Introduction
Breast cancer is one of the most common malignant
tumours in female patients, with an annually increasing trend. In
addition, breast cancer is a heterogeneous disease with multiple
subtypes; triple-negative breast cancer (TNBC) accounts for 15-20%
of cases, worldwide (1). The
typical characteristics of TNBC are the absence of oestrogen
receptor (ER), progesterone receptor and human epidermal growth
factor receptor 2 expression (1).
TNBC is highly invasive, has a poor clinical prognosis and rapidly
recurs (2,3). Consequently, it is necessary to
further explore the biological characteristics of TNBC and then
identify novel, effective and safe antitumor drugs to improve the
survival rate of patients.
At present, the treatment for TNBC is still
radiotherapy and chemotherapy, which is due to the lack of reliable
specific targets to develop targeted drugs (2,4). The
reprogramming of energy metabolism can be used as a sign of the
physiology of several cancer types, including TNBC (5). In normal cells, most pyruvate enters
mitochondria and is oxidized through the tricarboxylic acid cycle
to produce adenosine triphosphate and meet the energy needs of the
cell. However, in cancer cells, most pyruvate is reduced to lactic
acid by lactate dehydrogenase rather than entering the
mitochondria. This process is called ‘aerobic glycolysis’ and is
referred to as the ‘Warburg effect’ (6). Like most cancer cells, breast cancer
cells also have abnormal glucose metabolism with high glucose
absorption and glycolysis rates (7).
Glucose transporters (GLUTs) are transmembrane
transporters that are necessary for the entry of glucose into cells
(8). Fourteen types of GLUTs are
expressed in humans. GLUT1, GLUT2, GLUT3, GLUT4, GLUT5 and GLUT12
have been successively identified in breast cancer (9-12).
As the most invasive breast cancer subtype, TNBC exhibits higher
levels of GLUT1 compared with other subtypes of breast cancer;
however, diclofenac can significantly decrease GLUT1 expression and
glucose uptake (13). In addition,
c-Myc is a driving factor of glucose uptake and aerobic glycolysis
(14,15). The most recent research shows that
diclofenac can control glycolysis in melanoma cells by inhibiting
c-Myc, downregulating the expression of GLUT1 and suppressing
glucose metabolism (16). In
addition, hexokinase (HK) participates in the first step of
glycolysis as the key rate-limiting enzyme. Therefore, the decrease
in HK expression and activity can inhibit glycolysis. Studies have
shown that HK is also a target of c-Myc (17,18).
Recently, a team found that compared with other
types of breast cancer cells, TNBC cells have a unique molecular
mechanism; the high levels of c-Myc and low levels of TXNIP in TNBC
can promote cancer cell proliferation. High c-Myc/low TXNIP gene
expression is associated with a lower overall survival and
metastasis-free survival rates of patients with TNBC. Furthermore,
c-Myc and TXNIP can compete with each other; c-Myc promotes glucose
uptake and its use in tumor cells to maintain proliferation, while
TXNIP does the opposite (2). Based
on these results, diclofenac is expected to inhibit c-Myc
transcription and downregulate GLUT1 expression, subsequently
suppressing glycolysis and inducing apoptosis in TNBC cells.
To verify this hypothesis, the present study
investigated the effects of diclofenac on aerobic glycolysis in
TNBC by establishing cell models with human TNBC cell lines
(MDA-MB-231 and HCC1937) and a non-TNBC cell line (MCF-7). After
the cells were treated with diclofenac for 24 and 48 h, the effects
of diclofenac were evaluated, including alterations in cell
proliferation and apoptosis, using Cell Counting Kit-8 (CCK-8) and
flow cytometric assays. In addition, the expression levels of GLUT1
and c-Myc were analysed by western blotting to further elucidate
the underlying mechanism by which diclofenac inhibits TNBC cell
proliferation and induces TNBC cell apoptosis.
