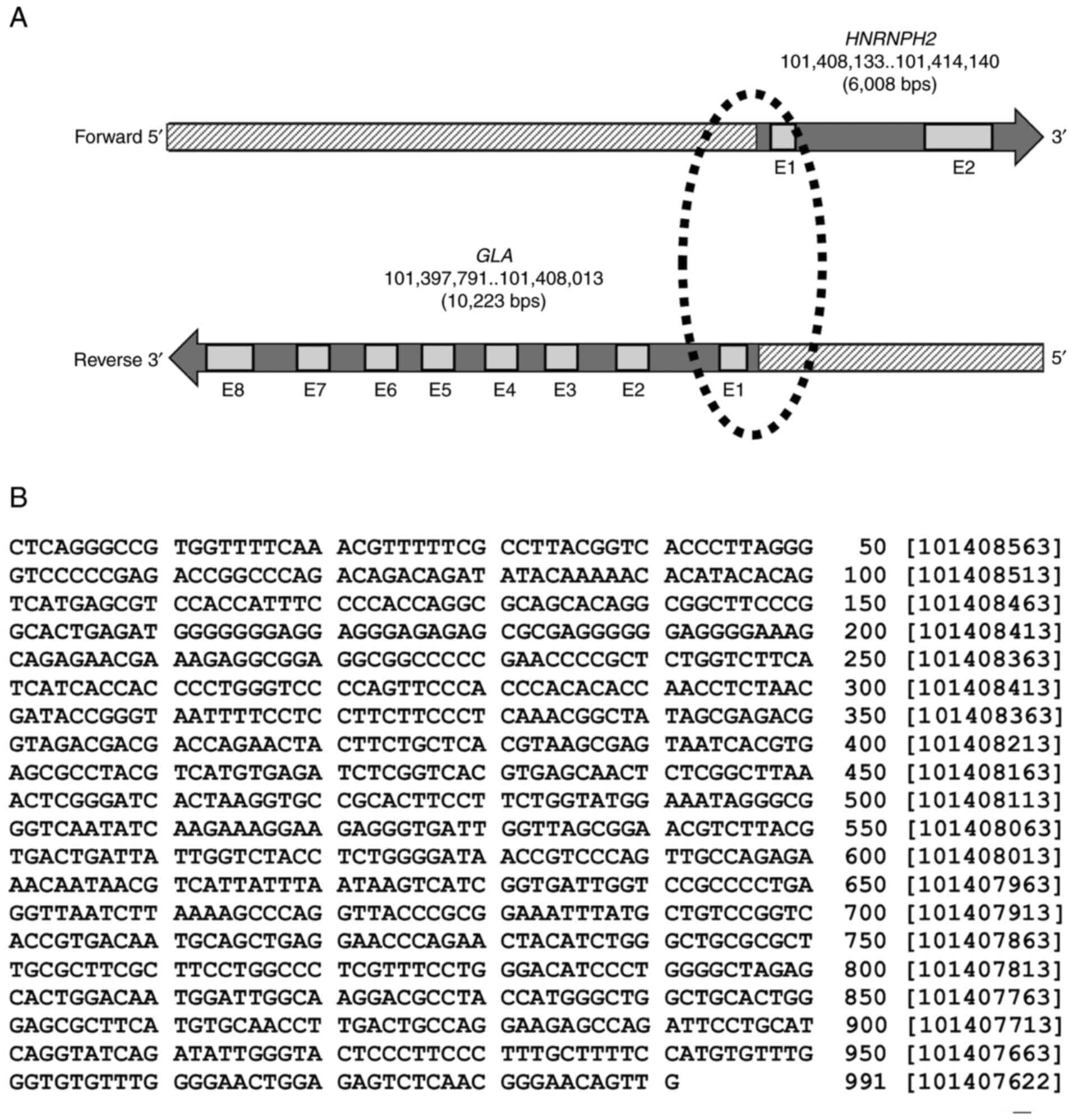
The genomic setting of the paired GLA
(ENSG00000102393; NCBI-Gene ID: 2717) and HNRNPH2
(ENSG00000126945; NCBI-Gene ID: 3188) genes is divergent, with a
head-to-head formation on chromosome X. The GLA locus is
situated on the reverse strand, whereas the HNRNHP2 locus appears
on the forward strand (Fig. 1A).
The Ensembl database revealed that GLA and HNRNPH2
shared a 991 nucleotide sequence between the two loci at the 5'-end
(Fig. 1B). Pairwise sequence
alignment analysis performed using the EMBOSS Needle tool also
determined that the shared sequence between GLA and
HNRNPH2 exhibited 100% similarity.
