1. Introduction
Cardiac fibrosis is a hallmark of cardiac remodeling
associated with nearly all forms of heart disease, such as heart
failure (HF), myocardial infarction (1), dilated and ischemic cardiomyopathies
(ICMs) (2), arrhythmia (3) and hyperaldosteronism (4). The adult mammalian heart has
negligible regenerative capacity, and cardiac repair is dependent
on the clearance of dead cells and the formation of scar tissue to
help preserve myocardial structural and functional integrity
(5). Meanwhile, unrestrained tissue
repair can result in pathological fibrosis, causing detrimental
abnormalities (5,6). Pathological cardiac fibrosis is
orchestrated predominantly by myofibroblasts, which are activated
fibroblasts characterized by overproduction of growth factors,
cytokines, chemokines, proteases and extracellular matrix (ECM)
proteins. Moreover, myofibroblasts are particularly responsive to
proinflammatory cytokines, including TNF-α, IL-1, IL-6 and TGF-β;
vasoactive peptide angiotensin II (Ang II), endothelin-1, atrial
natriuretic peptide and brain natriuretic peptide (5-7).
The sources of these activated fibroblasts that accumulate in
response to various pathological insults, such as myocardial
injury, oxidative stress, mechanical stretch, autocrine-paracrine
mediators and inflammatory stimuli, remain under active
investigation (8). Lineage tracing
has recently been utilized in cardiac fibrosis studies, and
numerous cells, including resident fibroblasts (9), endothelial cells (ECs) (10) or epicardial cells (11), hematopoietic bone marrow-derived
macrophages (12) and perivascular
cells (13), have been proposed as
precursors of the fibroblast population in the injured heart. Thus,
the differentiation of quiescent fibroblasts into active
matrix-producing myofibroblasts is a key step in disease
progression (5). Cardiac fibrosis
induces adverse structural remodeling of the myocardium, resulting
in abnormalities in cardiac conduction, loss of contractility, and
hardening of the ventricular walls (5). Thus, understanding the underlying
mechanisms of cardiac fibrosis will facilitate the development of
innovative treatment strategies to hinder pathological
fibrosis.
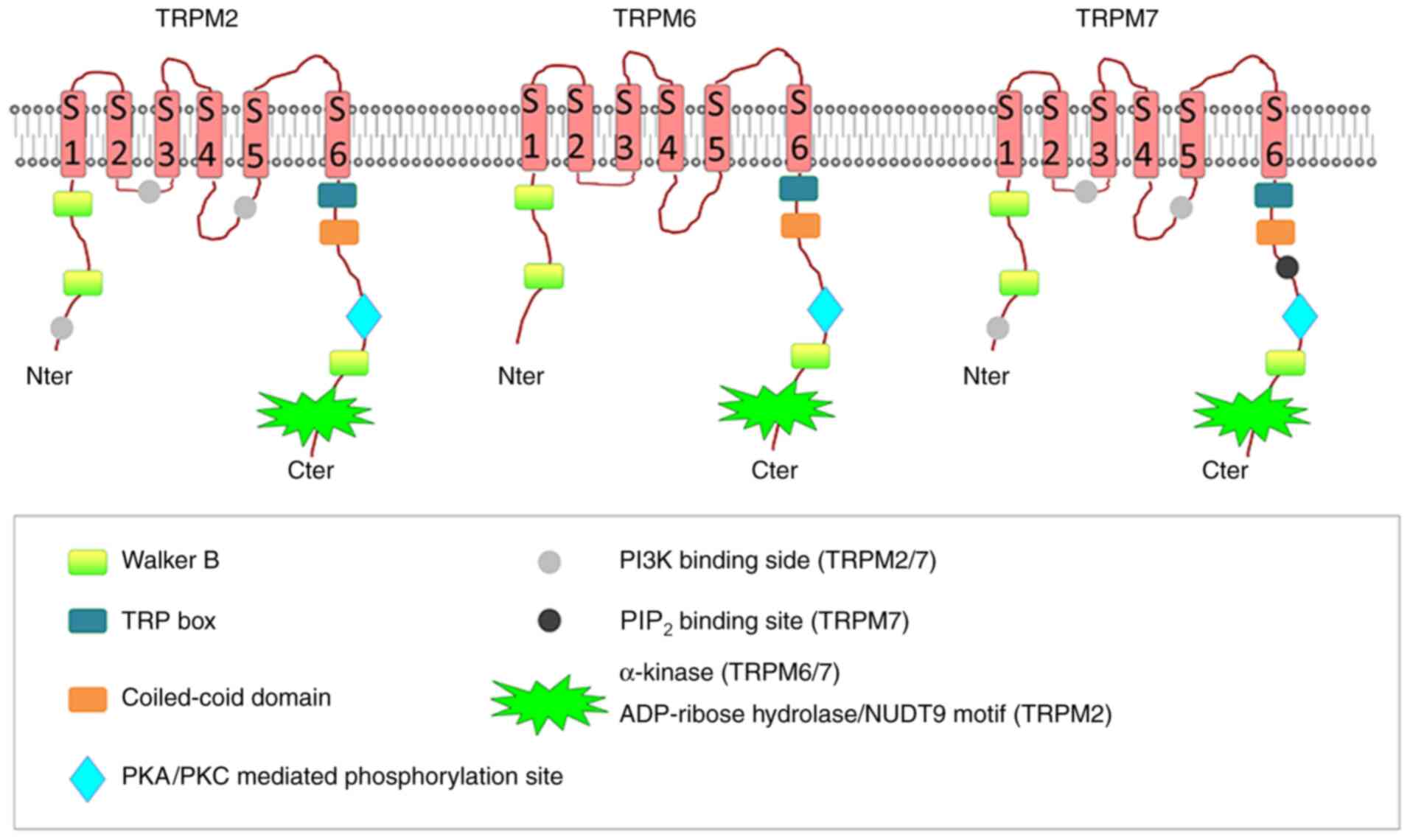
The TRPM subfamily plays a crucial role in various
physiological and pathological conditions such as those related to
sensory or renal physiology, cancer, cardiac health and neuronal
development (14). The subfamily
contains eight isoforms that form four subfamilies of TRPM
channels: TRPM1/3, TRPM2/8, TRPM4/5, and TRPM6/7. Among these,
TRPM7, TRPM2 and TRPM6 are unique because they are bifunctional
heteromeric ion channels containing functional enzymatic domains in
their highly varied C-terminal segments (15). The TRPM2 channel has an enzymatic
domain similar to Nudix hydrolase, cleaving monodinucleotidic
polyphosphates (15). On the other
hand, the TRPM6 and TRPM7 channels contain kinase domains and are
classified as atypical alpha protein kinases (15). The double activity as channels and
enzymes of these two proteins classifies them either as chanzymes
or channel-kinases (16) (Fig. 1). TRPM7 and its close homologue
TRPM6 are present in the tetrameric form, with each subunit
consisting of six transmembrane segments, and are both permeable to
calcium (Ca2+), magnesium (Mg2+) and zinc
cations (Zn2+) (17).
The disruption of TRPM6 kinase phosphorylation activity
reintroduces MgATP sensitivity to the heteromeric channel, which is
similar to that of TRPM7, supporting the notion that TRPM6 kinase
plays a critical role in the control of the TRPM7/6 channel complex
(18).
The TRPM subfamily has been shown to have highly
differing modes of activation, cation selectivity and tissue
distribution (19) and play crucial
roles in various physiological and pathological conditions such as
those related sensory or renal physiology, cancer, cardiac health
and neuronal development (14).
TRPM6 is mainly expressed in intestinal and renal epithelia, and
TRPM2 expression is highest in the brain and bone marrow (20). In contrast to TRPM6 and TRPM2, which
have specific expression patterns, TRPM7 is widely distributed in
the central nervous system as well as in the periphery, with the
highest expression levels in the heart, pituitary, bone and adipose
tissue (20). TRPM7 expression in
cardiac fibroblasts (CFs) has been verified by immunocytochemistry
(21). Two of the characteristics
of TRPM7 channels, specifically, their non-voltage-gated design and
Ca2+ permeability, suggest that they may have
significant pathological and physiological functions in various
cells, particularly in non-excitable cells such as CFs (21).
