Introduction
Cerebral infarction (commonly known as stroke) is a
leading cause of disability (1). In
recent years, the development of acute medical treatments for
stroke has led to a reduction in stroke mortality, but serious
disability remains high in many stroke survivors (2). Neurological deficits after a stroke
seriously affect quality of life (3).
Neuronal cell death occurs immediately after stroke.
Stroke results in massive cell death in the infarct core and severe
damage to neurons in the periinfarct area (4). Although periinfarct and penumbral
tissues are present in a structurally intact state, they possess
dysfunctional metabolism and ion homeostasis (5). Excitatory neurotransmitters released
after stroke overexcite tissues around the brain lesion, making the
surrounding neurons vulnerable to external stimuli (6). Glutamate is the major excitatory
neurotransmitter of the brain and binds to N-methyl-D-aspartate
(NMDA) receptors. It is also involved in secondary processes that
lead to neuronal death after traumatic insult (7,8).
Neuroprotective agents for treatment of acute stroke have received
much attention and are expected to effectively protect vulnerable
neurons and save ischemic penumbra (9). Several neuroprotective strategies
target acute processes, including excitotoxicity, glutamate
release, activation of NMDA receptors, calcium ion influx, and
apoptosis leading to early cell death in the infarct core and
periinfarct area (4).
Gamma aminobutyric acid (GABA) is a major inhibitory
neurotransmitter in the central nervous system (CNS) that inhibits
depolarization- and ischemia-induced glutamate release (9). GABA receptor agonists are conventional
sedatives with neuroprotective effects in decreasing infarct size
and improving functional outcomes in animal models of
cerebraovascular disease. However, their sedative effects may be
harmful, so their wider application in patients with acute stroke
have been limited (9).
Pregabalin is structurally derived from GABA, and
its mechanism of action is associated with binding to the
alpha2-delta (α2-δ) subunit of voltage-gated calcium
channels (VGCC). Pregabalin reduces abnormal neuronal excitability
by reducing the release of neurotransmitters including glutamate
from synapses in neural tissue (10). As a medication, it is used to treat
epilepsy and causes of neurogenic pain, such as fibromyalgia,
diabetic neuropathy, and complex regional pain syndrome (10,11).
The neuroprotective effects of pregabalin on stroke
outcome have been reported in a mouse model (11). Intraperitoneal administration of
pregabalin within 30-90 min of stroke reduces cerebral infarction
and improves neurological function after 6 h (11). However, the administration of drugs
within 90 min post-stroke is difficult to effectively translate
clinically. In addition, their study focused on data collection too
early to assess stroke function, making it difficult to determine
drug effectiveness on stroke recovery or possibly delayed
neurological damage. Therefore, we designed a study to evaluate the
effect of pregabalin administered 1 day after cerebral ischemia on
cerebral outcome in rats.
The aim of this study was to investigate the effect
of pregabalin on cerebral outcome after cerebral ischemia through a
middle cerebral artery occlusion (MCAO) rat model by histologic
examination, behavioral tests, and magnetic resonance imaging
(MRI).
Materials and methods
Animals
All animal procedures were performed with approval
by the Institutional Animal Care and Use Committee in Asan Medical
Center (2014-12-212). Male Sprague-Dawley rats weighing 250-280 g
and 8 weeks of age were allowed food and water ad libitum
and habituated under conditions of a 12 h light/dark cycle (lights
on at 07:00 a.m.) at a temperature of 22˚C with appropriate
humidity. Rats were randomized into one of four groups: Oral
administration of 5 mg/kg pregabalin (Pfizer Pharmaceuticals, New
York, NY, USA) for 1 day (PD1, n=10), or 5 days (PD5, n=10) after
MCAO surgery or an equal amount of normal saline administered 1 day
(SD1, n=7) or 5 days (SD5, n=7) after MCAO surgery.