Materials and methods
Cells and cell culture
TNBC cell lines (MDA-MB-231 and HCC1937) and a
non-TNBC cell line (MCF-7) were purchased from the The Cell Bank of
Type Culture Collection of The Chinese Academy of Sciences
(Shanghai, China). The cryovials containing the frozen cells were
removed from liquid nitrogen storage and immediately placed into a
37˚C water bath. Then, complete growth medium, consisting of DMEM
(Thermo Fisher Scientific, Inc.) supplemented with 10% (v/v) fetal
bovine serum (Biological Industries Israel Beit Haemek Ltd.) and 1%
(v/v) penicillin-streptomycin (Beijing Solarbio Science &
Technology Co., Ltd.), was added to resuspend the cells, and the
suspension was centrifuged at ~300 x g for 5-10 min at 37˚C. The
supernatant was decanted, and the cells were gently resuspended in
5 ml of complete growth medium. Then, the cells were transferred
into a culture flask and incubated at 37˚C in 5%
CO2.
Assessment of cell proliferation
The concentration of the three cell suspensions was
adjusted to 5x104 cells/ml, and 100 µl of each
suspension was added to 96-well plates and cultured in an incubator
at 37˚C and 5% CO2. Cells were treated with diclofenac
(Sigma-Aldrich; Merck KGaA) at the concentrations of 0, 0.2, 0.4,
0.8 mM for 24, 48, 72, 96, 120 h. Then, cells were incubated at
37˚C and 5% CO2 for another 2 h, following the addition
of CCK-8 (Beyotime Institute of Biotechnology) at 5 time points.
After brief shaking, the absorbance values at 450 nm were
immediately measured using a microplate reader.
Assessment of apoptosis
The three cell lines were treated with 0, 0.2, 0.4
and 0.8 mM diclofenac for 24 and 48 h and were then stained with 5
µl of Annexin-V-Fluorescein Isothiocyanate (Annexin-V-FITC) and 10
µl of Propidium Iodide (PI) (both obtained from BD Biosciences)
according to the manufacturer's instructions. The number of sample
cells was 1x105. Before the assay, three groups of
negative control samples, including blank, Annexin V-FITC-stained
and PI-stained, were analysed; this was repeated 3 times. Flow
cytometric analyses were performed with a Fluorescence activated
Cell Sorting (FACS) Calibur (BD Biosciences) using BD Cell Quest
Pro 5.1 software for data acquisition and analysis.
Measurement of HK activity
Cells in logarithmic growth phase were inoculated in
6-well plates at 2.5x106 cells/well and were then
incubated overnight with diclofenac (0, 0.2, 0.4 and 0.8 mM) at
37˚C in 5% CO2. For the detection of HK activity, each
group was cultured for both 24 and 48 h. Detection was performed
with a Hexokinase Activity Detection kit (Beijing Solarbio Science
& Technology Co., Ltd.) according to the manufacturer's
instructions. The reagents were mixed in proportional amounts into
the solution and preheated at 37˚C for 10 min. The absorbance
values at 340 nm at 20 sec (A1) after sample addition and 5 min
after water bath immersion (A2) were measured in a
spectrophotometer (Thermo Fisher Scientific, Inc.). These data were
input into the formula HK(U/104 cell)=[ΔAxV
total/(εxd)x109]÷(500xV sample/V sample
total)/T=2.572xΔA (ΔA=A2-A1; V Total, total volume of reaction
system, 2x10-4 l; ε, NADPH molar extinction coefficient,
6.22x103 l/mol/cm; d, 96-well plate optical path, 0.5
cm; V sample, sample volume 0.01 ml; V sample total, extract volume
1 ml; T, reaction time 5 min) to calculate HK activity. U is
defined as 1 nmol of NADPH produced per minute per 10,000 bacteria
or cells, and is considered to be an enzyme activity unit.