Their two-in-one protein structure, ubiquitous
expression profile and unique biophysical characteristics that
enable divalent ion transport results in the involvement of TRPM7
involvement in a number of pathophysiological processes (22-34),
including Mg2+ (25,26)
and Ca2+ (27)
homeostasis, immune system homeostasis (28), embryonic development (25,29),
hyperaldosteronism (4),
cardiovascular inflammation and fibrosis (22), hypertension (23), diabetes (30,31),
cerebral ischemia and hypoxia (32), airway remodeling (33) and tumorigenic activity (34,35). A
lack of either TRPM6 or TRPM7 was found to be embryonically lethal,
and several Mg-related diseases have been linked to mutations in
these channels (22,23,29,36,37).
Inhibition of TRPM2has been indicated to protect against renal
fibrosis and inflammation (38),
whereas chanzyme TRPM7 has been demonstrated to protect against
cardiovascular fibrosis and inflammation (22). Since TRPM7 expression is highest in
the heart, previous studies have demonstrated that TRPM7 plays a
crucial role in cardiac fibrosis-related diseases (21,22).
The present review will focus on recent developments in
understanding the role of TRPM7 in cardiac fibrogenesis and the
potential of TRPM7 as a therapeutic target for antifibrotic drug
development.
2. Interacting proteins
TRPM7 represents a constitutively active ion channel
that is heavily regulated by a variety of physiological feedback
mechanisms. One of the most important regulatory factors of channel
activity is free intracellular Mg2+ (39), which opens channels and thus result
in ion movements. Detailed biophysical examination revealed that
native TRPM7 in excised patches has two conductance states at 39
picosiemens (pS) and 186 pS, with both reversibly inhibited by
Mg2+ (40).
Information on proteins interacting with TRPM7
remain scarce, even for the kinase domain. A study has shown that
receptor-stimulated activation of phospholipase (PLC causes
inhibition of TRPM7 channel activity through localized
phosphatidylinositol 4,5-bisphosphate (PIP2) hydrolysis
(41). Furthermore, hypomagnesemic
conditions increased TRPM7-kinase-regulated Ser/Thr phosphorylation
in the C2 domain of PLCγ2, leading to reduced Ca2+
signaling (42), whereas the
α-kinase domain activates downstream target proteins involved in
cytoskeleton organization, cell proliferation, inflammatory
responses and vascular contraction, among other properties
(39,43). The involvement of TRPM7 kinase in
cell motility and adhesion has been linked to its ability to
phosphorylate the assembly domains of non-muscle myosin IIA, IIB,
and IIC and ATP-dependent motor proteins involved in
actomyosin-based cell motility (44-46).
Annexin A1, a Ca2+-dependent membrane-binding protein
with the ability to promote membrane fusion, is also phosphorylated
by the TRPM7 kinase, providing a possible link to TRPM7's known
involvement in cell growth and apoptosis (47,48).
TRPM7 has autophosphorylation residues, and cleavage
of the α-kinase results in the release of fragments that bind to
transcription factors, leading to epigenetic modifications
(49). Phosphomapping by mass
spectrometry identified 47 autophosphorylation sites on TRPM7, the
majority of which are located in the Ser/Thr-rich domain N-terminal
of the kinase region (16). This
part of the TRPM7 region is thought to control kinase substrate
binding (16). The TRPM7 kinase
specifically phosphorylates Ser and Thr residues in a
Mg2+-dependent manner (50). It autophosphorylates itself and
phosphorylates myelin basic protein as well as histone H3(51). At least two of the identified
autophosphorylation sites (S1511 and S1567) do not seem to
influence channel behavior (51).
These proteolytically TRPM7-cleaved kinase fragments (M7CKs)
translocate to the nucleus and bind multiple components of
chromatin remodeling complexes, leading to epigenetic modifications
and thereby influencing gene expression profiles (49). Furthermore, free cytosolic
Zn2+ is TRPM7-dependent and regulates M7CK binding to
transcription factors containing zinc-finger domains (49). These findings suggested that
TRPM7-mediated modulation of intracellular Zn2+
concentration couples ion channel signaling to epigenetic chromatin
covalent modifications that affect gene expression patterns
(49). Deletion of TRPM7 in T-cells
reduces apoptosis in response to Fas stimulation, while
caspase-dependent cleavage at D1510 leads to separation of TRPM7's
kinase domain from the channel without affecting the functionality
of the kinase but enhancing ion channel activity (52).
The channel and/or enzyme domain of TRPM7 is
relevant to cardiac fibrosis. TRPM7 is involved in Ang II-induced
cardiac fibrosis development by mediating Mg2+ and
Ca2+ influx (53).
TRPM7+/∆kinase mice exhibit cardiovascular inflammation
and fibrosis associated with [Mg2+]i
deficiency, abnormal macrophage activation, inflammatory cell
infiltration, and increased signaling through Smad3, calpain-II and
Stat1(22). The kinase domain may
influence the channel domain supported by the tissue, and cellular
Mg2+ levels were lower, while TRPM7 channel
phosphorylation was reduced in TRPM7+/∆kinase mice
compared with control counterparts (22). Other studies have also suggested
that the coexistence of the TRPM7 channel and kinase is
functionally relevant; for example, channel-mediated
Mg2+ influx is required for TRPM7 kinase activity, and
thus regulates RhoA activity and subsequent transcriptional
activation in hepatocellular carcinoma cells (34). The TRPM7 kinase inhibitor TG100-115
suppressed ion channel activity in breast cancer cells (54).
3. Physiological roles of TRPM7 in the
heart
TRPM7 is highly expressed in the adult heart and is
first expressed in the embryonic myocardium (25). The physiological consequences of
cardiac-targeted TRPM7 deletion depend on the timing of TRPM7
disruption during cardiogenesis (29). Early cardiac TRPM7 deletion before
embryonic day 9 causes HF and death by embryonic day 11.5 due to
myocardial fragility (29).
Remarkably, mice with TRPM7 deletion late in cardiogenesis, at
approximately embryonic day 13, exhibit normal adult ventricular
structures and functions (29).
Genetic deletion of TRPM7 at an intermediate developmental
timepoint during mouse cardiogenesis alters the myocardial
transcriptional profile and variably damages adult ventricular
function, inducing atrioventricular block, impaired repolarization
and ventricular arrhythmias (29).
Interestingly, cardiac-targeted TRPM7 deletion during the
intermediate and late stages (approximately embryonic days 11.5-13)
results in significant interstitial ECM deposition and interstitial
fibrosis in hearts at 6-8 months (29). Consistent with the cardiomyopathic
phenotype, researchers also observed expected enrichment in genes
upregulated in the ECM, ECM receptor interactions and
pressure-overload murine heart disease (29). These results indicated that subtle
differences in the timing of TRPM7 disruption led to significantly
different cardiac phenotypes. TRPM7 is dispensable in the adult
ventricular myocardium under basal conditions but is critical for
myocardial proliferation during early cardiogenesis and fibrosis
during intermediate-and-late cardiogenesis.
In addition, TRPM7 in other organs can also affect
heart function, especially in inflammation and fibrosis (4,22).
TRPM7-dificient mice with deletion of the kinase domain
(TRPM7+/∆kinase) showed significant cardiac hypertrophy,
fibrosis and inflammation (22).
Furthermore, TRPM7+/∆kinase mice exhibit distinct
pro-inflammatory and pro-fibrotic cardiovascular and renal
phenotype linked to macrophage activation, increased signaling
through Smad3, calpain-II and Stat1 and cellular hypomagnesaemia
(22). The different physiological
consequences between cardiac-targeted TRPM7 deletion and systemic
deletion of the kinase domain alone may be due to macrophages from
TRPM7+/∆kinase mice producing soluble factors that
promote a pro-fibrotic phenotype in CFs through
Mg2+-dependent mechanisms (22).