Occlusion of the middle cerebral
artery in rat
To generate a rat model of ischemic stroke, we
performed transient occlusion of the right middle cerebral artery
(MCA) using the intraluminal filament technique as previously
described (12). Inhalation
anesthesia was induced in rats with 5% isoflurane (JW Pharm, Seoul,
South Korea) and maintained with 3% isoflurane. Body temperature
was continuously monitored and maintained at 37˚C ± 0.5˚C using a
heating pad during surgery. An incision was made along the ventral
midline of the neck, and the right common carotid artery (CCA),
external carotid artery, and internal carotid artery (ICA) were
exposed. A silicone-coated monofilament nylon suture was inserted
into the right CCA through a small incision made in the proximity
of the carotid bifurcation, passed through the ICA, and advanced to
the right MCA. After 30 min of transient MCAO, reperfusion was
achieved by filament withdrawal.
Behavioral tests
Behavioral tests were performed at 1 and 7 days
after MCAO/reperfusion surgery. Tests were conducted by a rater who
was blind to the treatment. A wire hang test is used to evaluate
motor function in rodent models of CNS disorders (13). The test began with the animal
hanging from an elevated wire mesh grid (15x25 cm). The wire mesh
grid was then inverted, so that the rat was suspended from the wire
mesh. The latency to fall was recorded. Five repetitions were
performed with a 3-min interval between repeats and averaged. We
also performed the Garcia test, which is a composite neurological
test to evaluate various sensorimotor deficits after MCAO in rats
(14,15), and includes the following six tests:
Spontaneous activity, symmetry in four limb movement, forepaw
outstretching, climbing, body proprioception, and response to
vibrissae touch. Each test item was scored between 0-3 points and
summed to a maximum score of 18 points, with a higher score
indicating better function. For the balance beam test (16,17),
two square bars 1.5 cm in width and 40 cm in height were used as
