Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune
disease characterized by pronounced synovial hyperplasia and joint
synovitis, a consequence of bone and cartilage destruction induced
by the marked overproduction of inflammatory cytokines (1-3).
Fibroblast-like synoviocytes (FLSs) are a critical component in the
development of RA (4,5) and are reportedly associated with
inflammation and joint destruction in RA (6,7).
Therefore, investigating the key properties of RA FLS is being
increasingly applied in the search for novel therapies to block the
development of RA.
MicroRNA (miRNA), a category of small, non-coding
RNA, was initially identified in 1993(8). Presently, more than thirty thousand
mature miRNA sequences have been discovered in 206 species
(9). There is increasing evidence
to show that miRNAs have a variety of roles in many physiological
and pathological processes, including in the development of RA
(10). For example, Li et al
(11) found a downregulation of
miRNA (miR)-192 in RA, which is caused by caveolin 1
(CAV1)-mediated suppression in RA FLS. Kurowska-Stolarska et
al (12) reported that miR-34a
may sustain homeostatic control of CD1c+ dendritic cells
(DC) activation by regulating the expression of Axl in RA patients.
Gene-silencing of miR-34a decreases the content of pro-inflammation
factors (12). These investigations
provide evidence in support of miRNAs as potential targets for the
treatment of RA.
The Janus kinase (JAK)/signal transducer and
activator of transcription (STAT) pathway is critical in the
occurrence and development of RA (13-15).
Specifically, the involvement of STAT3 has been reported in the
progression of human malignancies (16,17),
angiogenesis (18) and metastasis
(19). Interactions between
associated pro-inflammatory cytokines, including IL-1β, IL-6,
TNF-alpha and their receptors results in the phosphorylation of Tyr
on the corresponding JAKs and the phosphorylation and dimerization
of STATs. In its dimeric form, STAT is delivered to the nuclei for
transcription. The JAK-STAT pathway is reportedly blocked by
cytokine-signaling protein inhibitors (20,21)
such as CP-690550, a JAK/STAT pathway-specific inhibitor, which has
demonstrated significant American College of Rheumatology 50/70
responses in phase II/III clinical trials (22,23).
In the present study, the focus was on the interplay
between RA and miR-140, which is widely reported to be variable
depending on the specific different biological processes in various
cell lines (24-33).
For instance, miR-140 may influence proliferation, apoptosis and
autophagy by targeting the fucosyltransferase 1 (FUT1) gene in
chondrocytes during the development of osteoarthritis (25). miR-140 also has a regulatory role on
proliferation, apoptosis and inflammation in non-small cell lung
cancer cells (24-29),
osteosarcoma cells (30-32)
and synovial fibroblasts (33).
Therefore, the present study conducted a novel investigation into
the difference in expression levels of miR-140 in RA patients and
their healthy counterparts. Considering the influence of miR-140 in
regulating various biological process (34-36),
the aim of the present study was to understand how miR-140 affects
the biological events in RA FLS and to elucidate the mechanism of
action behind how miR-140 regulates STAT3 expression in RA FLS.
Materials and methods
Sample origins
Synovial tissue samples were collected from 33 RA
patients after knee replacement surgeries from the Affiliated
Hospital of Zunyi Medical University, of which 17 were male and 16
were female, with an average age of 55.1±13.8 years and an age
range of 40.1-61.0 years. RA diagnoses were made based on previous
reference standards (37). Normal
synovial biopsies from 8 patients with traumatic knee injuries were
used as healthy controls, of which 5 were male and 3 were female,
with an average age of 39 (±10) years. All study participants had
given their written informed consent before participating in the
study and the study was approved by the Ethics Committee of Zunyi
Medical University.
Cell culture
Healthy and RA human FLSs were obtained from 3
healthy volunteer and RA patients, which were treated with 2.5 g/l
trypsin at 37˚C for 2 h. Culture of RA and healthy FLSs was carried
out in DMEM (Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% (v/v) heat-inactivated fetal bovine serum (FBS; Gibco;
Thermo Fisher Scientific, Inc.), 100 U/ml penicillin (Beyotime
Institute of Biotechnology) and 100 mg/ml streptomycin (Beyotime
Institute of Biotechnology), followed by 3-6 passages. Cells were
cultured in an incubator at 37˚C, 5% CO2 with saturated
humidity. Following 48 h of incubation, the medium was changed, and
cells were passaged when reaching 80-90% confluence.
Plasmid construction and
transfection
miR-140-5p and miR-NC was obtained from Origene
Technologies, Inc. The mature-miR-140-5p and miR-NC sequence was
cloned into pCDHCMV-MCS-EF1-coGFP constructs (Invitrogen; Thermo
Fisher Scientific, Inc.). For STAT3 overexpression, the coding
sequences of STAT3 were amplified from cDNA isolated from RA FLS
using a PCR kit (AP111-11; TransGen Biotech Co., Ltd.). The
thermocycling conditions were as follows: 95˚C for 10 min, followed
by 40 cycles of 95˚C for 15 sec, 60˚C for 30 sec and 72˚C for 30
sec. Sequences for primers were as follows: STAT3 forward,
5'-GACTTAGTCCCAGGTACT-3' and reverse, TTCAACTGACCTAGGACGTGGTCG-3'.
The product was inserted into the pcDNA3.1 vector (Invitrogen;
Thermo Fisher Scientific, Inc.) to generate STAT3 expression
vectors pcDNA3.1-SOX11. Cells were transfected with
pCDHCMV-MCS-EF1-coGFP-miR-140-5p and/or pcDNA3.1-SOX11 using
Lipofectamine® RNAiMAX/2000 transfection reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) and then collected at
48 h after transfection. All constructions were confirmed using
plasmid DNA sequencing using the dideoxy chain-termination (Sanger)
method with Applied Biosystems 3730XL (Genescript).
Cell Counting Kit (CCK)-8 assay
Viability of FLSs was determined using CCK-8 assays
(Sigma-Aldrich; Merck KGaA) in 96-well plates. The kit was used
according to the manufacturer's protocol. Cells were seeded in the
96-well plates at a density of 5x103 cells/well and
cultured until they reached 80% confluence and the aforementioned
plasmids were transfected into the FLS cells. At 72, 48, 24 and 0 h
post-transfection, CCK-8 reagents were added to each well and
incubated for 1 h.
An independent standard curve was drawn to calculate
the cell numbers. Briefly, 1x107 cells were counted
using a Countess II (Thermal Fisher Scientific, Inc.) and then
these cells were diluted 2-fold and the absorbance at 450 nm was
detected. Cell numbers in the other wells was calculated from the
standard curve.
BrdU incorporation assay
After a total of 48 h after transfection of the
FLSs, the cells were incubated for 1 h in BrdU stock solution,
diluted 1/1,000 in PBS, followed by a wash in PBS. Cells underwent
a subsequent fixation in 4% paraformaldehyde for 20 min at room
temperature, followed by permeabilization using Triton X-100 for 10
min at room temperature. After subsequent blocking with 10% FBS for
1 h at 37˚C, cells underwent overnight incubation with primary
rabbit antibodies against BrdU (1:200; cat. no. ab152095; Abcam) at
4˚C, followed by a PBS wash and incubation using the corresponding
secondary antibodies bound to Cy3 (1:500, cat. no. ab6939; Abcam)
for 2 h. An independent standard curve was generated to measure FLS
viability according to the method discussed previously.
MTT assay
After transfection for 24 h, cells were seeded at a
density of 5x103 cells/well in the 96-well plates and
incubated for 48 h at 37˚C. Three replicates were used for each
experimental group. MTT substrates (Promega Corporation) were then
added and incubated for 4 h at 37˚C. The proliferative ability of
cells was reflected in the absorbance at 570 nm.