Other studies suggested that TRPM7 activation causes
dysregulated immune responses and inflammation, and that TRPM7
inhibition may have therapeutic potential in pro-inflammatory
diseases and immune hypersensitivity (55,56).
These discrepancies likely depend on the relative contributions of
the TRPM7 channel vs. the kinase domain and highlight the
complexity of the system. These results provided diagnostic insight
into putative mutations in the TRPM7 gene in patients with
unexplained structural or electrophysiological heart disease.
4. Pathological functions of TRPM7 in
cardiac fibrosis
Pathological cardiac fibrosis in response to
myocardial injury and/or chronic alterations of myocardial loading
conditions increase myocardial wall stiffness and disrupt the order
of myocardial structure, which is a requisite for normal cardiac
output and electrical conduction, culminating in chamber
dilatation, cardiomyocyte hypertrophy and apoptosis and ultimately
leading to the development of HF and increased arrhythmogenicity
(57). Atrial fibrosis is also a
common pathological change in elderly people that is strongly
associated with the perpetuation of atrial fibrillation (AF)
(58). The biochemical mechanisms
of cardiac fibrosis involve impaired Ca2+ or
Mg2+ homeostasis, oxidative stress, hemodynamic
abnormalities and activation of neurohormones, including Ang II,
TGF-β1 and endothelin-1 (2,5,6,22,58,59).
The production of increased quantities of ECM proteins by CFs leads
to tissue fibrosis, which can impair both mechanical and electrical
function of the heart, contributing to AF and HF (60). Previous studies have shown that
TRPM7 is the only calcium channel expressed on the cell membrane of
CFs (61), and that Ca2+
signaling is closely associated with the initiation of fibrosis
(62-64).
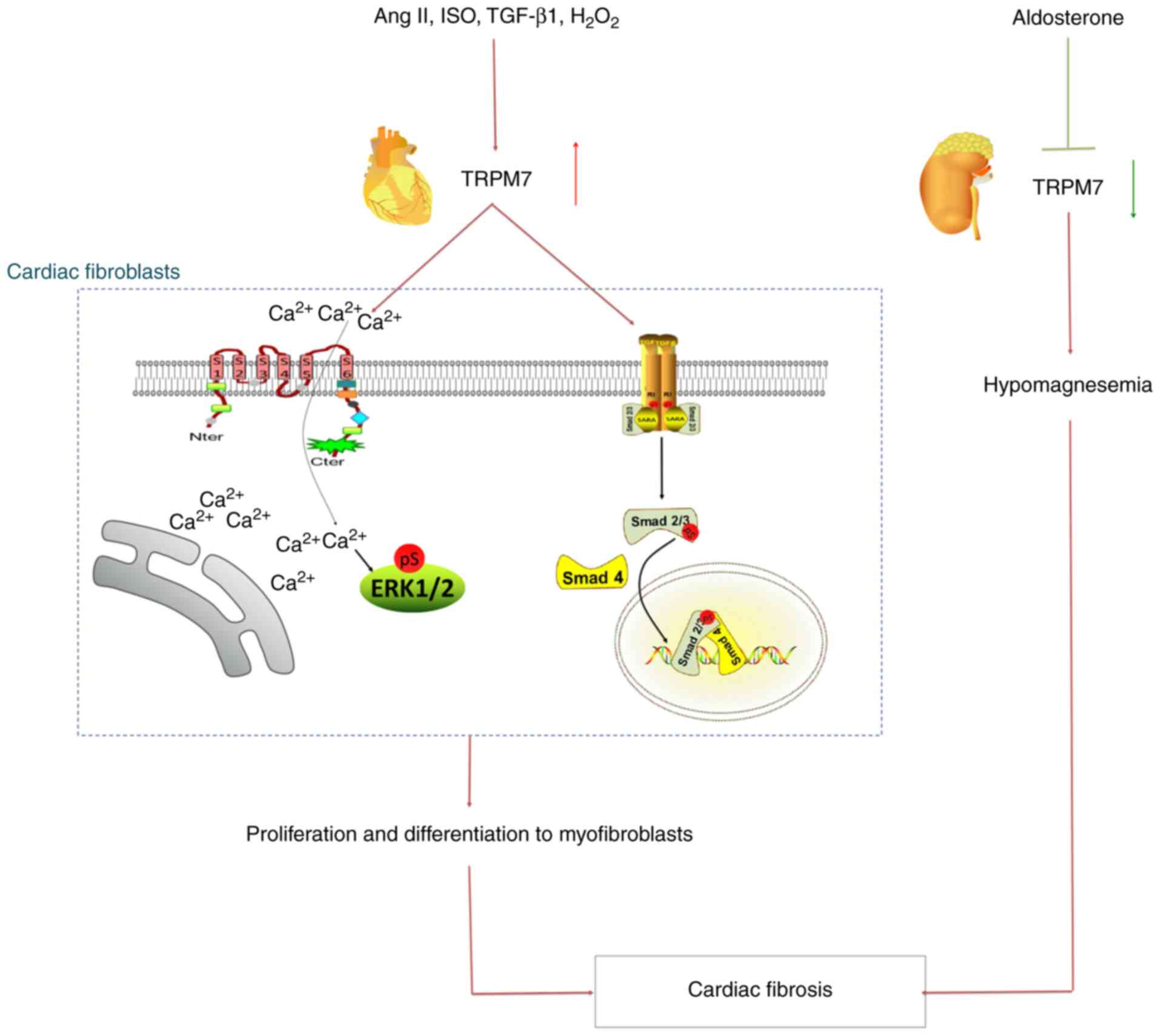
These findings suggested that TRPM7 may be the one of the most
potent fibrosis factors, playing an important role in the molecular
mechanism and pathological processes in cardiac fibrosis (Fig. 2).
TRPM7-mediated Ca2+
signaling during cardiac fibrosis
Ca2+, which is influenced by TRPM7, is
critically involved in controlling cell function including cell
proliferation, growth, secretion, migration, differentiation and
death (65). Ca2+ influx
is necessary for the biological functions of CFs during collagen
synthesis (66). The expression and
current of TRPM7, but not of TRPM4, are increased in human atrial
specimens from patients with AF compared with healthy controls
(61). TRPM7-mediated
Ca2+ influx is also significantly increased in atrial
fibroblasts from patients with AF (61). TRPM7 is the molecular basis of the
main Ca2+ permeable channel in human AF, which is the
hallmark of atrial structural remodeling (67). Atrial structural remodeling is one
of the most important fundamental mechanisms underlying the
perpetuation of AF and contributes synergistically with electrical
and neural remodeling to the AF substrate (3,58).
Previous studies have shown that TRPM7 is the only calcium channel
expressed on the cell membrane of CFs (61). Knocking down TRPM7 largely
eliminates endogenous TRPM7 currents and Ca2+ influx in
AF (67).
Mechanistically, TRPM7-mediated Ca2+
influx mediates atrial fibroblasts differentiation into
myofibroblasts, thus promoting atrial fibrosis (67). Myofibroblasts present with the
characteristics of both smooth muscle cells and fibroblasts
(66). In normal hearts,
myofibroblasts are found only in valves (66). During tissue damage repair,
myofibroblasts participate in two stages: Fiber formation and ECM
remodeling in the fibrosis cascade reaction (66). TGF-β1 can induce the transformation
of atrial fibroblasts into myofibroblasts, simultaneously
upregulating TRPM7(67). Inhibition
of TRPM7-mediated Ca2+ influx renders fibroblasts less
sensitive to TGF-β1-induced proliferation and differentiation,
indicating that TRPM7-mediated Ca2+ signal is necessary
for TGF-β1 elicited fibrogenesis (67). Inhibition of TRPM7 may prove to be
an effective approach to reduce fibroblast differentiation and
therefore attenuate fibrosis during AF.