Western blot analysis
The three cell lines were treated with 0, 0.4 and
0.8 mM diclofenac for 24 and 48 h, washed twice with cold phosphate
buffer saline (PBS) after collection, and lysed with radio
immunoprecipitation assay (RIPA) buffer (Beyotime Institute of
Biotechnology). Then, the lysates were centrifuged at 12,000 x g
and 4˚C for 30 min. The supernatant was collected, and sodium
dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE)
sample loading buffer (Beyotime Institute of Biotechnology) was
added at a buffer:lysate ratio of 1:4, and proteins were denatured
in a thermal cycler at 100˚C for 10 min. The protein concentration
was determined using a BCA Protein Assay kit (cat. no. 23227;
Thermo Fisher Scientific, Inc.). The proteins (30 µg per lane) were
separated on a denaturing 12% SDS-PAGE gel and transferred to a
polyvinylidene fluoride (PVDF) membrane for western blotting. The
membrane was sequentially probed with antibodies against GLUT1
(1:1,000; cat. no. 12939; Cell Signaling Technologies, Inc.), MYC
(1:1,000; cat. no. 9402; Cell Signaling Technologies, Inc.), and
β-actin (1:1,000; cat. no. 3700; Cell Signaling Technologies, Inc.)
diluted by Primary Antibody Dilution buffer (Beyotime Institute of
Biotechnology). After incubation with the primary antibodies, the
membrane was washed 3 times for 5 min by tris-buffered saline with
0.1% tween20 (TBST) and was then incubated with the secondary
antibody (1:15,000; IRDye® 800CW Goat anti-Rabbit IgG
Secondary Antibody cat. no. 926-32211; IRDye® 800CW Goat