Western blotting
Cells were suspended in RIPA buffer supplemented
with protease suppressor (Roche Diagnostics) for lysis and a BCA
Protein Quantitation kit was used for protein quantification. 10%
SDS-PAGE was used to isolate protein specimens (100 µg), which were
subsequently transferred onto 0.45 µm PVDF membranes. After
blocking in 5% BSA for 1 h, proteins on the membrane were incubated
with their respective primary antibodies at 4˚C overnight. The
primary antibodies used were as follows: Beta-Actin (1:5,000; cat.
no. ab8227; Abcam), IL-1β (1:1,000; cat. no. ab2105; Abcam), IL-6
(1:1,000; cat. no. ab9324; Abcam), IL-8 (1:1,000; cat. no. ab18672;
Abcam), TNF-alpha (1:1,000; cat. no. ab9635; Abcam), STAT3
(1:2,000; cat. no. ab5073; Abcam), Bcl-2 (1:2,000; cat. no.
ab59348; Abcam), Bax (1:5,000; cat. no. ab53154; Abcam), and
cleaved caspase-3 (1:2,000; cat. no. 9664; CST Biological Reagents
Co., Ltd.). Following three washes in TBS-T, proteins were probed
with secondary antibodies for 1 h at room temperature. Analyses of
protein expression levels were performed with β-Actin an internal
reference using the SuperSignal West Femto Maximum Sensitivity
Substrate kit (Thermo Fisher Scientific, Inc.) and a C-DiGit Blot
Scanner and Analyser (LI-COR Biosciences).
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Trizol® (Thermo Fisher Scientific, Inc.)
was used to isolate total RNA from FLSs. First-strand cDNA was
synthesized using total RNA with a PrimeScript RT Master Mix
(Takara Bio, Inc.). The samples were initially incubated at 25˚C
for 10 min, then subsequently incubated at 42˚C for 1 h. SYBR-Green
was incorporated to evaluate transcription and GAPDH used as
internal reference for the various genes evaluated.
Quantitative PCR was conducted in 20 µl volumes,
with the following temperature protocol: 95˚C for 10 min, followed
by 40 cycles of 95˚C for 15 sec, 60˚C for 30 sec, and 72˚C for 30
sec. Quantification was performed using the 2-ΔΔCq
method (38), normalizing to GAPDH.
The primer sequences used for detected genes were: miR-140 forward,
5'-TGCGGCAGTGGTTTTACCCTATG-3' and reverse,
5'-CCAGTGCAGGGTCCGAGGT-3'; IL-1β forward,
5'-TCAGGCAGATGGTGTCTGTC-3' and reverse, 5'-GGTCTATATCCTCCAGCTGC-3';
IL-6 forward, 5'-AACGCCTGGAAGAAGATGCC-3' and reverse,
5'-CTCAGGCTGAACTGCAGGAA-3'; IL-8 forward,
5'-GAAGATAGATTGCACCGATG-3' and reverse, 5'-CATAGCCTCTCACACATTTC-3';
TNF-alpha forward, 5'-GCTGTACCTCATCTACTCCC-3' and reverse,
5'-TAGACCTGCCCAGATTCAGC-3'; STAT3 forward, 5'-GGAACAAGCCCCAACCGG-3'
and reverse, 5'-CTAAAATCAGGGGTCCCAACTG-3'; Bcl-2 forward,
5'-CATTTCCACGTCAACAGAATTG-3' and reverse,
5'-AGCACAGGATTGGATATTCCAT-3'; Bax forward,
5'-AGCTGAGCGAGTGTCTCAAG-3' and reverse 5'-GTCCAATGTCCAGCCCATGA-3';
U6 forward, 5'CCTTATGCAGTTGCTCTCC-3' and reverse,
5'-CAGGAAACAGCTATGAC-3'); and GAPDH forward,
5'-GCTCAGACACCATGGGGAAGGT-3' and reverse
5'-GTGGTGCAGGAGGCATTGCTGA-3'.
Hoechst 33342 staining
FLSs at a density of 1x105 cells/ml were
inoculated in 12-well plates prior to transfection. Following 48 h
post-transfection, washing in PBS was carried out in triplicates,
followed by fixing with 4% paraformaldehyde for 15 min at room
temperature and incubation with 500 µl Hoechst 33342 solution for
30 min at 37˚C in the dark. A fluorescence microscope (Olympus
Corporation) was used to observe DNA staining. Ten fields were
randomly selected and a total of 200 cells were counted. The
following formula was used to calculate percentages of positively
stained cells: Apoptotic cells/the total number of counted
cells.
Annexin V-FITC/PI analysis
FLSs were harvested at 48 h post-transfection. An
annexin V-FITC/propidium iodide (PI) apoptosis detection kit
(Abcam) was used to detect cell apoptosis according to the
manufacturer's guidelines. Beckman Coulter FACSCalibur (version
6.0, Beckman Coulter, Inc.) was used to calculate the percentage of
apoptotic cells, using flow cytometry. Flow cytometry density plots
showing annexin V (X-axis) and PI (Y-axis) staining of cells were
generated using FACStation software (version 6.0; BD Bioscience).
The right lower quadrant represents annexin V positive/PI negative
staining, which indicates early apoptosis. The right upper quadrant
represents both high annexin V and PI staining, indicating late
apoptosis and the left upper quadrant represents low annexin V and
high PI staining, indicating necrosis. The left lower quadrant
indicates viable cells.
In vitro caspase-3 activity assay
Caspase-3 activity was determined using colorimetric
assay kits, which utilized synthetic tetrapeptides [Asp-Glu-Val-Asp
(DEAD) for caspase-3] (cat. no. A1086; Sigma-Aldrich; Merck KGaA)
labeled with p-nitroaniline (pNA; Abcam; cat. no. ab39401). The
kits were used according to the manufacturer's protocols. Briefly,
cells were lysed in the supplied lysis buffer for 30 min at 4˚C.
Supernatants were collected and incubated with the supplied
reaction buffer containing dithiothreitol and DEAD-pNA as
substrates at 37˚C. The reactions were measured by changes in
absorbance at 405 nm using the VERSAmax tunable microplate
reader.
Dual-luciferase reporter assay
The STAT3 gene was used as a target and
amplification of the STAT3 gene 3'-UTR was carried out, followed by
fusion in psiCHECK-2 vector (Promega Corporation) with the GV126
luciferase gene (Promega Corporation). The binding sites of the
STAT3 gene and miR-140 were eliminated by site-directed mutation,
producing a mutant which was used as a control. Thymidine kinase
promoters (pRL-TK vectors; Takara Bio, Inc.) and the plasmid
containing Renilla luciferase were used to adjust for
transfection efficiency. Co-transfection of control and miR-140
with luciferase reporter vectors into RA FLSs was carried out using
Lipofectamine 2000 (Thermo Fisher Scientific, Inc.). At 36 h post
transfection, the cells were cultured in myllicin-containing DMEM,
which was supplemented with 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.) and cells then placed into an incubator with 5%
carbon dioxide at 37˚C. The liquid was replaced every 48 h. After
treatment with trypsin, cells were subcultured until they reached
80% confluency. The relative light unit of the firefly luciferase
was recorded with a GloMax 96 microplate luminometer (Promega
Corporation). The ratio of firefly to Renilla was used to
normalize the firefly luciferase values.
TargetScan prediction
The prediction algorithm database, TargetScan
(www.targetscan.org) was used to nominate
targets of miR-140. Using the database, predictions were ranked
based on the predicted efficacy of targeting as calculated using
the cumulative weighted context ++ scores of the sites (39). As an option, predictions are also
ranked by their probability of conserved targeting (40).
Statistical method
Data are expressed as the mean ± SD. Intergroup
distinctions were evaluated using one-way ANOVAs with Tukey's post
hoc tests, or two-tailed Student's t-tests. P<0.05 was
considered to indicate a statistically significant difference.