Similar to the aforementioned roles in AF,
TRPM7-mediated Ca2+ signaling may also mediate
ventricular fibrosis. Previous studies have shown that TRPM7
expression and currents were upregulated in isoproterenol
(ISO)-induced cardiac fibrosis in vitro and in vivo
(68). Recently, TRPM7 was
demonstrated to be increased in left ventricular tissue samples
isolated from the explanted hearts of end-stage patients with HF
(69). HF is characterized by
impaired Ca2+ homeostasis/contraction, markedly
prolonged Ca2+ transients and impaired restoration of
low diastolic Ca2+ concentrations (70). Increased internal flow of
extracellular Ca2+ to the cell activates and initiates
biological signaling cascades, leading to the secretion of a large
quantity of pro-inflammatory and pro-proliferative cytokines and
ECM proteins, ultimately giving rise to cardiac interstitial
fibrosis (70). In adult rat
ventricular CFs, 2-aminoethyl diphenylborinate (2-APB, an TRPM7
inhibitor) or TRPM7 knockdown by short hairpin RNA abolished
Ca2+ influx induced by Ang II (53). Furthermore, 2-APB inhibited the
increase of myocardial connective tissue growth factor, α-smooth
muscle actin (SMA) expression and CF proliferation induced by Ang
II (53). To date, the mechanism of
TRPM7-mediated Ca2+ signaling in cardiac fibrosis in HF
has not been reported and requires further investigation.
TRPM7 is involved in hypomagnesemia
during cardiac fibrosis
Mg2+ is the second-most abundant cation
in mammalian cells and an essential cofactor in numerous enzymatic
reactions. Mg2+ influences cell growth processes
associated with remodeling and fibrosis, which are characteristic
features of vascular damage in hypertension, atherosclerosis and
diabetes (71). At the subcellular
level, these effects occur at least partly via
Mg2+-dependent regulation of mitogen-activated protein
kinases, tyrosine kinases and reactive oxygen species, which are
important signaling molecules involved in vascular smooth muscle
cell proliferation, fibrosis and inflammation (72). Human microvascular endothelial cell
growth inhibition by low magnesium is correlated with an increase
in P21 or P27 kip, which are inhibitors of
cyclin-dependent kinase (73).
TRPM7 is a key modulator of Mg2+
homeostasis, whose basal activity is regulated by intracellular
levels of Mg2+ and MgATP (37). The role of TRPM7-mediated
Mg2+ signaling in vascular homeostasis has been widely
reported (74) but is less reported
in the heart, especially in cardiac fibrosis. Recently, researchers
utilized a particular DMEM (Ca2+- and
Mg2+-free DMEM) supplemented with only calcium or only
magnesium. The increase in α-SMA and fibronectin expression induced
by Ang II were abolished without calcium or magnesium, and the same
outcome was observed for CF proliferation, indicating that both
cations are required for the fibrosis response induced by Ang II,
and that neither cation can be omitted (53). However, the association between
TRPM7 and Mg2+ signaling in CFs requires further
investigation.
In TRPM7 kinase-deficient mice, Ang II-induced
cardiac hypertrophy, interstitial fibrosis and left ventricular
dysfunction are amplified and associated with hypomagnesemia,
inhibited TRPM7 kinase expression/signaling and pro-inflammatory
vascular responses (23). Unlike
the pro-cardiac fibrosis role of TRPM7 in mediating Ca2+
signaling, findings from animal models indicated that TRPM7
activation and increased Mg2+ influx protected against
vascular and cardiac fibrosis (75). Hyperaldosteronism is associated with
hypertension, cardiovascular fibrosis and electrolyte disturbances,
including hypomagnesemia (76).
Aldosterone modulates renal TRPM7 expression, which could be
important in altered Mg2+ homeostasis associated with
hyperaldosteronism (76). In a
hyperaldosteronistic mouse model, aldosterone decreased the
expression of renal TRPM7 without affecting TRPM6 and mediated
blood pressure-independent renal and cardiovascular fibrosis and
inflammation via Mg2+-sensitive pathways (4). Mg2+ supplementation
normalizes TRPM7 mRNA expression and attenuates cardiac fibrosis
(4). Therefore, Mg2+
supplementation may have an underlying therapeutic benefit in
aldosterone-associated cardiac fibrosis.
TRPM7 is involved in oxidative stress
during cardiac fibrosis
TRPM7 can be activated by oxidative stress-related
pathologies, such as Alzheimer's disease, anoxia,
ischemia/reperfusion injury and diabetes (24,32,77).
Reactive oxygen species-mediated oxidative stress is also well
known to be the main contributor to cardiac injury and is involved
in cardiac remodeling (78). In
response to pathological stimuli such as oxidative stress and
inflammation, CFs can differentiate into myofibroblasts to initiate
myocardial fibrogenesis (79). In
explanted human hearts with idiopathic dilated cardiomyopathy,
ventricular tachycardia is associated with greater cardiomyocyte
hypertrophy, myocardial fibrosis, oxidative stress and increased
expression of TRPM7(80). Previous
studies have shown that TRPM7 is upregulated in rat models of
myocardial ischemia and reperfusion (81). TRPM7 mRNA levels are downregulated
in left atrial and left ventricular samples from patients with ICM,
and this change has an inverse relationship with ventricular
dysfunction (82). TRPM7 also
contributed to H2O2-induced cardiac fibrosis
by mediating Ca2+ influx and ERK1/2 activation in rat
primary cardiac fibroblasts, and the blockade or silencing of TRPM7
inhibited myocardial fibrogenesis (83). Wu et al (68) found that microRNA (miR)-135a
protects against ISO-induced cardiac fibrosis by downregulating
TRPM7 expression and currents. Astragaloside Ⅳ treatment also
inhibited ISO-induced cardiac fibrosis by targeting the
miR-135a-TRPM7-TGF-β/Smad pathway (84,85).
These results provided a better understanding of the potential
roles of TRPM7 in oxidative stress-induced cardiac fibrosis and
suggested that TRPM7 channels can be regarded as a therapeutic
target.
TRPM7 is involved in neurohormone
activation during cardiac fibrosis
Ang II, a pro fibrogenic cell growth factor, plays a
significant role in the occurrence and development of cardiac
fibrosis (86). The relationship
between TRPM7 and Ang II is complex. Ang II reduced TRPM7 kinase
expression in the heart and aorta of mice (23), whereas it increased TRPM7 expression
in rat ventricular CFs in a concentration-dependent manner
(53). In TRPM7 kinase-deficient
mice, Ang II-induced cardiac hypertrophy, interstitial fibrosis and
left ventricular dysfunction are amplified (23). However, downregulation of TRPM7
attenuated Ang II-induced CF proliferation, differentiation, ECM
production and accumulation (53,87).
The TRPM7 Ca2+ current, as well as the protein
expression levels of TRPM7 and collagen III, initially increases
and then later decreases under optimal Ang II concentrations for
inducing cardiac fibrosis (88).
Therefore, in different stages of Ang II-induced cardiac fibrosis,
the role of TRPM7 may be different.
Similar effects in fibrotic changes in a rat sick
sinus syndrome (SSS) model have been observed (89). The sinoatrial node and atria of SSS
rats exhibit more fibers and higher expression levels of Ang II,
TRPM7 and phosphorylated (p)-Smad2 and produce more collagen than
sham rats (89). TRPM7 small
interfering RNA (siRNA) inhibited Ang II-induced p-Smad2 expression
and collagen synthesis in cardiac fibroblasts in vitro
(89). The considerable inward flow
of Ca2+ activated signaling pathways such as the TGF
β/Smad pathway, causing inflammation and cellular differentiation
and thereby resulting in an acceleration of fibrosis (67). When exposure to Ang II is prolonged,
the induction of cardiac fibroblast apoptosis and necrosis
increases, possibly generating ulterior inflammatory reactions and
fibrogenesis.
TGF-β1 is a profibrotic factor which can stimulate
the proliferation of fibroblasts. TGF-β/Smad signaling is regarded
to be the main pathway leading to tissue fibrosis in a number of
diseases (90-92).