anti-Mouse IgG Secondary Antibody cat. no. 926-32210; LI-COR
Biosciences) for 2 h. Protein bands were visualized using Odyssey
Infrared Imaging System (LI-COR Biosciences). Finally, the
greyscale values of the protein bands were determined by Image
Studio Lite 5.2.5 (LI-COR Biosciences).
Statistical analysis
All results are presented as the means ± standard
deviations and were analysed with SPSS 17.0 statistical software
(SPSS, Inc.). Statistical analysis was performed with one-way ANOVA
with post hoc contrasts by Bonferroni's test. P≤0.05 was considered
to indicate a statistically significant difference. The experiments
were performed in triplicate.
Results
Diclofenac inhibits breast cancer cell
proliferation in vitro
The addition of diclofenac, a member of the
arylacetic acid group of non-steroidal anti-inflammatory drugs
(NSAIDs), at clinically relevant concentrations (see http://www.drugs.com/pro/diclofenac.html) led to
significant effects on TNBC cell lines starting at concentrations
as low as 0.2 mM.
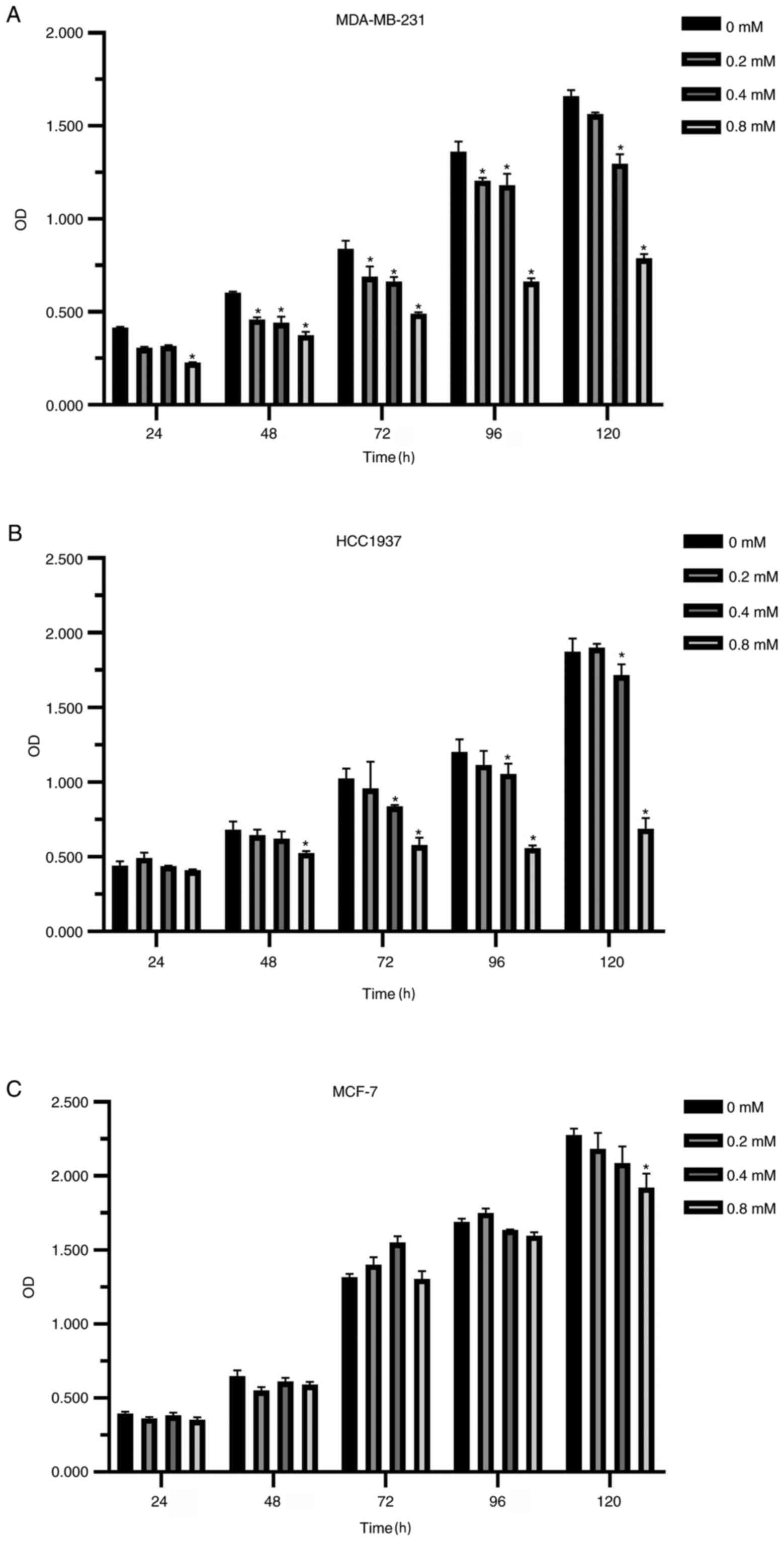
Compared with the non-medicated control group, the
proliferation of breast cancer cells was inhibited after diclofenac
was added for 24 h. The higher the concentration and the longer the
treatment time, the more notable the inhibitory effect was
(Fig. 1A-C). Interestingly, the
inhibitory effect of diclofenac on the TNBC cell lines (MDA-MB-231
and HCC1937) was stronger compared with that of the non-TNBC cell
line MCF-7 (Fig. 1A and B).