bilateral pillars, and a cylindrical rod 2 cm in diameter and 60 cm
in length was placed horizontally 40 cm above the floor, which was
covered by a foam pad. Rats were placed in the center of the
cylindrical rod, and the time to fall from the rod was measured up
to a maximum of 30 sec. Testing was repeated four times and
averaged for each rat.
Brain MRI
Brain MRI was performed at days 1 and 7 after
MCAO/reperfusion. For MRI, inhalation anesthesia was performed for
all rats with 2.0-2.5% isoflurane in a 1:2 mixture of
O2:N2O using a mask. A 9.4T/160 mm animal MRI
system (Agilent Technologies, Santa Clara, CA, USA) was used. A 72
mm birdcage volume coil was used for excitation, and a four-channel
phased array surface coil was used as a receiving coil. Rat body
temperature was monitored and maintained at 37˚C ± 0.5˚C using an
air heater system.
Infarction of the right hemisphere was confirmed by
T2-weighted and diffusion-weighted MRI. T2-weighted imaging was
obtained with spin-echo sequence and the following parameters:
Field of view (FOV)=30x30 mm; slice thickness=1.0 mm; no gap; time
to repeat (TR)=4,000 ms; effective time to echo (TE)=32.95 ms;
k-zero=3; echo spacing=10.98 ms; 32 segments; echo train length=8;
average=1; and matrix size=256x256. Diffusion-weighted imaging was
obtained by a four-shot spin-echo-based echo planar imaging
sequence with the following parameters: TR=3,750 ms; TE=46.22 ms;
96x96 matrix; and an encoding scheme of 30 gradient directions with
a b-value of 1,000 s/mm2. T2-weighted and
diffusion-weighted images confirmed right hemisphere infarction. By
measuring the volume of infarct-induced right cerebral hemisphere
(infarct, or inf) and normal-side cerebral hemisphere
(contralateral, or CL) on T2-weighted images, the degree of stroke
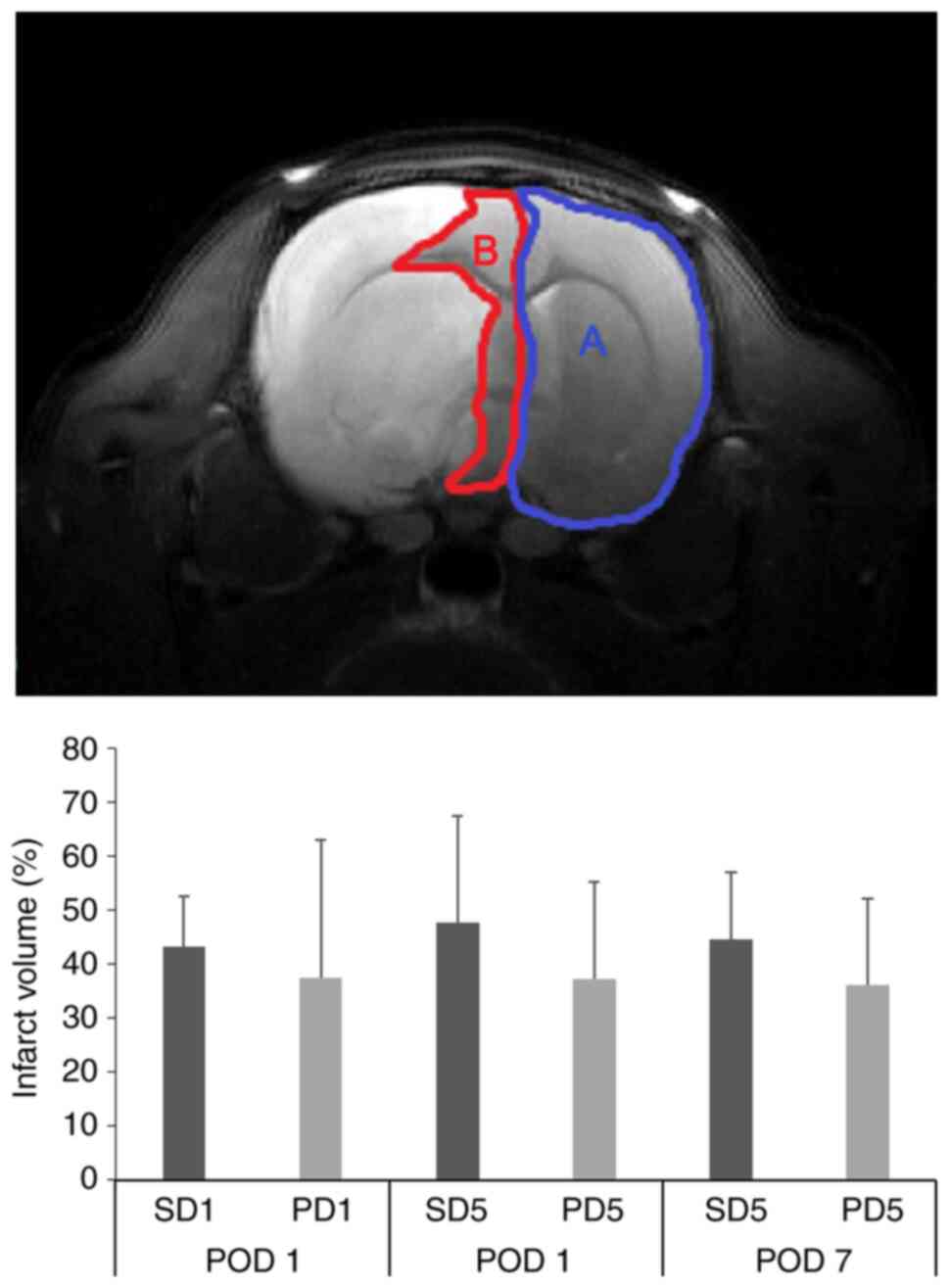
was calculated by the following formula (Fig. 1) (18): Infarct volume (%)=volume (inf-volume
(CL)/volume (CL) x100.
Immunohistochemistry
Rats were sacrificed 7 days after MCAO/reperfusion.