Results
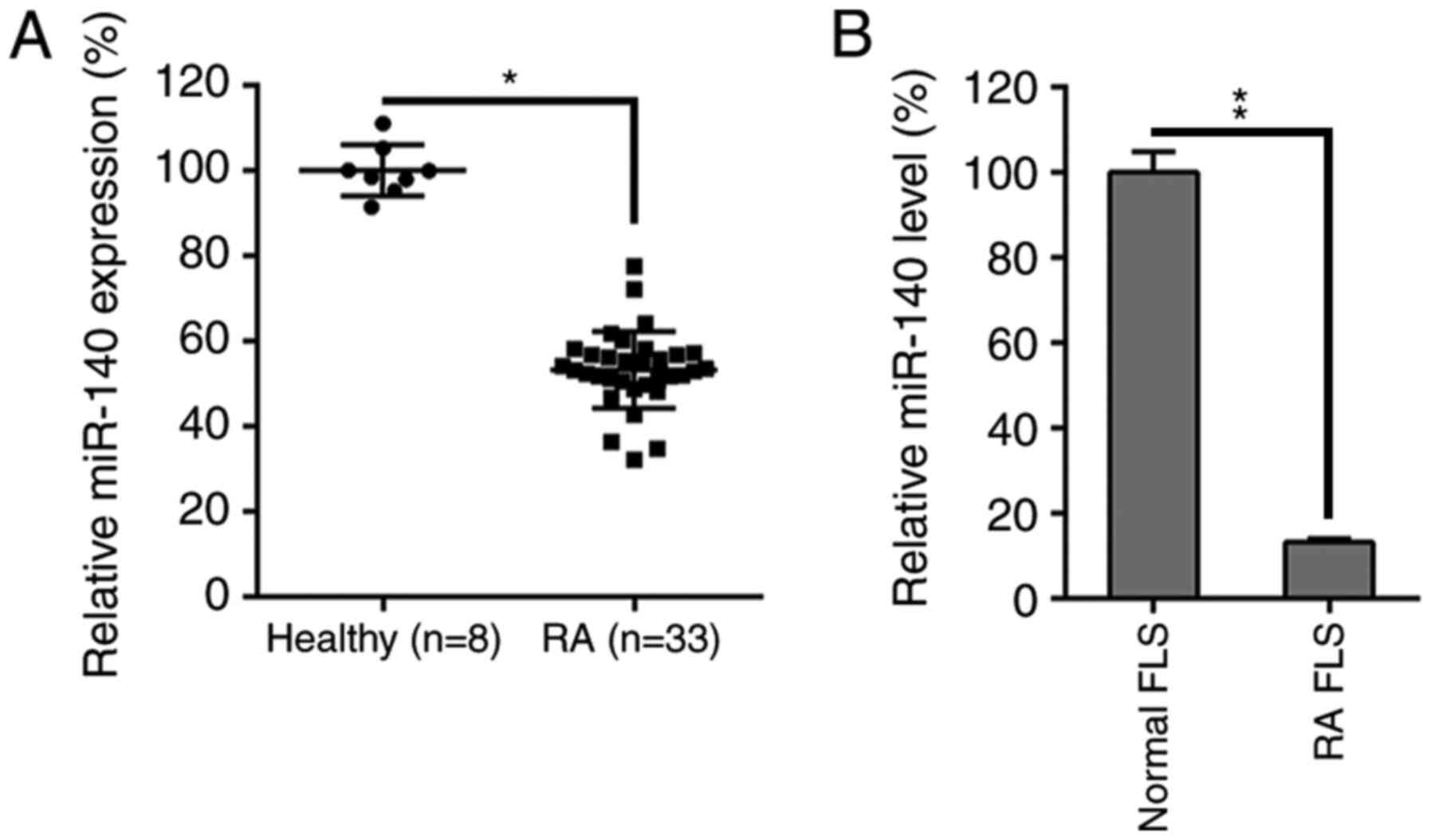
miR-140 is downregulated in the
synovial tissue of RA patients and RA FLSs
To evaluate how miR-140 was influenced by RA
progression and FLS functions, the miR-140 expression levels in RA
patients and isolated FLS lines were measured. miR-140 expression
was first evaluated in 33 RA specimens and 8 healthy specimens. A
decrease was identified in the miR-140 levels in the RA samples
compared to healthy controls (Fig.
1A). Furthermore, miR-140 expression levels were also
remarkably lower in the RA FLSs compared to healthy controls
(Fig. 1B).
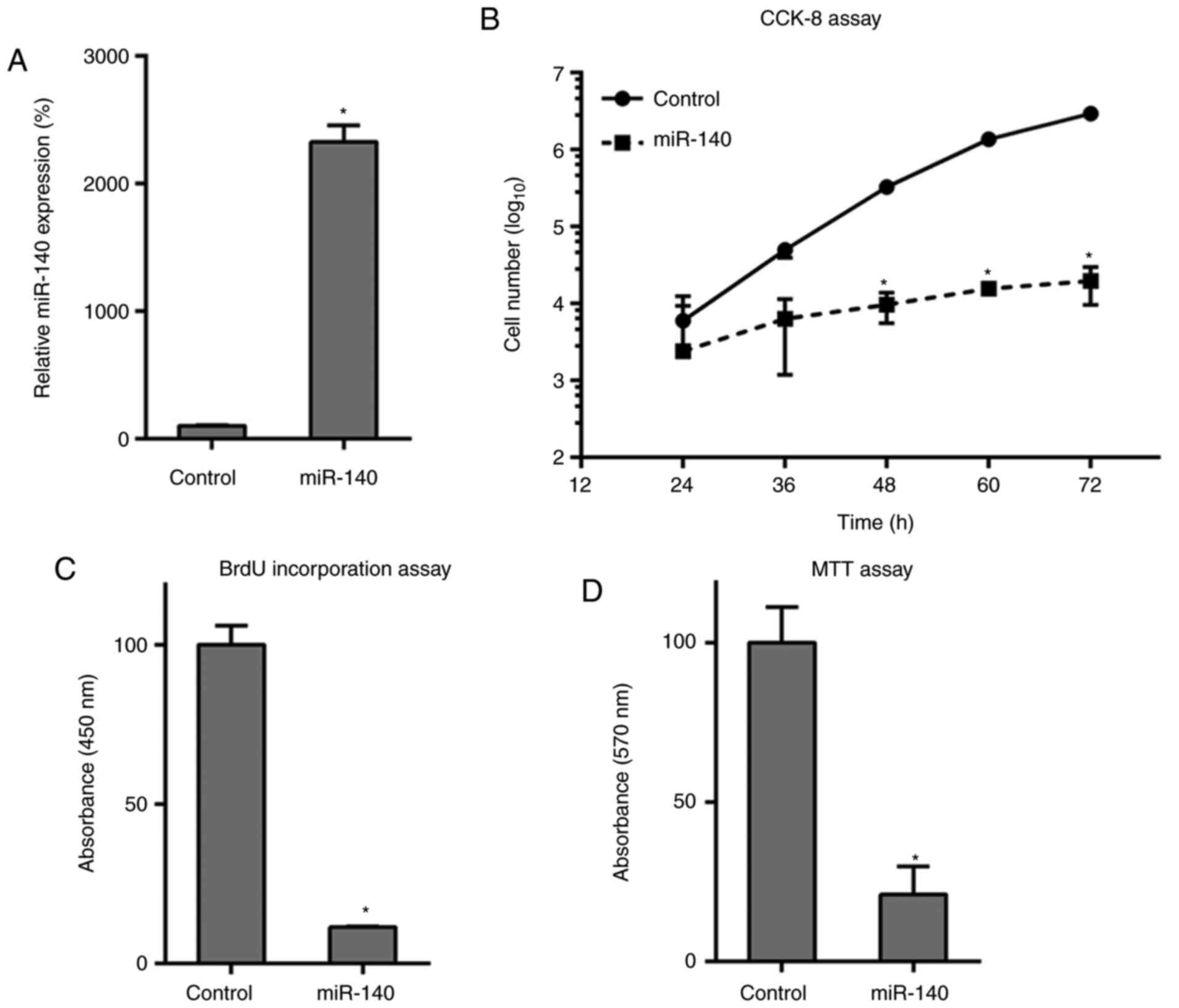
Overexpression of miR-140 impairs
proliferation of RA FLSs
To assess how miR-140 affects RA FLS growth and
proliferation, miR-140 expression was restored through
transfections. This upregulation of miR-140 was first confirmed in
RA FLSs after transfection with the miR-140 mimics, compared to
that in miR-NC-treated cells, as found by RT-qPCR (Fig. 2A). Transfection and rescue
experiments with miR-140 demonstrated a decreased cell growth rate
in the RA FLSs at 48-72 h post-transfection compared to that in the
NC group (Fig. 2B). BrdU
immunofluorescence assay and MTT assays were also performed to
measure cell proliferation at 48 h post-transfection. BrdU staining
revealed that, compared with that of the control group, miR-140
transfections inhibited cell DNA synthesis in RA FLSs by
approximately 80% (Fig. 2C) and
results from the MTT assay further supported this result, with
miR-140 transfection suppressing the cell proliferation of RA FLSs
(Fig. 2D). Overall, the
overexpression of miR-140 had an inhibitory effect on these FLS
cells.
miR-140 overexpression caused
apoptosis of RA FLSs
The miR-140-expressing cells showed that in
comparison with the control, the number of positive Hoechst
33342-stained cells was higher at 48 h after transfection (Fig. 3A). Moreover, the flow cytometry
results demonstrated that overexpression of miR-140 caused
increased apoptotic activity in RA FLSs (Fig. 3B). Since miR-140 suppressed cell
viability and promoted RA FLS apoptosis, its role in regulating the
expression of apoptosis-related proteins was further investigated.