Shared regulatory sequences between
GLA and HNRNHP2 loci
The 991 nucleotide sequence shared between
GLA and HNRNHP2 loci lacked a TATA box motif and was rich in
binding sites for several transcription factors that are found in
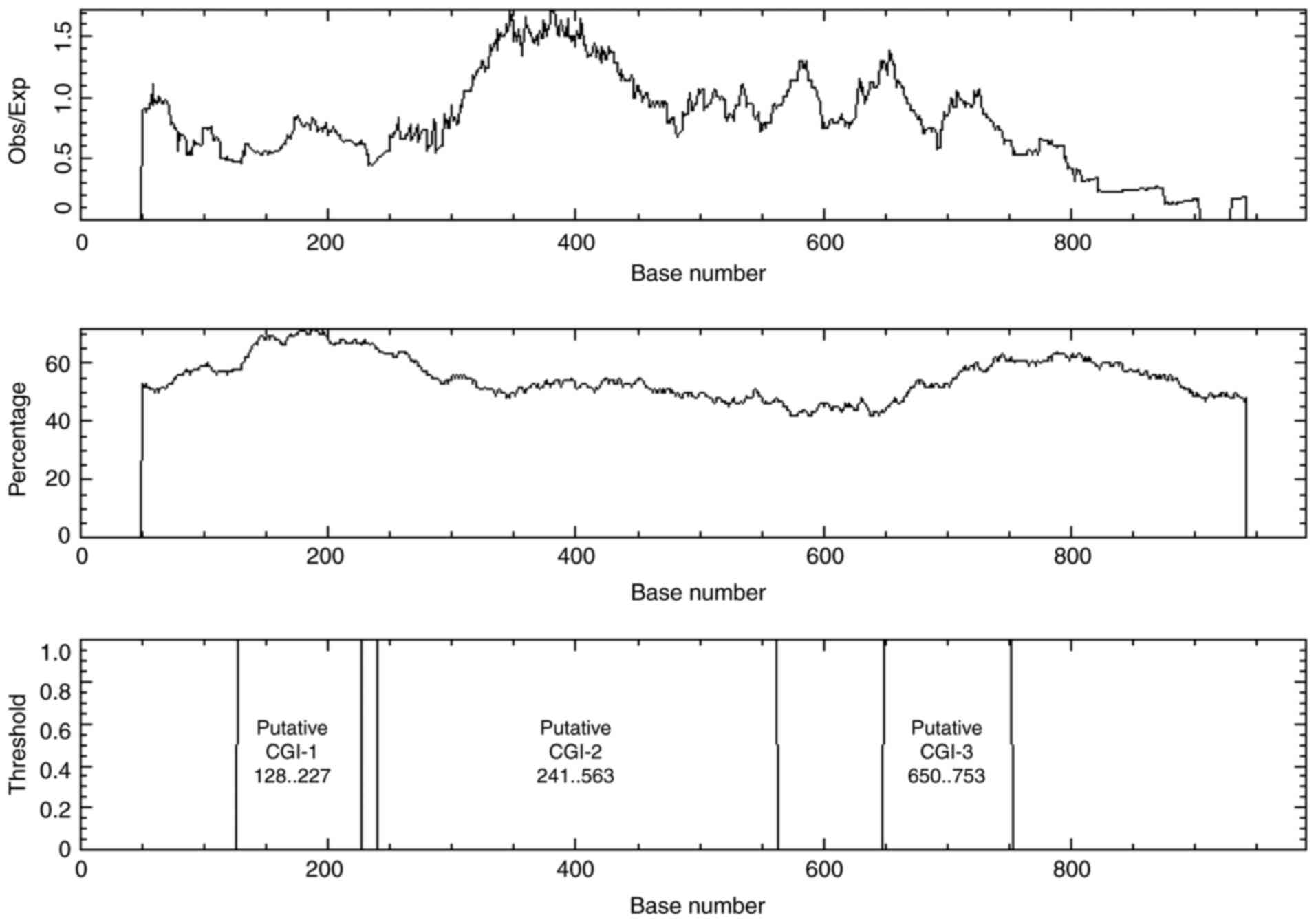
BDP, including YY1, NRF1, GABPA and E2F1 (Table I). Additionally, bioinformatics
analysis demonstrated that the shared sequence was CG rich,
containing CGIs. EMBOSS Cpgplot software revealed one CGI when the
following options were searched: Window size, 100; minimum sequence
length, 200 bases; minimum Obs/Exp CpG, >0.6; %C+%G, >50.00%.