TGF-β1 can promote cardiac fibrosis by phosphorylating downstream
Smad2/3, while activated Smad7 can ameliorate fibrosis by
triggering TGF-β receptor I and Smad protein degradation (93). Studies have found that the TRPM7
channel is also an important downstream target of TGF-β1 induced
liver (92), lung (94) and atrial fibrosis (67). TGF-β1 induced differentiation of
cultured human atrial fibroblasts is correlated with an increase of
TRPM7 expression induced by TGF-β1(67). In addition, inhibition of
TRPM7-mediated Ca2+ influx renders fibroblasts less
sensitive to TGF-β1 induced proliferation and differentiation
(67), indicating that
TRPM7-mediated Ca2+ signal is necessary for TGF-β1
elicited fibrogenesis. Interestingly, latest research confirmed
that TRPM7 gene silencing significantly suppressed the expression
of TGF-β1 and p-smad3, while the expression of Smad7 protein was
increased (84). In addition,
SB431542 (a TGF-β1 blocker) significantly inhibited the expression
of TRPM7 protein and currents in CFs (84). Thus, positive feedback occurs
between enhanced TRPM7 function and TGF-β/Smad pathway activation,
which promotes cardiac fibrosis progression.
5. Modulators for TRPM7
Due to the wide range of physiological and
pathophysiological roles ascribed to TRPM7, reliable drug-like
molecules allowing distinction of the channel versus kinase
activity in situ and under in vivo conditions are
urgently needed.
Several potent inhibitors of the TRPM7 channel have
been identified, including a group of non-specific channel blockers
such as SKF-96365 and 2-APB, natural compounds and metabolites
including waixenicin A, quinine and sphingosine and several
synthetic drug-like compounds (95). In adult rat ventricular CFs, 2-APB
inhibited Ang II-induced Ca2+ influx and the expression
of myocardial connective tissue growth factor, α-SMA and CF
proliferation (53).
Pharmacological targeting of the TRPM7 channel in conjunction with
genetic silencing of the entire TRPM7 protein or the comparative
analysis of effects induced by structurally unrelated TRPM7
modulators were shown to be instrumental in uncovering new cellular
functions of TRPM7 and assessing the therapeutic potential of
anti-TRPM7 drugs (95,96).
The first positive gating modulator of the TRPM7
channel, naltriben, an antagonist of δ-opioid receptors, reversibly
activated the TRPM7 channel without prior depletion of
intracellular Mg2+ and even under conditions of low
PIP2 (96). Naltriben is
the prototype of type 1 activators, allowing induction of the TRPM7
channel independently of [Mg2+]i (95). Moreover, mibefradil is able to
stimulate TRPM7-mediated Ca2+ entry as well as TRPM7
currents with an EC50 of 53 M (97). Mibefradil is a type 2 agonist acting
on the TRPM7 channel in a [Mg2+]i dependent
manner (97). Type 1 agonists
(naltriben) will stimulate Mg2+ and Ca2+
influx irrespective of cytosolic Mg2+ levels, whereas
type 2 agonists (mibefradil) will act preferentially on cells with
reduced intracellular Mg2+ content. Hence, it is worth
investigating whether putative endogenous TRPM7 ligands act in a
similar manner (95).
A set of small organic modulators of TRPM7 have
been identified (95-97).
These new compounds allow for the activation or inhibition of TRPM7
currents or modulate TRPM7 kinase activity (95-97).
The newly identified TRPM7 modulators will undoubtedly be
instrumental in deciphering the cellular functions of TRPM7. Small
molecules that have been identified to date might be regarded as
lead structures for further development of high-affinity in
vivo drugs targeting TRPM7(95).
6. Implications and conclusion
Myocardial fibrosis is a hallmark of cardiac
remodeling and functionally contributes to the development of HF, a
leading cause of death worldwide (98). Clinically, no valid therapeutic
agents target activated cardiac fibroblasts, owing to the
extraordinary etiological heterogeneity and sophisticated
pathogenesis of this disease. Blockade of TGF-β1 signaling through
pharmacological inhibition and magnetic nanoparticle targeted
delivery of TGF-β1 siRNA to ECs can relieve pathological cardiac
fibrosis and hypertrophy in EC-forkhead box P1 deletion mice
(6).
This growing evidence demonstrated that TRPM7 plays
a nonredundant and vital regulatory role in activated cardiac
fibroblasts through its ion channel function and/or kinase activity
(Table I). In addition, the heart
is a very compact organ comprising diverse cell types, and its
pluricellularity offers the opportunity for intercellular
communication within the heart. Fibroblasts create and sustain the
biochemical and mechanical environment of the heart through their
complex interactions with cardiomyocytes. Further investigations
are needed to assess the interaction between TRPM7 in cardiac
fibroblasts and cardiomyocytes or coronary microcirculatory ECs
under different pathological disease models related to cardiac
fibrogenesis.
 | Table IRole of TRPM7 in cardiac
fibrosis. |
Table I
Role of TRPM7 in cardiac
fibrosis.
| | TRPM7
alteration | |
|---|
| Modulators | Expression | Current | Divalent
cations | Experimental
models | Signaling
pathway | Refs. | Authors (year) |
|---|
| None | / | Ito ↓;
If ↓ | Unchanged
myocardial Mg2+ and Zn2+ | TRPM7
deletion mice at an intermediate developmental time point
(approximately embryonic day 11.5 to 13) during mouse
cardiogenesis | TGF-β1, SMAD6,
SMARCE1 and PDGF pathways | (29) | Sah et al
(2013) |
| None | / | / | Hypomagnesemia |
TRPM7+/∆kinase mice;
coculture of cardiac fibroblasts with TRPM7+/∆kinase
macrophages | Calpain-II and
TGF-β1 pathways | (22) | Rios et al
(2020) |
| None | ↑ | ↑ | Ca2+
influx ↑ | Atrial fibroblasts
from patients with AF | TGF-β1 | (68) | Du et al
(2010) |
| Ang II | ↑ | / | / | CFs from rat Ang
II-induced SAN fibrosis tissues | Ang II/TRPM7/Smad
pathways | (90) | Zhong et al
(2018) |
| None | ↑ | / | / | Left ventricular
free wall samples from patients with NIDCM and VT | / | (81) | Parajuli et
al (2015) |
| No | ↓ | / | / | Left ventricular
samples from patients with ICM | / | (83) | Ortega et al
(2016) |
| No | ↑ | / | / | Left ventricular
samples from patients with end-stage HF | / | (70) | Dragún et al
(2019) |
|
H2O2 | ↑ | / | Ca2+
influx ↑ | Neonatal rat
CFs | ERK1/2 pathway | (84) | Guo et al
(2014) |
| Isoproterenol | ↑ | ↑ | / | Neonatal rat
CFs |
miR-135a-TRPM7-TGF-β/Smad pathway | (69), (85), (86) | Wu et al
(2018), Wei et al (2020), Lu et al (2017) |
| Ang II | ↑ | ↑ | Ca2+ and
Mg2+ influx ↑ | Neonatal rat
CFs | / | (54), (85), (86) | Yu et al
(2014), Li et al (2017), Zhou et al (2015) |
| Ang II | / | / | Hypomagnesemia | Ang II-infused
TRPM7+/∆kinase mice | Calpain and
annexin-1 | (23) | Antunes et
al (2016) |
| Aldosterone | Renal ↓ | / | Hypomagnesemia | Aldosterone-infused
mice |
Mg2+-sensitive pathways | (4) | Sontia et al
(2008) |
In conclusion, these in vitro and in
vivo findings suggested that TRPM7 is a potential
pharmacological target for halting the development of fibrotic
cardiac diseases. Future studies will provide more mechanistic
insights into the role of TRPM7 in cardiac fibrosis.
Acknowledgements
Not applicable.
Funding
The project was supported by funding from the
National Natural Science Foundation of China (grant nos. 82000062,
81960015, 81960074 and 81500233), Science and Technology Planning
Project of Jiangxi Province (grant no. 20161ACG70012), Natural
Science Foundation of Jiangxi Province (grant no. 20192BAB205033)
and Jiangxi Province academic and technical leaders Training
Plan-Young Talents Project.
Availability of data and materials
Not applicable.