Bcl-2 and Bax are typical anti- and pro-apoptotic proteins,
respectively. In Fig. 3C-E,
transfection of RA FLSs with miR-140 caused a decrease in the
expression levels of Bcl-2, but increased Bax expression levels,
compared with observations from the control group, at both the
protein and mRNA levels. The cleavage of caspase-3, a typical
apoptotic marker (41), and its
activity in RA FLSs were examined to identify the role of miR-140
in apoptosis. The data revealed that miR-140 could induce the
expression of cleaved caspase-3 and facilitated the pro-apoptotic
activity at 3 days post-transfection (Fig. 3F and G). These findings pointed to a mechanism
of action by which miR-140 regulated apoptosis, further confirming
the previous findings.
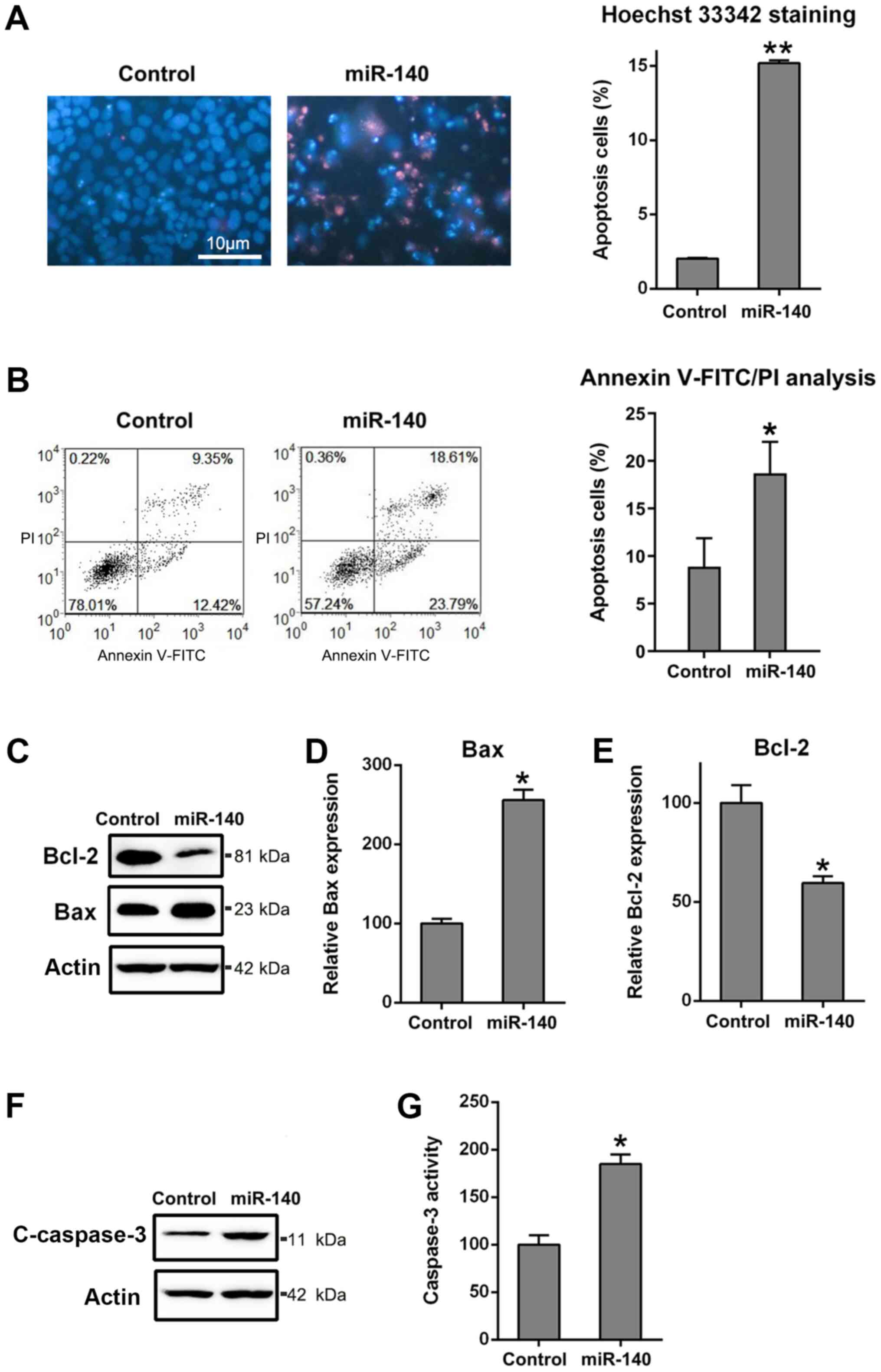 | Figure 3Ectopic overexpression of miR-140
enhances apoptosis of RA FLSs. (A) Hoechst 33342 staining was
carried out on the miR-140 overexpressing or control RA FLSs.
Magnification x200. The apoptotic rate of HepG2 cells, those
stained positive with Hoechst 33342, are displayed in the right
panel. (B) Annexin V-FITC/propidium iodide staining and flow
cytometry were performed to evaluate the number of apoptotic cells.
Early apoptotic cells and later apoptotic cells are indicated in
the top right and bottom right quadrants, respectively, in each
plot. Analysis of the apoptotic rate of RA FLSs in the two groups
is displayed in the right panel. (C) Western blotting for Bcl2 and
Bax, as well as reverse transcription-quantitative PCR for (D) Bax
and (E) Bcl2 were performed to assess the effects of miR-140 on the
Bcl-2 and Bax expression levels at the protein and mRNA levels. (F)
Western blotting was carried out to examine cCaspase-3 levels
following miR-140 transfection. (G) miR-140 reduced caspase-3
activity in RA FLSs. All groups were normalized to the control
group (100%). Data represents the mean ± SD. *P<0.05,
**P<0.01 vs. the control group. Cleaved caspase-3,
cCaspase-3; FLS, fibroblast-like synoviocyte; miR, microRNA; PI,
propidium iodide; RA, rheumatoid arthritis. |
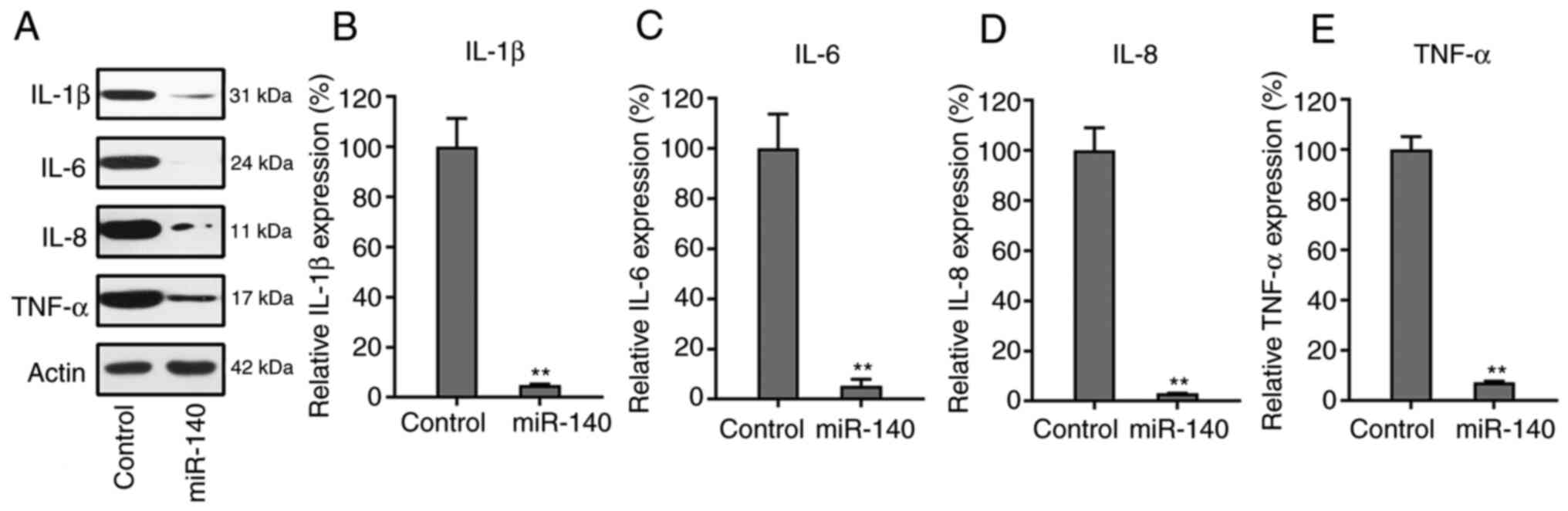
miR-140 suppresses the production of
pro-inflammatory cytokines
Western blotting and RT-qPCR were used to examine
the expression levels of each cytokine in the control and miR-140
overexpression groups. Notably, it was observed that the
inflammatory cytokines in the miR-140-expressing cells were clearly
decreased after transfection, compared to the control group
(Fig. 4A). The RT-qPCR data also
showed that the mRNA expression levels of the four cytokines
significantly decreased when RA FLSs were transfected with the
miR-140 precursor (Fig. 4B-E).