Rats were anesthetized with 5% isoflurane and transcardially
perfused with 100 ml phosphate buffered saline (PBS) and 100 ml of
4% paraformaldehyde in 0.1 M PBS. Brains were removed and fixed in
4% paraformaldehyde. Fixed specimens were embedded in paraffin and
were cut into 4 µm-thick sections. Sections were then
deparaffinized in xylene, hydrated in a series of graded alcohol,
washed with distilled water, and subjected to microwave in citrate
buffer (pH 6.0) for 10 min. Endogenous peroxidase activity was
inhibited by 3% hydrogen peroxide for 15 min. Primary antibody
against BDNF (1:100, Abcam, Cambridge, MA, USA) was incubated
overnight at 4˚C and detected with a Dako EnVision System HRP/DAB
kit (Dako, Glostrup, Denmark). Secondary antibodies including a
1:200 dilution of goat anti-rabbit immunoglobulin (Thermo Fisher
Scientific, Waltham, MA, USA) were incubated for 1 h at room
temperature. A 40x objective (DFC290; Leica, Heerbrugg, Germany)
with Leica Application Suite (version 3.3.0; Leica) were used to
digitize immunostained sections.
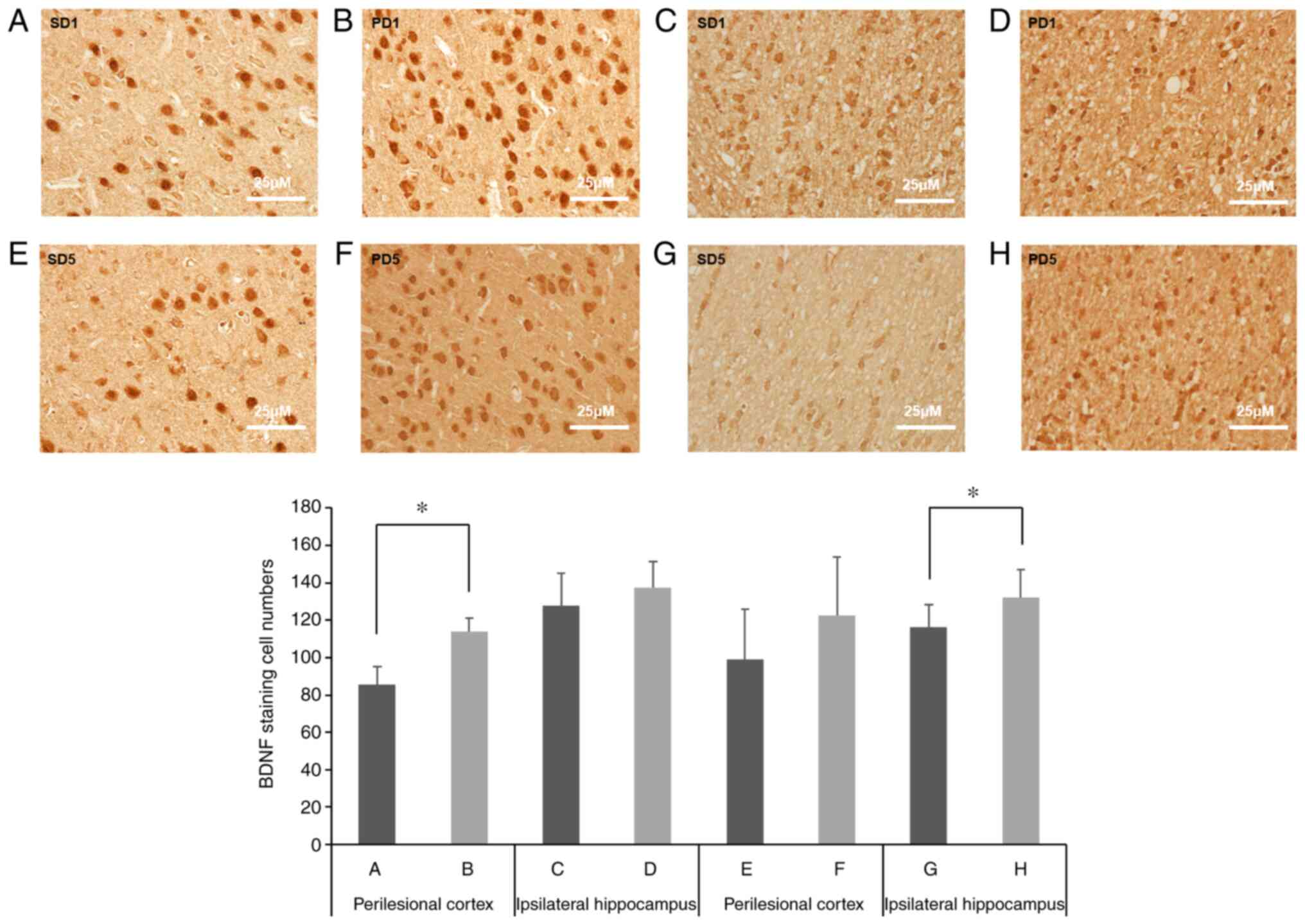
To determine the number of BDNF-positive stained
cells in the brain, five views of the perilesional cortex and the
ipsilateral hippocampus were captured in grayscale. Optical
densities were measured in five rectangles of 10,000 µm2
using MCID analysis (evaluation version 7.0; Imaging Research Inc.,
ON, Canada), and the values were averaged.