Authors' contributions
FHu participated in the review design and drafted
the manuscript. ML revised the manuscript critically for important
intellectual content and illustrated the figures. FHa revised the
manuscript critically for important intellectual content. QZ and YZ
contributed to preparation, editing and review of the manuscript.
WZ and XC participated in review design and contributed to quality
control of data and images. XC gave final approval of the version
to be published. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Weber KT, Sun Y and Díez J: Fibrosis: A
living tissue and the infarcted heart. J Am Coll Cardiol.
52:2029–2031. 2008.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Khan R and Sheppard R: Fibrosis in heart
disease: Understanding the role of transforming growth factor-beta
in cardiomyopathy, valvular disease and arrhythmia. Immunology.
118:10–24. 2006.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Klesen A, Jakob D, Emig R, Kohl P, Ravens
U and Peyronnet R: Cardiac fibroblasts: Active players in (atrial)
electrophysiology? Herzschrittmacherther Elektrophysiol. 29:62–69.
2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Sontia B, Montezano AC, Paravicini T,
Tabet F and Touyz RM: Downregulation of renal TRPM7 and increased
inflammation and fibrosis in aldosterone-infused mice: Effects of
magnesium. Hypertension. 51:915–921. 2008.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Leask A: Getting to the heart of the
matter: New insights into cardiac fibrosis. Circ Res.
116:1269–1276. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Liu J, Zhuang T, Pi J, Chen X, Zhang Q, Li
Y, Wang H, Shen Y, Tomlinson B, Chan P, et al: Endothelial Forkhead
Box Transcription Factor P1 Regulates Pathological Cardiac
Remodeling Through Transforming Growth Factor-β1-Endothelin-1
Signal Pathway. Circulation. 140:665–680. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Frangogiannis NG: Fibroblasts and the
extracellular matrix in right ventricular disease. Cardiovasc Res.
113:1453–1464. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Travers JG, Kamal FA, Robbins J, Yutzey KE
and Blaxall BC: Cardiac Fibrosis: The Fibroblast Awakens. Circ Res.
118:1021–1040. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Moore-Morris T, Guimarães-Camboa N,
Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu
Y, Dalton ND, Cedenilla M, et al: Resident fibroblast lineages
mediate pressure overload-induced cardiac fibrosis. J Clin Invest.
124:2921–2934. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Zeisberg EM, Tarnavski O, Zeisberg M,
Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT,
Roberts AB, et al: Endothelial-to-mesenchymal transition
contributes to cardiac fibrosis. Nat Med. 13:952–961.
2007.PubMed/NCBI View
Article : Google Scholar
|
|
11
|
Quijada P, Trembley MA and Small EM: The
Role of the Epicardium During Heart Development and Repair. Circ
Res. 126:377–394. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Simões FC, Cahill TJ, Kenyon A,
Gavriouchkina D, Vieira JM, Sun X, Pezzolla D, Ravaud C, Masmanian
E, Weinberger M, et al: Macrophages directly contribute collagen to
scar formation during zebrafish heart regeneration and mouse heart
repair. Nat Commun. 11(600)2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sundberg C, Ivarsson M, Gerdin B and Rubin
K: Pericytes as collagen-producing cells in excessive dermal
scarring. Lab Invest. 74:452–466. 1996.PubMed/NCBI
|
|
14
|
Samanta A, Hughes TET and Moiseenkova-Bell
VY: Transient Receptor Potential (TRP) Channels. Subcell Biochem.
87:141–165. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Kraft R and Harteneck C: The mammalian
melastatin-related transient receptor potential cation channels: An
overview. Pflugers Arch. 451:204–211. 2005.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Clark K, Middelbeek J, Morrice NA, Figdor
CG, Lasonder E and van Leeuwen FN: Massive autophosphorylation of
the Ser/Thr-rich domain controls protein kinase activity of TRPM6
and TRPM7. PLoS One. 3(e1876)2008.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Li M, Jiang J and Yue L: Functional
characterization of homo- and heteromeric channel kinases TRPM6 and
TRPM7. J Gen Physiol. 127:525–537. 2006.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Zhang Z, Yu H, Huang J, Faouzi M, Schmitz
C, Penner R and Fleig A: The TRPM6 kinase domain determines the
Mg·ATP sensitivity of TRPM7/M6 heteromeric ion channels. J Biol
Chem. 289:5217–5227. 2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Hofmann T, Chubanov V, Gudermann T and
Montell C: TRPM5 is a voltage-modulated and
Ca(2+)-activated monovalent selective cation channel.
Curr Biol. 13:1153–1158. 2003.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Fonfria E, Murdock PR, Cusdin FS, Benham
CD, Kelsell RE and McNulty S: Tissue distribution profiles of the
human TRPM cation channel family. J Recept Signal Transduct Res.
26:159–178. 2006.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Yue Z, Zhang Y, Xie J, Jiang J and Yue L:
Transient receptor potential (TRP) channels and cardiac fibrosis.
Curr Top Med Chem. 13:270–282. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Rios FJ, Zou ZG, Harvey AP, Harvey KY,
Nosalski R, Anyfanti P, Camargo LL, Lacchini S, Ryazanov AG,
Ryazanova L, et al: Chanzyme TRPM7 protects against cardiovascular
inflammation and fibrosis. Cardiovasc Res. 116:721–735.
2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Antunes TT, Callera GE, He Y, Yogi A,
Ryazanov AG, Ryazanova LV, Zhai A, Stewart DJ, Shrier A and Touyz
RM: Transient Receptor Potential Melastatin 7 Cation Channel
Kinase: New Player in Angiotensin II-Induced Hypertension.
Hypertension. 67:763–773. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Clapham DE: TRP channels as cellular
sensors. Nature. 426:517–524. 2003.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Duan J, Li Z, Li J, Hulse RE, Santa-Cruz
A, Valinsky WC, Abiria SA, Krapivinsky G, Zhang J and Clapham DE:
Structure of the mammalian TRPM7, a magnesium channel required
during embryonic development. Proc Natl Acad Sci USA.
115:E8201–E8210. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zou ZG, Rios FJ, Montezano AC and Touyz
RM: TRPM7, Magnesium, and Signaling. Int J Mol Sci.
20(1877)2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Suzuki S, Lis A, Schmitz C, Penner R and
Fleig A: The TRPM7 kinase limits receptor-induced calcium release
by regulating heterotrimeric G-proteins. Cell Mol Life Sci.
75:3069–3078. 2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Nadolni W and Zierler S: The
Channel-Kinase TRPM7 as Novel Regulator of Immune System
Homeostasis. Cells. 7(109)2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Sah R, Mesirca P, Mason X, Gibson W,
Bates-Withers C, Van den Boogert M, Chaudhuri D, Pu WT, Mangoni ME
and Clapham DE: Timing of myocardial trpm7 deletion during
cardiogenesis variably disrupts adult ventricular function,
conduction, and repolarization. Circulation. 128:101–114.
2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Huang Y, Leng TD, Inoue K, Yang T, Liu M,
Horgen FD, Fleig A, Li J and Xiong ZG: TRPM7 channels play a role
in high glucose-induced endoplasmic reticulum stress and neuronal
cell apoptosis. J Biol Chem. 293:14393–14406. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Chiang YF, Chen HY, Lee IT, Chien LS,
Huang JH, Kolisek M, Cheng FC and Tsai SW: Magnesium-responsive
genes are downregulated in diabetic patients after a three-month
exercise program on a bicycle ergometer. J Chin Med Assoc.
82:495–499. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Sun HS: Role of TRPM7 in cerebral
ischaemia and hypoxia. J Physiol. 595:3077–3083. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Chen M, Zhang W, Shi J and Jiang S:
TGF-β1-Induced Airway Smooth Muscle Cell Proliferation Involves
TRPM7-Dependent Calcium Influx via TGFβR/SMAD3. Mol Immunol.
103:173–181. 2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Voringer S, Schreyer L, Nadolni W, Meier
MA, Woerther K, Mittermeier C, Ferioli S, Singer S, Holzer K,
Zierler S, et al: Inhibition of TRPM7 blocks MRTF/SRF-dependent
transcriptional and tumorigenic activity. Oncogene. 39:2328–2344.