However, three CGIs were revealed in the shared sequence when the
search tool was set to a minimum sequence length of 100 bases
(Fig. 2).
 | Table ITranscription factor binding sites
predicted using the JASPAR tool in the GLA and
HNRNPH2 bidirectional promoter. |
Table I
Transcription factor binding sites
predicted using the JASPAR tool in the GLA and
HNRNPH2 bidirectional promoter.
| TFBSs | Number | Similarity
Score |
|---|
| YY1 | 10 | 0.81-0.93 |
| NRF1 | 8 | 0.80-0.89 |
| GABPA | 10 | 0.80-0.88 |
| E2F1 | 12 | 0.80-0.94 |
Two promoter prediction software tools determined
two transcriptional start sites (TSSs) along the 991 nucleotide
sequence (Table II), located at
the 5'-ends of GLA and HNRNPH2. Furthermore, the EPD
eukaryote promoter database revealed two main TSSs in the
GLA promoter and three main TSSs in the HNRNPH2
promoter within the predicted BDP. Bioinformatics analysis
suggested that the predicted 991 nucleotide sequence had the
molecular features of a BDP. The predicted BDP was therefore
presumed to direct the transcription of the GLA locus in a
divergent manner along with its counterpart locus, HNRNHP2.
| Table IITwo TSSs predicted by two promoter
prediction tools in the bidirectional promoter. |
Table II
Two TSSs predicted by two promoter
prediction tools in the bidirectional promoter.
| Promoter prediction
tool | TSS position | Scorea |
|---|
| Promoter 2.0
prediction server | 200 | 0.632 |
| Neural network
promoter prediction | 688 | 0.97 |
Methylation status of CGIs in kidney
cell lines
The CGIs associated with bidirectional promoters are
prone to DNA methylation (25). The
methylation status of the previously elucidated CGI-2 at position:
241..563 (Fig. 2) was determined in
four cell types, including human adult epidermal keratinocyte cells
(AEK), human renal glomerular endothelial cells (HRG), human renal
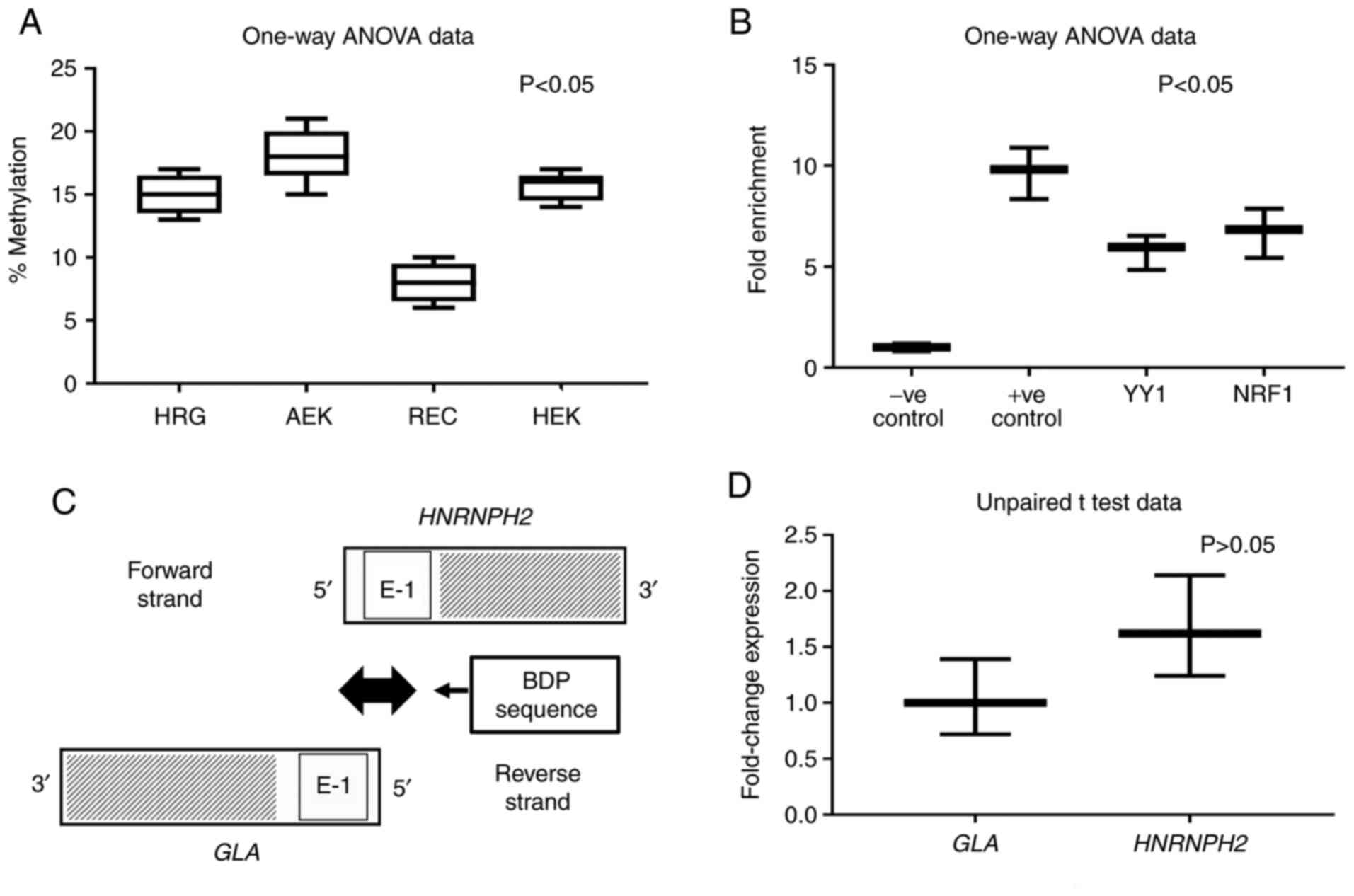
epithelial cells (REC) and 293T cells. The results revealed
variable levels and significant differences in the extent of DNA
methylation in AEK, HRG, REC and 293T cells, the highest result of
which was observed in AEKs (Fig.