Statistical analysis
Statistical analysis was performed using IBM SPSS
Statistics for Windows version 21.0 (IBM Corp., Armonk, NY, USA).
The Mann-Whitney U-test was used to compare BDNF expression of the
perilesional cortex and ipsilesional hippocampus, infarct volume,
and behavioral data between groups. Data are expressed as the mean
± standard deviation. The cutoff for statistical significance was
set at P<0.05.
Results
Behavioral tests and brain infarct
volume
Behavioral test scores of rats from the PD1 group
showed better results than those of rats from the SD1 group;
however, the differences were not statistically significant.
Further, no significant differences were found between the PD5 and
SD5 groups (Table I and Table II). No differences in brain infarct
volume were found between rats receiving pregabalin and their
respective saline controls for 1 or 5 days (PD1 vs. SD1 and PD5 vs.
SD5; Fig. 1, Table I, and Table II).
 | Table IInfarct volume, behavioral test
scores, and histologic data after 1 day administration of
pregabalin or normal saline. |
Table I
Infarct volume, behavioral test
scores, and histologic data after 1 day administration of
pregabalin or normal saline.
| Test | Normal saline 1 day
(n=7) | Pregabalin 1 day
(n=10) | P-value |
|---|
| Wire hang test (POD
1, sec) | 0.18±0.37 | 0.90±0.94 | 0.137 |
| Garcia test (POD
1) | 3.86±1.57 | 5.30±2.95 | 0.244 |
| Beam balance test
(POD 1, sec) | 0.75±1.14 | 2.55±3.32 | 0.153 |
| Infarct volume (POD
1, %) | 43.2±9.4 | 37.5±25.6 | 0.810 |
| Number of
BDNF-positive cells in perilesional cortex | 85.77±9.58 | 114.00±7.03 | 0.001a |
| Number of
BDNF-positive cells in ipsilateral hippocampus | 127.63±17.28 | 137.64±13.66 | 0.282 |
| Table IIInfarct volume, behavioral test
scores, and histologic data after 5 days administration of
pregabalin or normal saline. |
Table II
Infarct volume, behavioral test
scores, and histologic data after 5 days administration of
pregabalin or normal saline.