2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Yee NS: Role of TRPM7 in Cancer: Potential
as Molecular Biomarker and Therapeutic Target. Pharmaceuticals
(Basel). 10(39)2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lal N, Bhardwaj S, Lalgudi Ganesan S,
Sharma R and Jain P: Case of hypomagnesemia with secondary
hypocalcemia with a novel TRPM6 mutation. Neurol India.
66:1795–1800. 2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Ryazanova LV, Rondon LJ, Zierler S, Hu Z,
Galli J, Yamaguchi TP, Mazur A, Fleig A and Ryazanov AG: TRPM7 is
essential for Mg(2+) homeostasis in mammals. Nat Commun.
1(109)2010.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Wang Y, Chen L, Wang K, Da Y, Zhou M, Yan
H, Zheng D, Zhong S, Cai S, Zhu H, et al: Suppression of TRPM2
reduces renal fibrosis and inflammation through blocking
TGF-β1-regulated JNK activation. Biomed Pharmacother.
120(109556)2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Nadler MJ, Hermosura MC, Inabe K, Perraud
AL, Zhu Q, Stokes AJ, Kurosaki T, Kinet JP, Penner R, Scharenberg
AM, et al: LTRPC7 is a Mg.ATP-regulated divalent cation channel
required for cell viability. Nature. 411:590–595. 2001.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Chokshi R, Matsushita M and Kozak JA:
Sensitivity of TRPM7 channels to Mg2+ characterized in
cellfree patches of Jurkat T lymphocytes. Am J Physiol Cell
Physiol. 302:C1642–C1651. 2012.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Runnels LW, Yue L and Clapham DE: The
TRPM7 channel is inactivated by PIP(2) hydrolysis. Nat Cell Biol.
4:329–336. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
42
|
Deason-Towne F, Perraud AL and Schmitz C:
Identification of Ser/Thr phosphorylation sites in the C2-domain of
phospholipase C γ2 (PLCγ2) using TRPM7-kinase. Cell Signal.
24:2070–2075. 2012.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Abiria SA, Krapivinsky G, Sah R,
Santa-Cruz AG, Chaudhuri D, Zhang J, Adstamongkonkul P, DeCaen PG
and Clapham DE: TRPM7 senses oxidative stress to release
Zn2+ from unique intracellular vesicles. Proc Natl Acad
Sci USA. 114:E6079–E6088. 2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Clark K, Langeslag M, van Leeuwen B, Ran
L, Ryazanov AG, Figdor CG, Moolenaar WH, Jalink K and van Leeuwen
FN: TRPM7, a novel regulator of actomyosin contractility and cell
adhesion. EMBO J. 25:290–301. 2006.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Clark K, Middelbeek J, Dorovkov MV, Figdor
CG, Ryazanov AG, Lasonder E and van Leeuwen FN: The alpha-kinases
TRPM6 and TRPM7, but not eEF-2 kinase, phosphorylate the assembly
domain of myosin IIA, IIB and IIC. FEBS Lett. 582:2993–2997.
2008.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Clark K, Middelbeek J, Lasonder E,
Dulyaninova NG, Morrice NA, Ryazanov AG, Bresnick AR, Figdor CG and
van Leeuwen FN: TRPM7 regulates myosin IIA filament stability and
protein localization by heavy chain phosphorylation. J Mol Biol.
378:790–803. 2008.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Dorovkov MV and Ryazanov AG:
Phosphorylation of annexin I by TRPM7 channel-kinase. J Biol Chem.
279:50643–50646. 2004.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Dorovkov MV, Kostyukova AS and Ryazanov
AG: Phosphorylation of annexin A1 by TRPM7 kinase: A switch
regulating the induction of an α-helix. Biochemistry. 50:2187–2193.
2011.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Krapivinsky G, Krapivinsky L, Manasian Y
and Clapham DE: The TRPM7 chanzyme is cleaved to release a
chromatin-modifying kinase. Cell. 157:1061–1072. 2014.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Ryazanova LV, Dorovkov MV, Ansari A and
Ryazanov AG: Characterization of the protein kinase activity of
TRPM7/ChaK1, a protein kinase fused to the transient receptor
potential ion channel. J Biol Chem. 279:3708–3716. 2004.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Matsushita M, Kozak JA, Shimizu Y,
McLachlin DT, Yamaguchi H, Wei FY, Tomizawa K, Matsui H, Chait BT,
Cahalan MD, et al: Channel function is dissociated from the
intrinsic kinase activity and autophosphorylation of TRPM7/ChaK1. J
Biol Chem. 280:20793–20803. 2005.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Desai BN, Krapivinsky G, Navarro B,
Krapivinsky L, Carter BC, Febvay S, Delling M, Penumaka A, Ramsey
IS, Manasian Y, et al: Cleavage of TRPM7 releases the kinase domain
from the ion channel and regulates its participation in Fas-induced
apoptosis. Dev Cell. 22:1149–1162. 2012.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Yu Y, Chen S, Xiao C, Jia Y, Guo J, Jiang
J and Liu P: TRPM7 is involved in angiotensin II induced cardiac
fibrosis development by mediating calcium and magnesium influx.
Cell Calcium. 55:252–260. 2014.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Song C, Bae Y, Jun J, Lee H, Kim ND, Lee
KB, Hur W, Park JY and Sim T: Identification of TG100-115 as a new
and potent TRPM7 kinase inhibitor, which suppresses breast cancer
cell migration and invasion. Biochim Biophys Acta Gen Subj.
1861:947–957. 2017.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Yang CW, Liu H, Li XD, Sui SG and Liu YF:
Salvianolic acid B protects against acute lung injury by decreasing
TRPM6 and TRPM7 expressions in a rat model of sepsis. J Cell
Biochem. 119:701–711. 2018.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Liu A, Wu J, Yang C, Wu Y, Zhang Y, Zhao
F, Wang H, Yuan L, Song L, Zhu T, et al: TRPM7 in CHBP-induced
renoprotection upon ischemia reperfusion-related injury. Sci Rep.
8(5510)2018.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Al Hattab D and Czubryt MP: A primer on
current progress in cardiac fibrosis. Can J Physiol Pharmacol.
95:1091–1099. 2017.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Jalife J and Kaur K: Atrial remodeling,
fibrosis, and atrial fibrillation. Trends Cardiovasc Med.
25:475–484. 2015.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Schirone L, Forte M, Palmerio S, Yee D,
Nocella C, Angelini F, Pagano F, Schiavon S, Bordin A, Carrizzo A,
et al: A Review of the Molecular Mechanisms Underlying the
Development and Progression of Cardiac Remodeling. Oxid Med Cell
Longev. 2017(3920195)2017.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Yue L, Xie J and Nattel S: Molecular
determinants of cardiac fibroblast electrical function and
therapeutic implications for atrial fibrillation. Cardiovasc Res.
89:744–753. 2011.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Zhang YH, Sun HY, Chen KH, Du XL, Liu B,
Cheng LC, Li X, Jin MW and Li GR: Evidence for functional
expression of TRPM7 channels in human atrial myocytes. Basic Res
Cardiol. 107(282)2012.PubMed/NCBI View Article : Google Scholar
|
|
62
|
González A, López B and Díez J: Fibrosis
in hypertensive heart disease: Role of the
renin-angiotensin-aldosterone system. Med Clin North Am. 88:83–97.
2004.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Olson ER, Shamhart PE, Naugle JE and
Meszaros JG: Angiotensin II-induced extracellular signal-regulated
kinase 1/2 activation is mediated by protein kinase Cdelta and
intracellular calcium in adult rat cardiac fibroblasts.