3A; P<0.05). RECs exhibited the lowest level of methylation
compared with HRG and 293T cells, which themselves demonstrated
comparable methylation percentages. Multiple comparison results
presented statistical differences between HRG and AEK, HRG and REC,
AEK and REC, and REC and 293T (P<0.05), but not between HRG and
293T and between AEK and 293T (P>0.05).
ChIP assay predicts NRF1 and YY1
motifs in the BDP
The JASPAR 2018 tool (37,38)
identified the occurrence of YY1 and NRF1 binding sites in the
shared 991 nucleotide sequence (Table
I). The results of the ChIP assay, which was conducted using
293T cells, identified YY1 and NRF1 binding sites in the BDP
sequence (Fig. 3B). The designed
primers produced amplicons (~100 bp) containing YY1 and NRF1
motifs. One-way analysis showed significant statistical differences
in four tested groups, P<0.05. But multiple comparison results
displayed statistical differences between every possible pair of
all groups (P<0.05) but not between YY1 and NRF1, P>0.05.
Quantification of nascent GLA and
HNRNPH2 mRNA transcripts by NRO
Nascent NRO bromouridine-labeled mRNA derived from
the nuclei of 293T cells was measured via RT-qPCR. The primer sets
used during PCR were designed to amplify the adjacent sequences
specific to GLA and HNRNPH2 at the 5'-ends (Fig. 3C). The data obtained using the
relative 2-∆∆CT method (40) revealed an insignificant difference
in divergent transcription activity between GLA and
HNRNPH2 when using primer sets designed for exon 1 of
GLA and HNRNPH2, P>0.05 (Fig. 3D).
Pathogenic variants of the BDP
The NCBI-dbSNP database revealed 2,657 and 1,517
variants in the GLA and HNRNPH2 loci, respectively.
Additionally, pathogenic variants were identified in the BDP
sequence at chromosome X:101407622-101408612. Several of these
variants are presented in Table
III. For example, the position of rs104894829 (C>T) was at
chromosome X:101407773, whereas rs104894847 (C>G) was at
chromosome X:101407846. Hossain et al (36) reported a 37-year-old female carrier
of a heterozygous mutation with severe FD symptoms. The position of
the C>A variant was at chrX:101407903 in the BDP sequence.
| Table IIIPathogenic variants identified in the
bidirectional promotor. |
Table III
Pathogenic variants identified in the
bidirectional promotor.
| Variant ID | Chromosome
position | Type |
|---|
| NA | chrX:101407903 | C>A |
| rs104894829 | chrX:101407773 | C>T |
| rs104894831 | chrX:101407786 | G>A |
| rs104894835 | chrX:101407803 | T>C |
|
rs104894836/rs28935192 | | |
| rs104894847 | chrX:101407738 | A>C |
| rs104894848 | chrX:101407846 | C>G |
| rs1569306022 | chrX:101407710 | C>G |
| rs1569306168 | chrX:101407716 | C>T |
| | chrX:101407809 | A>G |
Discussion
FD is a clinically heterogeneous, slow and
progressive disease that shares common symptoms in the general
population (15-21).
Although patients with FD exhibit a wide range of clinical
symptoms, their detailed genetic presentation has not yet been
fully elucidated. Pathogenic mutations in GLA have been
reported as the main genetic cause of FD (11-13).
A common clinical presentation of patients with FD is neuropathic
pain, with both sexes exhibiting symptoms into adulthood (45,46).
However, neuropathic pain usually presents in the early years of
childhood (47,48). The majority of patients with FD may
experience chronic or episodic pain, which are known as FD crises
or acroparaesthesiae (49-52).
There is an indirect association between pain and
the alternate splicing of mRNA. Donaldson and Beazley-Long
(53) reviewed the known
alternative splice variants that are important to pain. They
suggested that these sites may serve as appropriate therapeutic
targets. Furthermore, their study demonstrated that pain may result
from indirect genetic conditions that arise from defects in the
alternative RNA splicing process of several genes. Proteins encoded
by the HNRNP gene family, including HNRNPH2, are RNA binding
proteins that are associated with the mRNA splicing process.