| Test | Normal saline 5 days
(n=7) | Pregabalin 5 days
(n=10) | P-value |
|---|
| Wire hang test,
sec | | | |
|
POD 1 | 0.50±0.85 | 0.65±0.56 | 0.249 |
|
POD 7 | 1.21±0.88 | 1.20±1.70 | 0.922 |
| Garcia test,
score | | | |
|
POD 1 | 4.57±3.15 | 5.90±3.18 | 0.457 |
|
POD 7 | 7.29±4.15 | 8.40±4.79 | 0.921 |
| Beam balance test,
sec | | | |
|
POD 1 | 1.61±2.82 | 0.80±0.98 | 0.597 |
|
POD 7 | 1.25±1.85 | 1.23±1.53 | 0.961 |
| Infarct volume,
% | | | |
|
POD 1 | 47.8±19.8 | 37.2±18.1 | 1.000 |
|
POD 7 | 44.7±12.3 | 36.1±16.1 | 0.610 |
| Number of
BDNF-positive cells in perilesional cortex | 99.00±26.83 | 122.34±31.24 | 0.097 |
| Number of
BDNF-positive cells in ipsilateral hippocampus | 116.23±11.91 | 132.36±14.56 | 0.040a |
Immunohistochemistry
Using immunohistochemistry (Fig. 2), we found more BDNF-positive cells
in the perilesional cortex of rats from the PD1 group compared with
that of the SD1 group (Table I,
P=0.001). In the ipsilateral hippocampus, we found that the number
of BDNF-positive cells in the PD1 group was higher than that of the
SD1 group, but the difference was not statistically significant
(P=0.282). We also found more BDNF-positive cells in the
ipsilateral hippocampus of rats from the PD5 group compared to the
SD5 group (Fig. 2 and Table II, P=0.04), whereas in the
perilesional cortex, although there were more BDNF-positive cells
in the PD5 group compared with the SD5 group, the difference was
not significant (P=0.097).
Discussion
In the current study, we evaluated the
neuroprotective effect of pregabalin administration 1 and 5 days
after MCAO in a rat model of cerebral infarction. Our results show
that pregabalin induces a neuroprotective effect by increasing BDNF
expression in our MCAO rat model.
Excessive glutamate release from presynaptic nerve
endings occurs during and after cerebral ischemia, resulting in
persistent calcium influx in cells through postsynaptic NMDA
receptors and VGCC. High calcium levels induce neuronal death in
the penumbra of ischemic brain (11). Pregabalin specifically binds to the
α2-δ subunit of VGCC, and its strong binding affinity to this site
reduces calcium influx in presynaptic nerve endings, which
subsequently leads to the reduced release of excitatory
neurotransmitters, such as glutamate and noradrenalin (11,19).
It also protects neurons from calcium ion overload during ischemic
stroke.
Several previous studies investigated the
neuroprotective effects of pregabalin (11,19-22).
In a rat model of ischemic stroke, pregabalin administration 30-90
min after transient MCAO and reperfusion reduces infarct volume,
neurological deficits, and neuronal damage by 24 h after stroke
(11). The neuroprotective action
of pregabalin also contributes to a reduction of
calcium/calpain-mediated proteolysis (11). Further, the beneficial effect of
pregabalin on functional and histologic cerebral outcome was
demonstrated in rats after cardiopulmonary bypass with deep
hypothermic circulatory arrest (19). In addition, pregabalin protects
against oxidative stress damage after cerebral ischemia and
reperfusion by reducing lipid peroxidation and activity of
antioxidant enzymes (21). When
administered upon reperfusion, pregabalin reduces neuronal death
and improves neurological function in a rat model of hyperglycemic
stroke (22). Thus, our data
support growing evidence of the neuroprotective effect and
potential application of pregabalin after cerebral ischemia.
BDNF is a potent modulator beneficial to neuronal
function (8). BDNF plays a crucial
role in synaptic plasticity in the CNS, stimulating neurons to
survive and promoting the growth and differentiation of new neurons
(23). The neuroprotective effect
of BDNF in stroke models is achieved by anti-apoptotic,
anti-inflammatory, and anti-neurotoxic effects, and promotion of
neural regeneration (8). In the
present study, BDNF was upregulated in the perilesional cortex
after administration of pregabalin for 1 day and in the ipsilateral
hippocampus after pregabalin administration for 5 days compared
with administration of normal saline, suggesting that pregabalin
has a neuroprotective effect in rat models of cerebral ischemia.
Furthermore, pregabalin may present new possibilities in the
promotion of neurotrophic factors in ischemic brain. In addition,
our finding that BDNF upregulation was significantly observed in
perilesional cortex after administration of pregabalin for 1 day
and in the ipsilateral hippocampus after pregabalin administration
for 5 days may reflect differences in pregabalin response in motor-
and cognition-related brain regions and duration of pregabalin
administration. However, further research is needed to investigate
this interpretation.