These results indicated that miR-140 inhibited the production of
cytokines during the development of RA.
STAT3 is a target of miR-140
Bioinformatics analysis revealed that miR-140
directly targeted STAT3, a key mediator of the JAK/STAT signaling
pathway (Fig. 5A). The interaction
between STAT3 and miR-140 was assessed using a dual-luciferase
reporter assay. The data revealed that in miR-140-transfected RA
FLSs, PTEN 3'-UTR-fused luciferase activity was reduced by 50%
compared with that of the control groups (Fig. 5B). Expression of STAT3 was also
examined in RA FLSs transfected with miR-140 and miR-NC using
western blotting and RT-qPCR. The results conclusively showed that
expression levels of STAT3 were suppressed due to miR-140
overexpression (Fig. 5C and
D). The mRNA levels of STAT3 in
both the normal FLSs and RA FLSs were also studied and it was found
that STAT3 in RA FLSs was significantly higher than that in normal
FLSs (Fig. 5E), also indicating the
potential of miR-140 in regulating RA FLS properties.
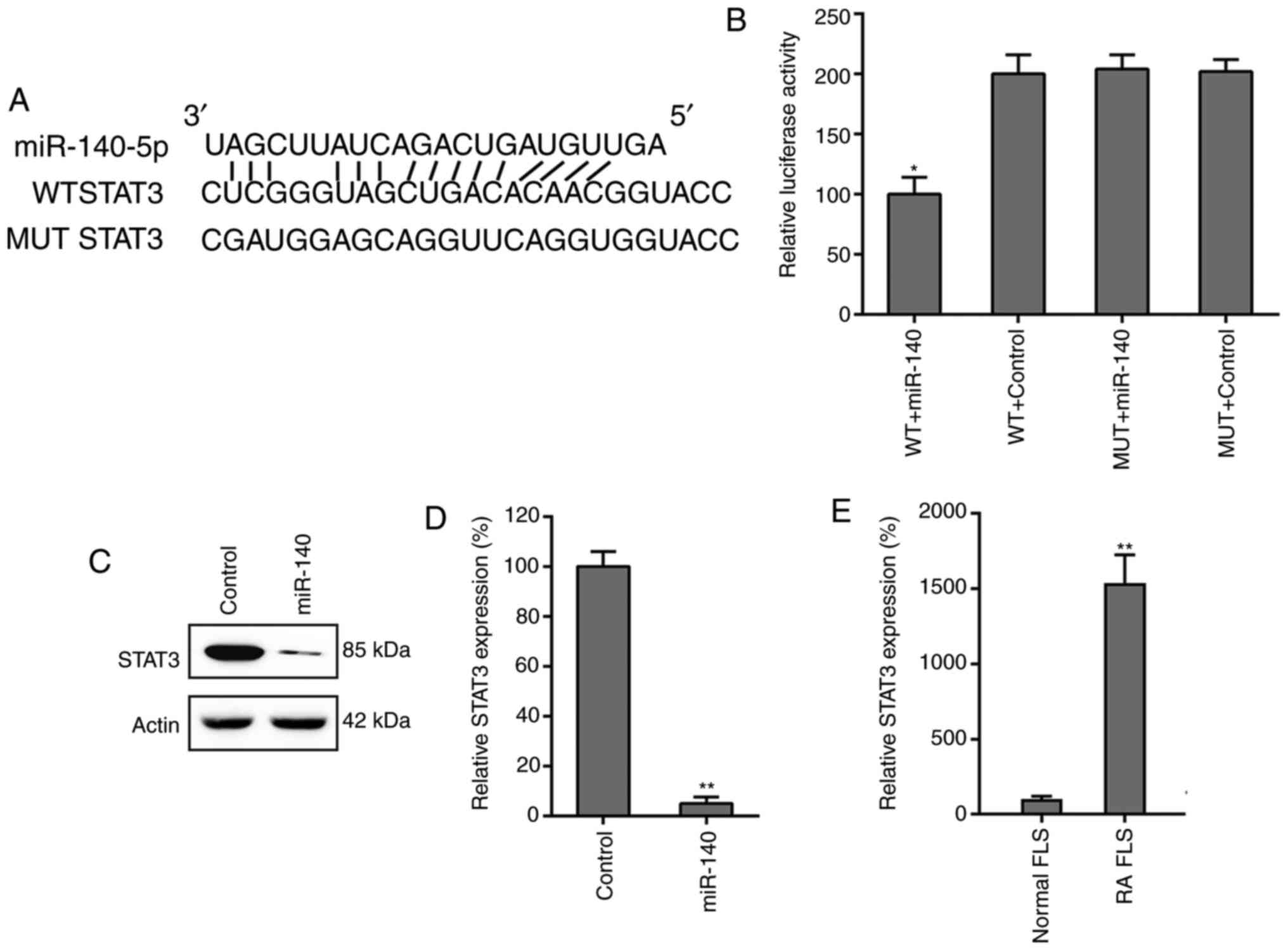 | Figure 5STAT3 is a direct target of miR-140.
(A) Graphical representation of the conserved miR-140 binding motif
at the STAT3 3'-UTR. The complementary sequences to the seed
regions of miR-140 and corresponding sequence of the 3'-UTR of
STAT3. (B) The luciferase activity exhibited by the reporter
constructs, containing either the WT or MUT human STAT3 3'-UTR
after miR-140 transfection. The observed luciferase activity was
normalized to that of β-galactosidase. Overexpression of miR-140
markedly decreased the relative luciferase activity in the WT
3'-UTR, but not the MUT 3'-UTR, of STAT3 mRNA. All groups were
normalized to the WT + control group (100%). (C) Western blotting
and (D) RT-qPCR analysis were used to evaluate the STAT3 protein
and mRNA expression levels after transfection of RA FLSs with
miR-140 or miR-negative control. All groups were normalized to the
control group (100%). (E) Increased expression levels of STAT3 in
RA FLSs was observed by RT-qPCR, compared to that of healthy FLSs.