Previous studies have demonstrated the association between HNRNP
genes and various clinical symptoms, such as pain. In this respect,
HNRNPH1 and HNRNPF can serve as post-transcriptional regulators of
opioid receptor expression (54,55).
HNRNPH2 and HNRNPF genes encode a similar protein structure
and bind to similar sequences in order to influence gene expression
(56). Since HNRNPH2 is a
member of the HNRNP gene family, this may indicate its involvement
in the pain experienced by patients with FD. The NCBI-Gene database
notes that HNRNPH2 may be implicated in FD. It is
hypothesized that aberrations in HNRNPH2 expression can
cause defects in mRNA splicing. Nearly all human multiexon genes
are subject to alternative splicing (57), which has been reported in GLA
(58-60).
A recent study has suggested that DNA methylation not only affects
transcription, but also regulates alternative splicing (61). Accordingly, the current study
hypothesized that defects in HNRNPH2 expression caused by
BDP mutations can influence symptoms associated with pre-mRNA
splicing malfunctions, including pain. Thus, defects in
HNRNPH2 expression can alter the normal mRNA splicing
process of multiexon genes including GLA and other genes
associated with diseases and pain (53).
At present, there is insufficient information
available on the regulatory mechanisms associated with the
expression of paired, divergent HNRNPH2 and GLA loci
on chromosome X. The present study provided bioinformatic and
experimental data that demonstrated the occurrence of a BDP
regulatory sequence at the 5'-end of GLA and HNRNPH2.
Bioinformatics analysis identified a shared 991 nucleotide sequence
in the BDP, which was associated with bidirectional transcription.
The main features of this sequence, which are characteristic of
BDPs (23-29),
was the absence of TATA-box binding motifs, alongside the presence
of a 323 bp CGI-2 and the occurrence of specific transcription
factor motifs, including, YY1 and NFR1. The results of the present
study also indicated the presence of YY1 and NFR1 transcription
factors' motifs in 323 bp CGI-2. The methylation status of four
human cell lines was also evaluated. The NRO assay performed in the
current study confirmed the divergent expression of GLA and
HNRNPH2 in the nascent nuclear transcripts of 293T
cells.
Several mammalian promoters demonstrate divergent
transcription. In the human genome, it is estimated that >10% of
genes are divergently transcribed, where a single promoter is
shared with their transcriptional start sites (23). BDPs are known to direct divergent
gene expression, which is the case for GLA and
HNRNPH2. Additionally, bidirectional promoters are
associated with various diseases, for example cancer (30-33),
cerebral cavernous malformations (34) and nicotine initiation or addiction
(35). The present data also
provided a reason for the unexplained clinical manifestations
observed in patients with FD. Although asymptomatic females with FD
are likely an exception, it has been reported that heterozygous
females may suffer from significant multisystemic clinical symptoms
(62).
Finally, although the results of the present study
indicated the potential involvement of the BDP, the HNRNPH2
gene and the GLA gene in FD, the precise mechanism that
regulates the bidirectional transcription of GLA and
HNRNPH2 is yet to be understood. Additional experimental
studies using novel methods, including high-throughput assays
(63) are required for the better
understanding of the architecture and cis-regulatory elements of
GLA and HNRNPH2. Furthermore, investigation into the
role of chromatin (64) on the
transcriptional status of the two genes may be required. Another
limitation to the present study is the lack of tissues or cells
from patients with FD.
In conclusion, the molecular characteristics of the
GLA and HNRNHP2 bidirectional promoter may define the role
of upstream pathogenic variants of the divergently paired
GLA and HNRNPH2 genes in FD. Accordingly, defects in
the BDP can simultaneously disturb the expression of GLA and
HNRNPH2 and cause diverse clinical manifestations associated
with FD.
Acknowledgements
The authors would like to thank Professor Kalkunte
S. Srivenugopal (School of Pharmacy, Texas Tech University Health
Sciences Center, Amarillo, Texas) and Dr Todd E. Bell (School of
Medicine, TTUHSC, Amarillo, Texas) for their support and for
providing laboratory facilities.
Funding
The present study was supported by the
Sanofi-Genzyme Corporation (project GZ-2017-11708).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MAIAO, IAO and TLV conceived and designed the
current study. MAIAO and IAO performed the molecular and
bioinformatic analysis. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Germain DP: Fabry disease. Orphanet J Rare
Dis. 5(30)2010.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Chan B and Adam DN: A review of Fabry
disease. Skin Therapy Lett. 23:4–6. 2018.PubMed/NCBI
|
|
3
|
Hsu TR and Niu DM: Fabry disease: Review
and experience during newborn screening. Trends Cardiovasc Med.