We found no significant difference in cerebral
infarct volume between pregabalin and control groups. For
behavioral tests, although not statistically significant, the
overall score was higher in the pregabalin-administered group than
in the control group, with a few exceptions. This results are
inconsistent with previous studies that found infarct volume and
neurological deficits were reduced 24 h post-stroke when 5-10 mg/kg
pregabalin was given intraperitoneally 30-90 min after MCAO and
reperfusion in rats (11).
Similarly, a different study found that intraperitoneal injection
of 30 mg/kg pregabalin 1 h after MCAO and reperfusion reduced
infarct size and improved neurological function as determined by
neurobehavioral assessment 24 h post-stroke in rats with
hyperglycemic stroke (22). It is
speculated that the dose of pregabalin, route of administration, or
time point of outcome measurement may have an effect on these
differing results in cerebral infarct volume and neurological
function between studies; however, further research is needed to
determine whether this is the case.
There are several limitations to this study. First,
we did not directly reveal the effect of pregabalin on each
specific pathway of neuronal cell death. Although we examined the
effects of pregabalin on BDNF expression in the brain of ischemic
rats from pathological and functional perspectives, we did not
assess the possible underlying molecular mechanism such as the
signaling pathway by which pregabalin increases BDNF expression in
the brain after ischemia. Second, immunohistochemical staining was
not further analyzed semi-quantitatively. Further research to
determine BDNF protein levels in the brain by western blotting is
needed to identify the mechanistic pathways directly involved in
cerebral outcomes as a result of the neuroprotective effects of
pregabalin. Third, we did not directly compare the effects of
pregabalin administration duration between 1 and 5 days; we only
compared the pregabalin-administered group to their respective
normal saline control group. However, previous studies have
evaluated the effects of single doses of pregabalin; therefore, our
study is still informative because we varied the duration of
pregabalin administration. Nevertheless, further research is needed
to investigate whether the duration of pregabalin administration
affects other responses. Finally, the hippocampus is involved in
cognitive function, but the behavioral tests performed in this
study did not include any relevant cognitive tests.
In conclusion, pregabalin treatment confers a
beneficial effect on histologic cerebral outcome in a rat model of
cerebral ischemia, and that this effect may be mediated by
increased BDNF expression. Our results support the neuroprotective
role of pregabalin and its potential use for ischemic
cerebrovascular disease. However, because pregabalin did not affect
infarct volume or performance on behavioral tests in our study,
additional research is warranted.
Acknowledgements
Not applicable.
Funding
This study was supported by a grant (2018-454) from
the Asan Institute for Life Sciences, Asan Medical Center, Seoul,
Korea.
Availability of data and materials
The datasets used and/or analyzed in the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JL analyzed the data and wrote the manuscript. CGK
designed the study and analyzed the data. CRP and IKH performed the
experiments. DYK designed the study and revised the manuscript. JK
and DKY confirm the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Institutional Animal
Care and Use Committee in Asan Medical Center in South Korea
(2014-12-212) and conducted in strict accordance with the
recommendations of the United States National Institutes of Health
Guide for the Care and Use of Laboratory Animals (Publication No.
85-23, revised 1996).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Duncan PW: Stroke disability. Phys Ther.
74:399–407. 1994.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Brewer L, Horgan F, Hickey A and Williams
D: Stroke rehabilitation: Recent advances and future therapies.
QJM. 106:11–25. 2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Sturm JW, Donnan GA, Dewey HM, Macdonell
RA, Gilligan AK, Srikanth V and Thrift AG: Quality of life after
stroke: The north East melbourne stroke incidence study (NEMESIS).
Stroke. 35:2340–2345. 2004.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kumar A and Kitago T: Pharmacological
enhancement of stroke recovery. Curr Neurol Neurosci Rep.
19(43)2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sist B, Fouad K and Winship IR: Plasticity
beyond peri-infarct cortex: Spinal up regulation of structural
plasticity, neurotrophins, and inflammatory cytokines during
recovery from cortical stroke. Exp Neurol. 252:47–56.