All groups were normalized to the normal FLS group (100%). Data
represents the mean ± SD. *P<0.05,
**P<0.01 vs. the corresponding control. FLS,
fibroblast-like synoviocyte; miR, microRNA; MUT, mutant; RA,
rheumatoid arthritis; RT-qPCR, reverse transcription-quantitative
PCR; UTR, untranslated region; WT, wild-type. |
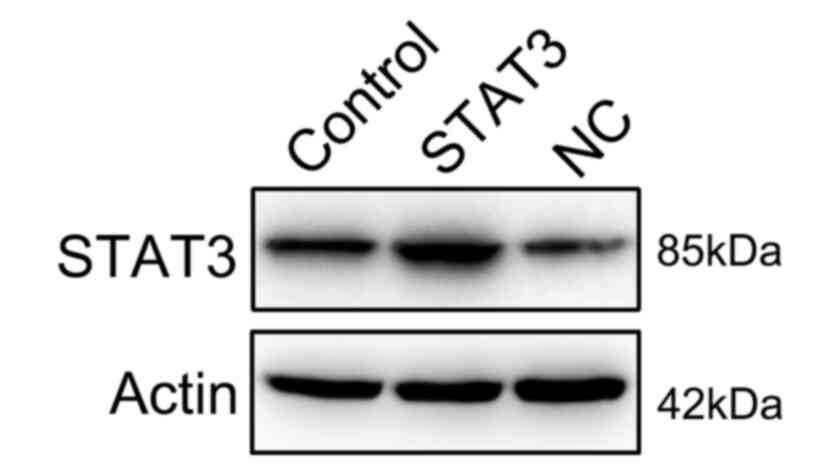
Overexpression of STAT3 reverses the
effect of miR-140 on RA FLSs
To further determine whether STAT3 restoration
rescued the regulatory effect of miR-140 on RA FLS properties,
STAT3 expression was rescued in RA FLSs expressing miR-140 through
transfections with a STAT3-expressing vector. First, the successful
expression of STAT3 was confirmed through the transfection of
STAT3-expressing vector into the cells (Fig. 6). Subsequently, STAT3 was found to
be overexpressed in RA FLSs co-expressing miR-140 at the protein
and mRNA levels (Fig. 7A and
B). BrdU and MTT assays showed that
enhanced expression of STAT3 restored the proliferation of RA FLS,
previously shown to be inhibited by miR-140 (Fig. 7C and D). The relationship between the apoptotic
activity of miR-140-expressing RA FLSs and STAT3 restoration was
determined using annexin V-FITC/PI flow cytometry. Cell apoptosis
was evidently reduced in the STAT3- and miR-140-expressing RA FLSs,
compared with that of cells which were overexpressing miR-140 alone
and with that of the control cells (Fig. 7E). Furthermore, western blotting and
RT-qPCR were used to investigate signs of inflammation of RA FLSs
expressing both STAT3 and miR-140. Overexpression of STAT3 was
observed to also cause a reverse effect on the downregulation of
inflammatory cytokines at the protein and mRNA level, which was
mediated by miR-140 transfection (Fig.
7F). Taken together, restoration of STAT3 expression may
recover the growth and apoptotic abilities, as well as restore the
expression of inflammatory cytokine production, of RA FLSs
disrupted by miR-140.
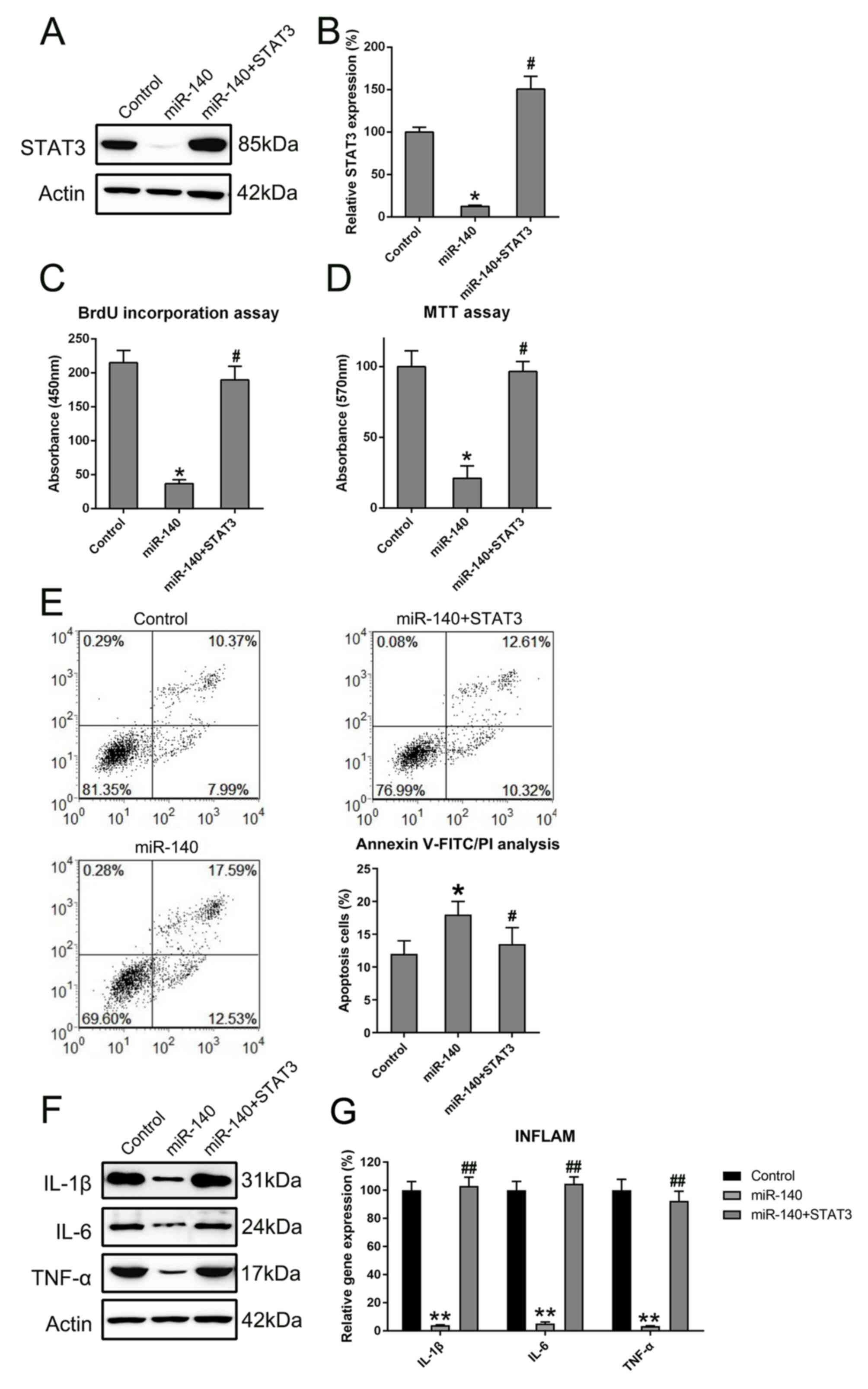 | Figure 7STAT3 restored the regulatory role of
miR-140 in RA FLSs. STAT3 expression levels were detected in cells
using (A) western blotting and (B) RT-qPCR following miR-140 and
STAT3 transfections. All groups were normalized to the control
group (100%). The proliferative ability of cells expressing STAT3
and miR-140, miR-140 alone, or control was measured at 48 h
post-transfection using the (C) BrdU incorporation assays and (D)
MTT assays. (E) The apoptotic rate of RA FLSs in each group was
determined by annexin V-FITC/PI flow cytometry. Early apoptotic
cells and later apoptotic cells are indicated in the top right and
bottom right quadrants, respectively, in each plot. Quantitative
analysis of the apoptotic rate of RA FLSs in two groups is
displayed in the right panel. (F) Western blotting was carried out
to assess the expression levels of pro-inflammatory cytokines
regulated by the miR-140 and STAT3 plasmid transfections. (G)
RT-qPCR was performed to examine the gene expression of cytokines
from the RA FLSs after transfection. All groups were normalized to
the control group (100%). Data represents the mean ± SD.
*P<0.05, **P<0.01 vs. the control
group; #P<0.05, ##P<0.01 vs. the
miR-140 group. FLS, fibroblast-like synoviocyte; IL, interleukin;
miR, microRNA; PI, propidium iodide; RA, rheumatoid arthritis;
RT-qPCR, reverse transcription-quantitative PCR; TNF, tumor
necrosis factor. |
Discussion
Increasing evidence has demonstrated that miRNAs are
closely related to RA progression (16). It has been reported that many miRNAs
such as miR-30a (42),
miR-155(43), miR-223(44) and miR-27a (45) are involved in modulating RA
occurrence and development. Hence, miRNAs have been posited as a
potential therapeutic strategy for RA treatment (10). For instance, it has been reported
that involvement of miR-21 facilitates the proliferation of FLSs in
RA models through NF-κB pathway (46). However, the relevant mechanism and
dysfunction of miR-140-5p in RA is not well established.