28:274–281. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Platt FM, d'Azzo A, Davidson BL, Neufeld
EF and Tifft CJ: Lysosomal storage diseases. Nat Rev Dis Primers.
4(27)2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Nair V, Belanger EC and Veinot JP:
Lysosomal storage disorders affecting the heart: A review.
Cardiovasc Pathol. 39:12–24. 2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Sun A: Lysosomal storage disease overview.
Ann Transl Med. 6(476)2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Desnick RJ, Ioannou YA and Eng CM:
Alpha-galactosidase A deficiency: Fabry disease. In: The metabolic
and molecular bases of inherited disease. Scriver CR, Beaudet AL,
Sly WS, Valle D, Childs B, Kinzler KW and Vogelstein B (eds).
McGraw Hill, New York, NY, pp3733-3774, 2001.
|
|
8
|
Gal A: Molecular genetics of Fabry disease
and genotype-phenotype correlation. In: Fabry disease. Elstein D,
Altarescu G and Beck M (eds). Springer, Dordrecht, pp3-19,
2010.
|
|
9
|
Miller JJ, Kanack AJ and Dahms NM:
Progress in the understanding and treatment of Fabry disease.
Biochim Biophys Acta, Gen Subj. 129437(2020)1864.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Saito S, Ohno K and Sakuraba H: urihttp://Fabry-database.orgsimpleFabry-database.org.
Database of the clinical phenotypes, genotypes and mutant
α-galactosidase A structures in Fabry disease. J Hum Genet.
56:467–468. 2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Oliveira JP and Ferreira S: Multiple
phenotypic domains of Fabry disease and their relevance for
establishing genotype- phenotype correlations. Appl Clin Genet.
12:35–50. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Cocozza S, Russo C, Pontillo G, Pisani A
and Brunetti A: Neuroimaging in Fabry disease: Current knowledge
and future directions. Insights Imaging. 9:1077–1088.
2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Cairns T, Müntze J, Gernert J, Spingler L,
Nordbeck P and Wanner C: Hot topics in Fabry disease. Postgrad Med
J. 94:709–713. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Cuestas D, Perafan A, Forero Y, Bonilla J,
Velandia A, Gutierrez A, Motta A, Herrera H and Rolon M:
Angiokeratomas, not everything is Fabry disease. Int J Dermatol.
58:713–721. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zarate YA and Hopkin RJ: Fabry's disease.
Lancet. 372:1427–1435. 2008.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Körver S, Geurtsen GJ, Hollak CE, van
Schaik IN, Longo MG, Lima MR, Vedolin L, Dijkgraaf MG and Langeveld
M: Depressive symptoms in Fabry disease: The importance of coping,
subjective health perception and pain. Orphanet J Rare Dis.
15(28)2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Schiffmann R: Fabry disease. Handb Clin
Neurol. 132:231–248. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Ortiz A, Germain DP, Desnick RJ, Politei
J, Mauer M, Burlina A, Eng C, Hopkin RJ, Laney D, Linhart A, et al:
Fabry disease revisited: Management and treatment recommendations
for adult patients. Mol Genet Metab. 123:416–427. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Germain DP, Elliott PM, Falissard B, Fomin
VV, Hilz MJ, Jovanovic A, Kantola I, Linhart A, Mignani R, Namdar
M, et al: The effect of enzyme replacement therapy on clinical
outcomes in male patients with Fabry disease: A systematic
literature review by a European panel of experts. Mol Genet Metab
Rep. 19(100454)2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
McCafferty EH and Scott LJ: Migalastat: A
review in Fabry disease. Drugs. 79:543–554. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Del Pino M, Andrés A, Bernabéu AA, de
Juan-Rivera J, Fernández E, de Dios García Díaz J, Hernández D,
Luño J, Fernández IM, Paniagua J, et al: Fabry Nephropathy: An
evidence-based narrative review. Kidney Blood Press Res.
43:406–421. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Müntze J, Gensler D, Maniuc O, Liu D,
Cairns T, Oder D, Hu K, Lorenz K, Frantz S, Wanner C, et al: Oral
chaperone therapy migalastat for treating Fabry disease: Enzymatic
response and serum biomarker changes after 1 year. Clin Pharmacol
Ther. 105:1224–1233. 2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Trinklein ND, Aldred SF, Hartman SJ,
Schroeder DI, Otillar RP and Myers RM: An abundance of
bidirectional promoters in the human genome. Genome Res. 14:62–66.
2004.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Dash A, Gurdaswani V, D'Souza JS and Ghag
SB: Functional characterization of an inducible bidirectional
promoter from Fusarium oxysporum f. sp. cubense. Sci Rep.