Hypertension. 51:704–711. 2008.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Manabe I, Shindo T and Nagai R: Gene
expression in fibroblasts and fibrosis: Involvement in cardiac
hypertrophy. Circ Res. 91:1103–1113. 2002.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Parekh AB: Calcium signalling in health
and disease. Semin Cell Dev Biol. 94:1–2. 2019.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Fan D, Takawale A, Lee J and Kassiri Z:
Cardiac fibroblasts, fibrosis and extracellular matrix remodeling
in heart disease. Fibrogenesis Tissue Repair. 5(15)2012.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Du J, Xie J, Zhang Z, Tsujikawa H, Fusco
D, Silverman D, Liang B and Yue L: TRPM7-mediated Ca2+
signals confer fibrogenesis in human atrial fibrillation. Circ Res.
106:992–1003. 2010.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Wu Y, Liu Y, Pan Y, Lu C, Xu H, Wang X,
Liu T, Feng K and Tang Y: MicroRNA-135a inhibits cardiac fibrosis
induced by isoproterenol via TRPM7 channel. Biomed Pharmacother.
104:252–260. 2018.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Dragún M, Gažová A, Kyselovič J, Hulman M
and Máťuš M: TRP Channels Expression Profile in Human End-Stage
Heart Failure. Medicina (Kaunas). 55(380)2019.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Morgan JP, Erny RE, Allen PD, Grossman W
and Gwathmey JK: Abnormal intracellular calcium handling, a major
cause of systolic and diastolic dysfunction in ventricular
myocardium from patients with heart failure. Circulation.
81:121–132. 1990.PubMed/NCBI
|
|
71
|
Altura BM, Kostellow AB, Zhang A, Li W,
Morrill GA, Gupta RK and Altura BT: Expression of the nuclear
factor-kappaB and proto-oncogenes c-fos and c-jun are induced by
low extracellular Mg2+ in aortic and cerebral vascular
smooth muscle cells: Possible links to hypertension, atherogenesis,
and stroke. Am J Hypertens. 16:701–707. 2003.
|
|
72
|
Touyz RM and Yao G: Up-regulation of
vascular and renal mitogen-activated protein kinases in
hypertensive rats is normalized by inhibitors of the
Na+/Mg2+ exchanger. Clin Sci (Lond).
105:235–242. 2003.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Baldoli E and Maier JA: Silencing TRPM7
mimics the effects of magnesium deficiency in human microvascular
endothelial cells. Angiogenesis. 15:47–57. 2012.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Bates-Withers C and Sah RandClapham DE:
TRPM7, the Mg(2+) inhibited channel and kinase. Adv Exp
Med Biol. 704:173–183. 2011.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Montezano AC, Zimmerman D, Yusuf H, Burger
D, Chignalia AZ, Wadhera V, van Leeuwen FN and Touyz RM: Vascular
smooth muscle cell differentiation to an osteogenic phenotype
involves TRPM7 modulation by magnesium. Hypertension. 56:453–462.
2010.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Schiffrin EL and Touyz RM: Calcium,
magnesium, and oxidative stress in hyperaldosteronism. Circulation.
111:830–831. 2005.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Miller BA and Zhang W: TRP channels as
mediators of oxidative stress. Adv Exp Med Biol. 704:531–544.
2011.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Sun Y: Myocardial repair/remodelling
following infarction: Roles of local factors. Cardiovasc Res.
81:482–490. 2009.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Swynghedauw B: Molecular mechanisms of
myocardial remodeling. Physiol Rev. 79:215–262. 1999.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Parajuli N, Valtuille L, Basu R, Famulski
KS, Halloran PF, Sergi C and Oudit GY: Determinants of ventricular
arrhythmias in human explanted hearts with dilated cardiomyopathy.
Eur J Clin Invest. 45:1286–1296. 2015.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Demir T, Yumrutas O, Cengiz B, Demiryurek
S, Unverdi H, Kaplan DS, Bayraktar R, Ozkul N and Bagcı C:
Evaluation of TRPM (transient receptor potential melastatin) genes
expressions in myocardial ischemia and reperfusion. Mol Biol Rep.
41:2845–2849. 2014.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Ortega A, Roselló-Lletí E, Tarazón E,
Gil-Cayuela C, Lago F, González-Juanatey JR, Martinez-Dolz L,
Portolés M and Rivera M: TRPM7 is down-regulated in both left atria
and left ventricle of ischaemic cardiomyopathy patients and highly
related to changes in ventricular function. ESC Heart Fail.
3:220–224. 2016.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Guo JL, Yu Y, Jia YY, Ma YZ, Zhang BY, Liu
PQ, Chen SR and Jiang JM: Transient receptor potential melastatin 7
(TRPM7) contributes to H2O2-induced cardiac
fibrosis via mediating Ca(2+) influx and extracellular
signal-regulated kinase 1/2 (ERK1/2) activation in cardiac
fibroblasts. J Pharmacol Sci. 125:184–192. 2014.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Wei Y, Wu Y, Feng K, Zhao Y, Tao R, Xu H
and Tang Y: Astragaloside IV inhibits cardiac fibrosis via
miR-135a-TRPM7-TGF-β/Smads pathway. J Ethnopharmacol.
249(112404)2020.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Lu J, Wang QY, Zhou Y, Lu XC, Liu YH, Wu
Y, Guo Q, Ma YT and Tang YQ: AstragalosideIV against cardiac
fibrosis by inhibiting TRPM7 channel. Phytomedicine. 30:10–17.
2017.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Olson ER, Shamhart PE, Naugle JE and
Meszaros JG: Angiotensin II-induced extracellular signal-regulated
kinase 1/2 activation is mediated by protein kinase Cdelta and
intracellular calcium in adult rat cardiac fibroblasts.
Hypertension. 51:704–711. 2008.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Li S, Li M, Yi X, Guo F, Zhou Y, Chen S
and Wu X: TRPM7 channels mediate the functional changes in cardiac
fibroblasts induced by angiotensin II. Int J Mol Med. 39:1291–1298.
2017.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Zhou Y, Yi X, Wang T and Li M: Effects of
angiotensin II on transient receptor potential melastatin 7 channel
function in cardiac fibroblasts. Exp Ther Med. 9:2008–2012.
2015.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Zhong H, Wang T, Lian G, Xu C, Wang H and
Xie L: TRPM7 regulates angiotensin II-induced sinoatrial node
fibrosis in sick sinus syndrome rats by mediating Smad signaling.
Heart Vessels. 33:1094–1105. 2018.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Xu F, Liu C, Zhou D and Zhang L:
TGF-β/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J
Histochem Cytochem. 64:157–167. 2016.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Liu KH, Zhou N, Zou Y, Yang YY, OuYang SX
and Liang YM: Spleen Tyrosine Kinase (SYK) in the Progression of
Peritoneal Fibrosis Through Activation of the TGF-β1/Smad3
Signaling Pathway. Med Sci Monit. 25:9346–9356. 2019.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Penke LR and Peters-Golden M: Molecular
determinants of mesenchymal cell activation in fibroproliferative
diseases. Cell Mol Life Sci. 76:4179–4201. 2019.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Overstreet JM, Samarakoon R, Meldrum KK
and Higgins PJ: Redox control of p53 in the transcriptional
regulation of TGF-β1 target genes through SMAD cooperativity. Cell
Signal. 26:1427–1436. 2014.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Yu M, Huang C, Huang Y, Wu X, Li X and Li
J: Inhibition of TRPM7 channels prevents proliferation and
differentiation of human lung fibroblasts. Inflamm Res. 62:961–970.
2013.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Chubanov V, Ferioli S and Gudermann T:
Assessment of TRPM7 functions by drug-like small molecules. Cell
Calcium. 67:166–173. 2017.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Hofmann T, Schäfer S, Linseisen M, Sytik
L, Gudermann T and Chubanov V: Activation of TRPM7 channels by
small molecules under physiological conditions. Pflugers Arch.
466:2177–2189. 2014.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Schäfer S, Ferioli S, Hofmann T, Zierler
S, Gudermann T and Chubanov V: Mibefradil represents a new class of
benzimidazole TRPM7 channel agonists. Pflugers Arch. 468:623–634.
2016.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Tanai E and Frantz S: Pathophysiology of
Heart Failure. Compr Physiol. 6:187–214. 2015.PubMed/NCBI View Article : Google Scholar
|