Accumulating miRNAs exert their biological roles by
regulating their target molecules. miR-140 has been documented to
target different genes, such as AKT3(47), Smad3(48) and E2F8(49), which are all involved in different
biological process. In the present study, using bioinformatic
analysis, the target of miR-140 and the conservative site of STAT3
were predicted. From the dual-luciferase reporter assay, results
showed that STAT3 directly interacted with miR-140 in RA FLS. STAT3
is correlated with multiple biological processes (50,51),
with phosphorylated STAT3 modulating various genes, including
Bcl-xL, Bcl-2, VEGF and MMPs (50-53).
For colorectal cancer cells, inhibition of STAT3 signaling induces
apoptosis, cell cycle arrest and reduces tumor cell invasion
(54). In human liver cancer cells,
inhibition of STAT3 signaling blocks the anti-apoptotic activity of
IL-6(55). Inhibition of STAT3
signaling induces apoptosis and decreases survivin expression in
primary effusion lymphoma (56).
These reports provide a rationale for explaining the findings of
the current study. Here, STAT3 was found to be decreased after
aberrant expression of miR-140, and restoration of STAT3 expression
in RA FLS restored the properties of FLS which were modified by
miR-140, especially in reducing the apoptotic rate. This evidence
suggested an important role of miR-140 in RA FLS, which is mediated
by STAT3.
Since a lower apoptosis rate is one of the essential
characteristics of FLS during RA development (57), a higher apoptotic rate is required
to ameliorate RA progression. In the present study, overexpression
of miR-140 showed a similar restorative effect by upregulating
caspase-3 and Bax, and downregulating the Bcl-2, ultimately
promoting apoptosis in RA FLS. miR-140 has been shown to exhibit
its regulatory role in chondrocytes (25), cardiomyocytes (58) and ovarian cancer cells (41). Therefore, the current data suggested
that miR-140 facilitates RA FLS apoptosis by both the mitochondria
pathway (through reduced Bcl-2) and the death receptor pathway
(through increased caspase-3).
In conclusion, this present study demonstrated that
miR-140 can suppress proliferation, promote apoptosis and suppress
inflammatory cytokines in RA FLSs, exerted through STAT3. As such,
the interplay between miR-140 and STAT3 may serve as an effective
therapeutic target for the treatment of RA in humans.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JZ conceived and designed the research, and
interpreted the results of the experiments. JW contributed to the
design of the study and the interpretation of experimental results.
JH, WD, YH, HP, and JL performed experiments, analyzed data,
prepared figures and drafted the manuscript. WD and YH also
approved the final version of manuscript. HP and JL also edited and
revised manuscript.
Ethics approval and consent to
participate
All participants gave written informed consent prior
to participating and the current study was approved by the Ethics
Committee of Zunyi Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Meier FM, Frerix M, Hermann W and
Muller-Ladner U: Current immunotherapy in rheumatoid arthritis.
Immunotherapy. 5:955–974. 2013.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Boissier MC, Semerano L, Challal S,
Saidenberg-Kermanac'h N and Falgarone G: Rheumatoid arthritis: From
autoimmunity to synovitis and joint destruction. J Autoimmun.
39:222–228. 2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Tobon GJ, Youinou P and Saraux A: The
environment, geo-epidemiology, and autoimmune disease: Rheumatoid
arthritis. J Autoimmun. 35:10–14. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Rockel JS and Kapoor M: Autophagy:
Controlling cell fate in rheumatic diseases. Nat Rev Rheumatol.
13(193)2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Bartok B, Hammaker D and Firestein GS:
Phosphoinositide 3-kinase delta regulates migration and invasion of
synoviocytes in rheumatoid arthritis. J Immunol. 192:2063–2070.
2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Qin B, Yang M, Fu H, Ma N, Wei T, Tang Q,
Hu Z, Liang Y, Yang Z and Zhong R: Body mass index and the risk of
rheumatoid arthritis: A systematic review and dose-response
meta-analysis. Arthritis Res Ther. 17(86)2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Qin B, Liang Y, Yang Z and Zhong R:
Response to: ‘Body mass index and the risk of rheumatoid arthritis:
A systematic review and dose-response meta-analysis’-authors'
reply. Arthritis Res Ther. 17(217)2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Lee RC, Feinbaum RL and Ambros V: The C.
Elegans heterochronic gene lin-4 encodes small RNAs with antisense
complementarity to lin-14. Cell. 75:843–854. 1993.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kozomara A and Griffiths-Jones S: miRBase:
Integrating microRNA annotation and deep-sequencing data. Nucleic
Acids Res. 39:D152–D157. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Filkova M, Jungel A, Gay RE and Gay S:
MicroRNAs in rheumatoid arthritis: Potential role in diagnosis and
therapy. BioDrugs. 26:131–141. 2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Li S, Jin Z and Lu X: MicroRNA-192
suppresses cell proliferation and induces apoptosis in human
rheumatoid arthritis fibroblast-like synoviocytes by downregulating
caveolin 1. Mol Cell Biochem. 432:123–130. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kurowska-Stolarska M, Alivernini S,
Melchor EG, Elmesmari A, Tolusso B, Tange C, Petricca L, Gilchrist
DS, Di Sante G, Keijzer C, et al: MicroRNA-34a dependent regulation
of AXL controls the activation of dendritic cells in inflammatory
arthritis. Nat Commun. 8(15877)2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Gao W, McCormick J, Connolly M, Balogh E,
Veale D and Fearon U: Hypoxia and STAT3 signalling interactions
regulate pro-inflammatory pathways in rheumatoid arthritis. Ann
Rheum Dis. 74:1275–1283. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Isomaki P, Junttila I, Vidqvist KL,
Korpela M and Silvennoinen O: The activity of JAK-STAT pathways in
rheumatoid arthritis: constitutive activation of STAT3 correlates
with interleukin 6 levels. Rheumatology (Oxford). 54:1103–1113.
2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Nowell MA, Richards PJ, Fielding CA,
Ognjanovic S, Topley N, Williams AS, Bryant-Greenwood G and Jones
SA: Regulation of pre-B cell colony-enhancing factor by
STAT-3-dependent interleukin-6 tranes-signaling: Implications in
the pathogenesis of rheumatoid arthritis. Arthritis Rheum.
54:2084–2095. 2006.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Turkson J and Jove R: STAT proteins: Novel
molecular targets for cancer drug discovery. Oncogene.
19:6613–6626. 2000.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Turkson J, Bowman T, Garcia R, Caldenhoven
E, De Groot RP and Jove R: Stat3 activation by src induces specific
gene regulation and is required for cell transformation. Mol Cell
Biol. 18:2545–2552. 1998.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chen Z and Han ZC: STAT3: A critical
transcription activator in angiogenesis. Med Res Rev. 28:185–200.
2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Devarajan E and Huang S: STAT3 as a
central regulator of tumor metastases. Curr Mol Med. 9:626–633.
2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Starr R, Willson TA, Viney EM, Murray LJ,
Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA
and Hilton DJ: A family of cytokine-inducible inhibitors of
signalling. Nature. 387:917–921. 1997.PubMed/NCBI View
Article : Google Scholar
|
|
21
|
Endo TA, Masuhara M, Yokouchi M, Suzuki R,
Sakamoto H, Mitsui K, Matsumoto A, Tanimura S, Ohtsubo M, Misawa H,
et al: A new protein containing an SH2 domain that inhibits JAK
kinases. Nature. 387:921–924. 1997.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Coombs JH, Bloom BJ, Breedveld FC,
Fletcher MP, Gruben D, Kremer JM, Burgos-Vargas R, Wilkinson B,
Zerbini CA and Zwillich SH: Improved pain, physical functioning and
health status in patients with rheumatoid arthritis treated with
CP-690,550, an orally active Janus kinase (JAK) inhibitor: Results
from a randomised, double-blind, placebo-controlled trial. Ann
Rheum Dis. 69:413–416. 2010.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Tanaka Y, Suzuki M, Nakamura H, Toyoizumi
S and Zwillich SH: Tofacitinib Study Investigators. Phase II study
of tofacitinib (CP-690,550) combined with methotrexate in patients
with rheumatoid arthritis and an inadequate response to
methotrexate. Arthritis Care Res (Hoboken). 63:1150–1158.
2011.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Dong W, Yao C, Teng X, Chai J, Yang X and
Li B: miR-140-3p suppressed cell growth and invasion by
downregulating the expression of ATP8A1 in non-small cell lung
cancer. Tumour Biol. 37:2973–2985. 2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wang Z, Hu J, Pan Y, Shan Y, Jiang L, Qi X
and Jia L: miR-140-5p/miR-149 affects chondrocyte proliferation,
apoptosis, and autophagy by targeting FUT1 in osteoarthritis.