10(2323)2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Orekhova AS and Rubtsov PM: Bidirectional
promoters in the transcription of mammalian genomes. Biochemistry
(Mosc). 78:335–341. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Li YY, Yu H, Guo ZM, Guo TQ, Tu K and Li
YX: Systematic analysis of head-to-head gene organization:
Evolutionary conservation and potential biological relevance. PLOS
Comput Biol. 2(e74)2006.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Anno YN, Myslinski E, Ngondo-Mbongo RP,
Krol A, Poch O, Lecompte O and Carbon P: Genome-wide evidence for
an essential role of the human Staf/ZNF143 transcription factor in
bidirectional transcription. Nucleic Acids Res. 39:3116–3127.
2011.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Davuluri RV, Suzuki Y, Sugano S, Plass C
and Huang TH: The functional consequences of alternative promoter
use in mammalian genomes. Trends Genet. 24:167–177. 2008.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Yang MQ and Elnitski LL: Diversity of core
promoter elements comprising human bidirectional promoters. BMC
Genomics. 9 (Suppl 2)(S3)2008.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Germot A and Maftah A: POFUT1 and PLAGL2
gene pair linked by a bidirectional promoter: The two in one of
tumour progression in colorectal cancer? EBioMedicine. 46:25–26.
2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Li D, Lin C, Li N, Du Y, Yang C, Bai Y,
Feng Z, Su C, Wu R, Song S, et al: PLAGL2 and POFUT1 are regulated
by an evolutionarily conserved bidirectional promoter and are
collaboratively involved in colorectal cancer by maintaining
stemness. EBioMedicine. 45:124–138. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Drak Alsibai K, Vacher S, Meseure D,
Nicolas A, Lae M, Schnitzler A, Chemlali W, Cros J, Longchampt E,
Cacheux W, et al: High positive correlations between ANRIL and
p16-CDKN2A/p15-CDKN2B/p14-ARF gene cluster overexpression in
multi-tumor types suggest deregulated activation of an ANRIL-ARF
bidirectional promoter. Noncoding RNA. 5(44)2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Al-Obaide MA, Alobydi H, Abdelsalam AG,
Zhang R and Srivenugopal KS: Multifaceted roles of 5'-regulatory
region of the cancer associated gene B4GALT1 and its comparison
with the gene family. Int J Oncol. 47:1393–1404. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Scimone C, Bramanti P, Ruggeri A, Donato
L, Alafaci C, Crisafulli C, Mucciardi M, Rinaldi C, Sidoti A and
D'Angelo R: CCM3/SERPINI1 bidirectional promoter variants in
patients with cerebral cavernous malformations: A molecular and
functional study. BMC Med Genet. 17(74)2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Aziz HA, Abdel-Salam AG, Al-Obaide MAI,
Alobydi HW and Al-Humaish S: Kynurenine 3-monooxygenase gene
associated with nicotine initiation and addiction: Analysis of
novel regulatory features at 5' and 3'-regions. Front Genet.
9(198)2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Hossain MA, Yanagisawa H, Miyajima T, Wu
C, Takamura A, Akiyama K, Itagaki R, Eto K, Iwamoto T, Yokoi T, et
al: The severe clinical phenotype for a heterozygous Fabry female
patient correlates to the methylation of non-mutated allele
associated with chromosome 10q26 deletion syndrome. Mol Genet
Metab. 120:173–179. 2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Khan A, Fornes O, Stigliani A, Gheorghe M,
Castro-Mondragon JA, van der Lee R, Bessy A, Chèneby J, Kulkarni
SR, Tan G, et al: JASPAR 2018: Update of the open-access database
of transcription factor binding profiles and its web framework.
Nucleic Acids Res. 46:D260–D266. 2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Fornes O, Castro-Mondragon JA, Khan A, van
der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M,
Baranašić D, et al: JASPAR 2020: Update of the open-access database
of transcription factor binding profiles. Nucleic Acids Res.
48:D87–D92. 2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Li LC and Dahiya R: MethPrimer: Designing
primers for methylation PCRs. Bioinformatics. 18:1427–1431.
2002.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Udvardi MK, Czechowski T and Scheible WR:
Eleven golden rules of quantitative RT-PCR. Plant Cell.
20:1736–1737. 2008.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Ng EK, Leung CP, Shin VY, Wong CL, Ma ES,
Jin HC, Chu KM and Kwong A: Quantitative analysis and diagnostic
significance of methylated SLC19A3 DNA in the plasma of breast and
gastric cancer patients. PLoS One. 6(e22233)2011.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Eads CA, Danenberg KD, Kawakami K, Saltz
LB, Blake C, Shibata D, Danenberg PV and Laird PW: MethyLight: A
high-throughput assay to measure DNA methylation. Nucleic Acids
Res. 28(E32)2000.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Roberts TC, Hart JR, Kaikkonen MU,
Weinberg MS, Vogt PK and Morris KV: Quantification of nascent
transcription by bromouridine immunocapture nuclear run-on RT-qPCR.