2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Risedal A, Zeng JS and Johansson BB: Early
training may exacerbate brain damage after focal brain ischemia in
the rat. J Cereb Blood Flow Metab. 19:997–1003. 1999.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Humm JL, Kozlowski DA, Bland ST, James DC
and Schallert T: Use-dependent exaggeration of brain injury: Is
glutamate involved? Exp Neurol. 157:349–358. 1999.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Chen A, Xiong LJ, Tong Y and Mao M: The
neuroprotective roles of BDNF in hypoxic ischemic brain injury.
Biomed Rep. 1:167–176. 2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Liu J, Zhang J and Wang LN: Gamma
aminobutyric acid (GABA) receptor agonists for acute stroke.
Cochrane Database Syst Rev. 10(CD009622)2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Taylor CP, Angelotti T and Fauman E:
Pharmacology and mechanism of action of pregabalin: The calcium
channel alpha2-delta (alpha2-delta) subunit
as a target for antiepileptic drug discovery. Epilepsy Res.
73:137–150. 2007.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Yoon JS, Lee JH, Son TG, Mughal MR, Greig
NH and Mattson MP: Pregabalin suppresses calcium-mediated
proteolysis and improves stroke outcome. Neurobiol Dis. 41:624–629.
2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sasaki M, Honmou O and Kocsis JD: A rat
middle cerebral artery occlusion model and intravenous cellular
delivery. Methods Mol Biol. 549:187–195. 2009.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Olivan S, Calvo AC, Rando A, Munoz MJ,
Zaragoza P and Osta R: Comparative study of behavioural tests in
the SOD1G93A mouse model of amyotrophic lateral sclerosis. Exp
Anim. 64:147–153. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Garcia JH, Wagner S, Liu KF and Hu XJ:
Neurological deficit and extent of neuronal necrosis attributable
to middle cerebral artery occlusion in rats. Statistical
validation. Stroke. 26:627–634; discussion 635. 1995.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hartman R, Lekic T, Rojas H, Tang J and
Zhang JH: Assessing functional outcomes following intracerebral
hemorrhage in rats. Brain Res. 1280:148–157. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Combs DJ and D'Alecy LG: Motor performance
in rats exposed to severe forebrain ischemia: Effect of fasting and
1,3-butanediol. Stroke. 18:503–511. 1987.PubMed/NCBI View Article : Google Scholar
|
|
17
|
DeGraba TJ, Ostrow P, Hanson S and Grotta
JC: Motor performance, histologic damage, and calcium influx in
rats treated with NBQX after focal ischemia. J Cereb Blood Flow
Metab. 14:262–268. 1994.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Frey LC, Hellier J, Unkart C, Lepkin A,
Howard A, Hasebroock K, Serkova N, Liang L, Patel M, Soltesz I and
Staley K: A novel apparatus for lateral fluid percussion injury in
the rat. J Neurosci Methods. 177:267–272. 2009.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Shim JK, Ma Q, Zhang Z, Podgoreanu MV and
Mackensen GB: Effect of pregabalin on cerebral outcome after
cardiopulmonary bypass with deep hypothermic circulatory arrest in
rats. J Thorac Cardiovasc Surg. 148:298–303. 2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ha KY, Kim YH, Rhyu KW and Kwon SE:
Pregabalin as a neuroprotector after spinal cord injury in rats.
Eur Spine J. 17:864–872. 2008.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Asci S, Demirci S, Asci H, Doguc DK and
Onaran I: Neuroprotective effects of pregabalin on cerebral
ischemia and reperfusion. Balkan Med J. 33:221–227. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Song Y, Jun JH, Shin EJ, Kwak YL, Shin JS
and Shim JK: Effect of pregabalin administration upon reperfusion
in a rat model of hyperglycemic stroke: Mechanistic insights
associated with high-mobility group box 1. PLoS One.
12(E0171147)2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Hu Y, Guo TC, Zhang XY, Tian J and Lu YS:
Paired associative stimulation improves synaptic plasticity and
functional outcomes after cerebral ischemia. Neural Regen Res.
14:1968–1976. 2019.PubMed/NCBI View Article : Google Scholar
|