Inflammation. 41:959–971. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Flamini V, Jiang WG and Cui Y: Therapeutic
role of miR-140-5p for the treatment of non-small cell lung cancer.
Anticancer Res. 37:4319–4327. 2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Li W and He F: Monocyte to macrophage
differentiation-associated (MMD) targeted by miR-140-5p regulates
tumor growth in non-small cell lung cancer. Biochem Biophys Res
Commun. 450:844–850. 2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Xie WB, Liang LH, Wu KG, Wang LX, He X,
Song C, Wang YQ and Li YH: miR-140 expression regulates cell
proliferation and targets PD-L1 in NSCLC. Cell Physiol Biochem.
46:654–663. 2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Yuan Y, Shen Y, Xue L and Fan H: miR-140
suppresses tumor growth and metastasis of non-small cell lung
cancer by targeting insulin-like growth factor 1 receptor. PLoS
One. 8(e73604)2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Song B, Wang Y, Xi Y, Kudo K, Bruheim S,
Botchkina GI, Gavin E, Wan Y, Formentini A, Kornmann M, et al:
Mechanism of chemoresistance mediated by miR-140 in human
osteosarcoma and colon cancer cells. Oncogene. 28:4065–4074.
2009.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Wei R, Cao G, Deng Z, Su J and Cai L:
miR-140-5p attenuates chemotherapeutic drug-induced cell death by
regulating autophagy through inositol 1,4,5-trisphosphate kinase 2
(IP3k2) in human osteosarcoma cells. Biosci Rep.
36(e00392)2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Xiao Q, Huang L, Zhang Z, Chen X, Luo J,
Zhang Z, Chen S, Shu Y, Han Z and Cao K: Overexpression of miR-140
inhibits proliferation of osteosarcoma cells via suppression of
histone deacetylase 4. Oncol Res. 25:267–275. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Li H, Guan SB, Lu Y and Wang F: miR-140-5p
inhibits synovial fibroblasts proliferation and inflammatory
cytokines secretion through targeting TLR4. Biomed Pharmacother.
96:208–214. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Liu X, Wang S, Yuan A, Yuan X and Liu B:
MicroRNA-140 represses glioma growth and metastasis by directly
targeting ADAM9. Oncol Rep. 36:2329–2338. 2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Rothman AM, Arnold ND, Pickworth JA,
Iremonger J, Ciuclan L, Allen RM, Guth-Gundel S, Southwood M,
Morrell NW, Thomas M, et al: MicroRNA-140-5p and SMURF1 regulate
pulmonary arterial hypertension. J Clin Invest. 126:2495–2508.
2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zhang R, Ma J and Yao J: Molecular
mechanisms of the cartilage-specific microRNA-140 in
osteoarthritis. Inflamm Res. 62:871–877. 2013.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Aggarwal R, Rider LG, Ruperto N, Bayat N,
Erman B, Feldman BM, Oddis CV, Amato AA, Chinoy H, Cooper RG, et
al: 2016 American college of rheumatology/european league against
rheumatism criteria for minimal, moderate, and major clinical
response in adult dermatomyositis and polymyositis: An
international myositis assessment and clinical studies
group/paediatric rheumatology international trials organisation
collaborative initiative. Ann Rheum Dis. 76:792–801.
2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
Elife. 12(4)2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Friedman RC, Farh KK, Burge CB and Bartel
DP: Most mammalian mRNAs are conserved targets of microRNAs. Genome
Res. 19:92–105. 2009.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Lan H, Chen W, He G and Yang S: miR-140-5p
inhibits ovarian cancer growth partially by repression of PDGFRA.
Biomed Pharmacother. 75:117–122. 2015.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Alsaleh G, Francois A, Philippe L, Gong
YZ, Bahram S, Cetin S, Pfeffer S, Gottenberg JE, Wachsmann D,
Georgel P and Sibilia J: miR-30a-3p negatively regulates BAFF
synthesis in systemic sclerosis and rheumatoid arthritis
fibroblasts. PLoS One. 9(e111266)2014.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Alivernini S, Kurowska-Stolarska M,
Tolusso B, Benvenuto R, Elmesmari A, Canestri S, Petricca L,
Mangoni A, Fedele AL, Di Mario C, et al: MicroRNA-155 influences
B-cell function through PU.1 in rheumatoid arthritis. Nat Commun.
7(12970)2016.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Chen SY: MicroRNA-223: A double-edged
sword in rheumatoid arthritis. Rheumatol Int. 34:285–286.
2014.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Shi DL, Shi GR, Xie J, Du XZ and Yang H:
MicroRNA-27a inhibits cell migration and invasion of
fibroblast-like synoviocytes by targeting follistatin-like protein
1 in rheumatoid arthritis. Mol Cells. 39:611–618. 2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Chen Y, Xian PF, Yang L and Wang SX:
MicroRNA-21 promotes proliferation of fibroblast-like synoviocytes
through mediation of NF-κB nuclear translocation in a rat model of
collagen-induced rheumatoid arthritis. Biomed Res Int.
2016(9279078)2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Zhang F and Wu Z: Significantly altered
expression of miR-511-3p and its target AKT3 has negative
prognostic value in human prostate cancer. Biochimie. 140:66–72.
2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Pais H, Nicolas FE, Soond SM, Swingler TE,
Clark IM, Chantry A, Moulton V and Dalmay T: Analyzing mRNA
expression identifies Smad3 as a microRNA-140 target regulated only
at protein level. RNA. 16:489–494. 2010.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Sun J, Shi R, Zhao S, Li X, Lu S, Bu H, Ma
X and Su C: E2F8, a direct target of miR-144, promotes papillary
thyroid cancer progression via regulating cell cycle. J Exp Clin
Cancer Res. 36(40)2017.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Masuda M, Suzui M, Yasumatu R, Nakashima
T, Kuratomi Y, Azuma K, Tomita K, Komiyama S and Weinstein IB:
Constitutive activation of signal transducers and activators of
transcription 3 correlates with cyclin D1 overexpression and may
provide a novel prognostic marker in head and neck squamous cell
carcinoma. Cancer Res. 62:3351–3355. 2002.PubMed/NCBI
|
|
51
|
Kim BH, Yi EH and Ye SK: Signal transducer
and activator of transcription 3 as a therapeutic target for cancer
and the tumor microenvironment. Arch Pharm Res. 39:1085–1099.
2016.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Cafferkey C and Chau I: Novel STAT 3
inhibitors for treating gastric cancer. Expert Opin Investig Drugs.
25:1023–1031. 2016.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Bosch-Barrera J, Queralt B and Menendez
JA: Targeting STAT3 with silibinin to improve cancer therapeutics.
Cancer Treat Rev. 58:61–69. 2017.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Xiong H, Zhang ZG, Tian XQ, Sun DF, Liang
QC, Zhang YJ, Lu R, Chen YX and Fang JY: Inhibition of JAK1,
2/STAT3 signaling induces apoptosis, cell cycle arrest, and reduces
tumor cell invasion in colorectal cancer cells. Neoplasia.
10:287–297. 2008.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Liu Y, Li PK, Li C and Lin J: Inhibition
of STAT3 signaling blocks the anti-apoptotic activity of IL-6 in
human liver cancer cells. J Biol Chem. 285:27429–27439.
2010.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Aoki Y, Feldman GM and Tosato G:
Inhibition of STAT3 signaling induces apoptosis and decreases
survivin expression in primary effusion lymphoma. Blood.
101:1535–1542. 2003.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Meng Q, Du X, Wang H, Gu H, Zhan J and
Zhou Z: Astragalus polysaccharides inhibits cell growth and
pro-inflammatory response in IL-1β-stimulated fibroblast-like
synoviocytes by enhancement of autophagy via PI3K/AKT/mTOR
inhibition. Apoptosis. 22:1138–1146. 2017.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Li J, Li Y, Jiao J, Wang J, Li Y, Qin D
and Li P: Mitofusin 1 is negatively regulated by microRNA 140 in
cardiomyocyte apoptosis. Mol Cell Biol. 34:1788–1799.
2014.PubMed/NCBI View Article : Google Scholar
|