Nat Protoc. 10:1198–1211. 2015.PubMed/NCBI View Article : Google Scholar
|
|
45
|
MacDermot KD, Holmes A and Miners AH:
Anderson-Fabry disease: Clinical manifestations and impact of
disease in a cohort of 98 hemizygous males. J Med Genet.
38:750–760. 2001.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Löhle M, Hughes D, Milligan A, Richfield
L, Reichmann H, Mehta A and Schapira AH: Clinical prodromes of
neurodegeneration in Anderson-Fabry disease. Neurology.
84:1454–1464. 2015.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Ries M, Ramaswami U, Parini R, Lindblad B,
Whybra C, Willers I, Gal A and Beck M: The early clinical phenotype
of Fabry disease: A study on 35 European children and adolescents.
Eur J Pediatr. 162:767–772. 2003.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Ramaswami U, Whybra C, Parini R,
Pintos-Morell G, Mehta A, Sunder-Plassmann G, Widmer U and Beck M:
FOS European Investigators. Clinical manifestations of Fabry
disease in children: Data from the Fabry Outcome Survey. Acta
Paediatr. 95:86–92. 2006.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Ramaswami U, Parini R, Pintos-Morell G,
Kalkum G, Kampmann C, Beck M and Investigators FOS: FOS
Investigators. Fabry disease in children and response to enzyme
replacement therapy: Results from the Fabry Outcome Survey. Clin
Genet. 81:485–490. 2012.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Magg B, Riegler C, Wiedmann S, Heuschmann
P, Sommer C and Üçeyler N: Self-administered version of the
Fabry-associated pain questionnaire for adult patients. Orphanet J
Rare Dis. 10(113)2015.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Gibas AL, Klatt R, Johnson J, Clarke JT
and Katz J: A survey of the pain experienced by males and females
with Fabry disease. Pain Res Manag. 11:185–192. 2006.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Crosbie TW, Packman W and Packman S:
Psychological aspects of patients with Fabry disease. J Inherit
Metab Dis. 32:745–753. 2009.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Donaldson LF and Beazley-Long N:
Alternative RNA splicing: Contribution to pain and potential
therapeutic strategy. Drug Discov Today. 21:1787–1798.
2016.PubMed/NCBI View Article : Google Scholar
|
|
54
|
de la Peña JB and Campbell ZT: RNA-binding
proteins as targets for pain therapeutics. Neurobiol Pain. 4:2–7.
2018.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Song KY, Choi HS, Law PY, Wei LN and Loh
HH: Post-transcriptional regulation of mu-opioid receptor: Role of
the RNA-binding proteins heterogeneous nuclear ribonucleoprotein H1
and F. Cell Mol Life Sci. 69:599–610. 2012.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Alkan SA, Martincic K and Milcarek C: The
hnRNPs F and H2 bind to similar sequences to influence gene
expression. Biochem J. 393:361–371. 2006.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Dvinge H: Regulation of alternative mRNA
splicing: Old players and new perspectives. FEBS Lett.
592:2987–3006. 2018.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Li P, Zhang L, Zhao N, Xiong Q, Zhou YA,
Wu C and Xiao H: A Novel α-galactosidase a splicing mutation
predisposes to fabry disease. Front Genet. 10(60)2019.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Chang WH, Niu DM, Lu CY, Lin SY, Liu TC
and Chang JG: Modulation the alternative splicing of GLA
(IVS4+919G>A) in Fabry disease. PLoS One.
12(e0175929)2017.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Ishii S, Nakao S, Minamikawa-Tachino R,
Desnick RJ and Fan JQ: Alternative splicing in the
alpha-galactosidase A gene: Increased exon inclusion results in the
Fabry cardiac phenotype. Am J Hum Genet. 70:994–1002.
2002.PubMed/NCBI View
Article : Google Scholar
|
|
61
|
Lev Maor G, Yearim A and Ast G: The
alternative role of DNA methylation in splicing regulation. Trends
Genet. 31:274–280. 2015.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Wang RY, Lelis A, Mirocha J and Wilcox WR:
Heterozygous Fabry women are not just carriers, but have a
significant burden of disease and impaired quality of life. Genet
Med. 9:34–45. 2007.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Weingarten-Gabbay S, Nir R, Lubliner S,
Sharon E, Kalma Y, Weinberger A and Segal E: Systematic
interrogation of human promoters. Genome Res. 29:171–183.
2019.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Jangid RK, Kelkar A, Muley VY and Galande
S: Bidirectional promoters exhibit characteristic chromatin
modification signature associated with transcription elongation in
both sense and antisense directions. BMC Genomics.
19(313)2018.PubMed/NCBI View Article : Google Scholar
|