Diclofenac induces TNBC cell apoptosis
in vitro
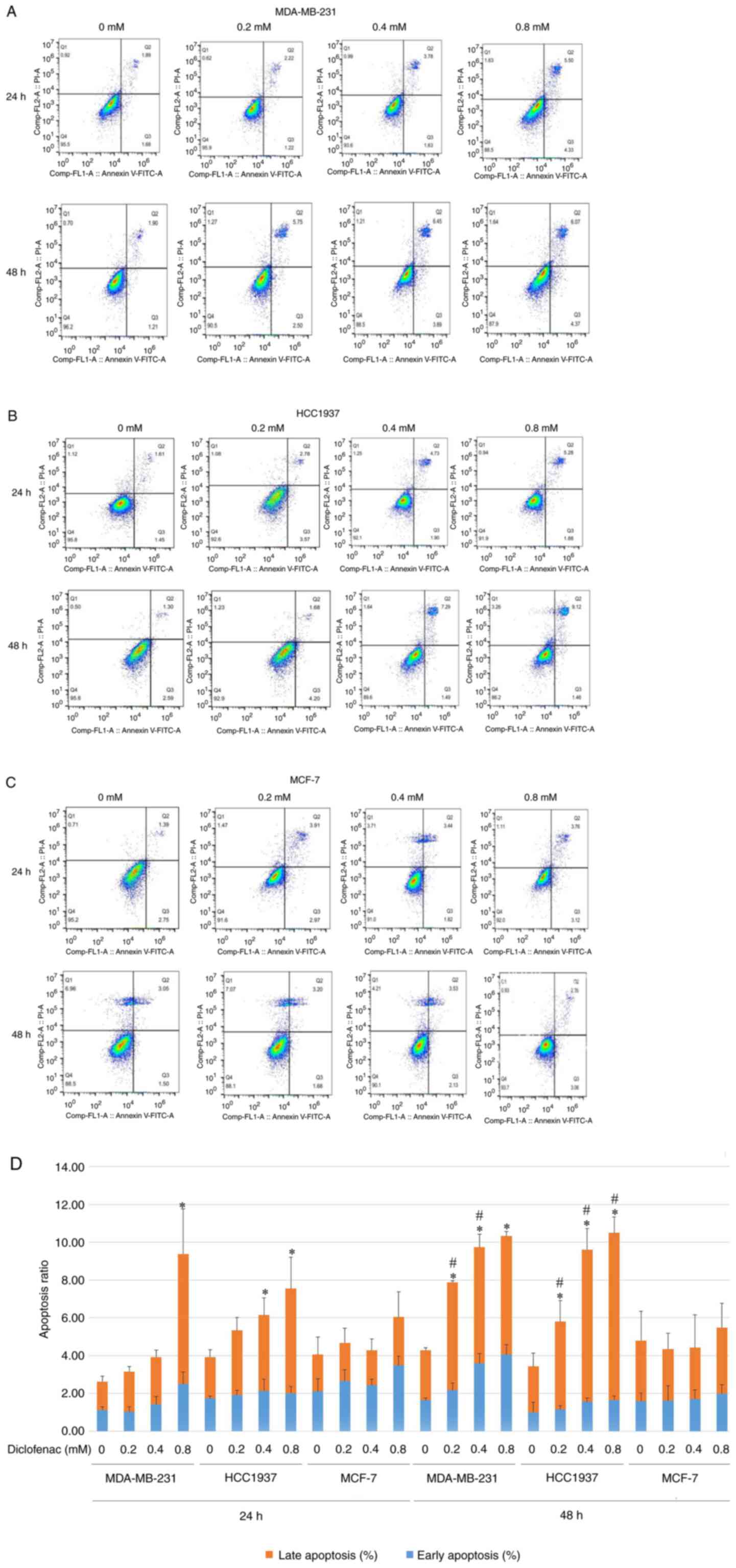
The TNBC cell lines (MDA-MB-231 and HCC1937) and the
non-TNBC cell line (MCF-7) were treated with diclofenac at
concentrations of 0.2, 0.4 and 0.8 mM for 24 and 48 h, and
apoptosis was then assessed by flow cytometry. Compared with that
of the corresponding untreated control cells, the apoptosis rate of
the TNBC cell lines (MDA-MB-231 and HCC1937) was significantly
increased. The increase in the apoptosis rate was most pronounced
in the diclofenac-treated group at 0.4 and 0.8 mM (Fig. 2A and B). Cells treated with the same drug
concentration for 48 h exhibited higher apoptosis rates than the
corresponding cells treated for 24 h (Fig. 2D). The results in Fig. 2A, B
and D show that diclofenac induced
apoptosis in the TNBC cell lines (MDA-MB-231 and HCC1937) in a
dose- and time-dependent manner. However, in the non-TNBC cell line
(MCF-7), diclofenac did not show similar results (Fig. 2C).
Diclofenac inhibits HK activity in
TNBC cells
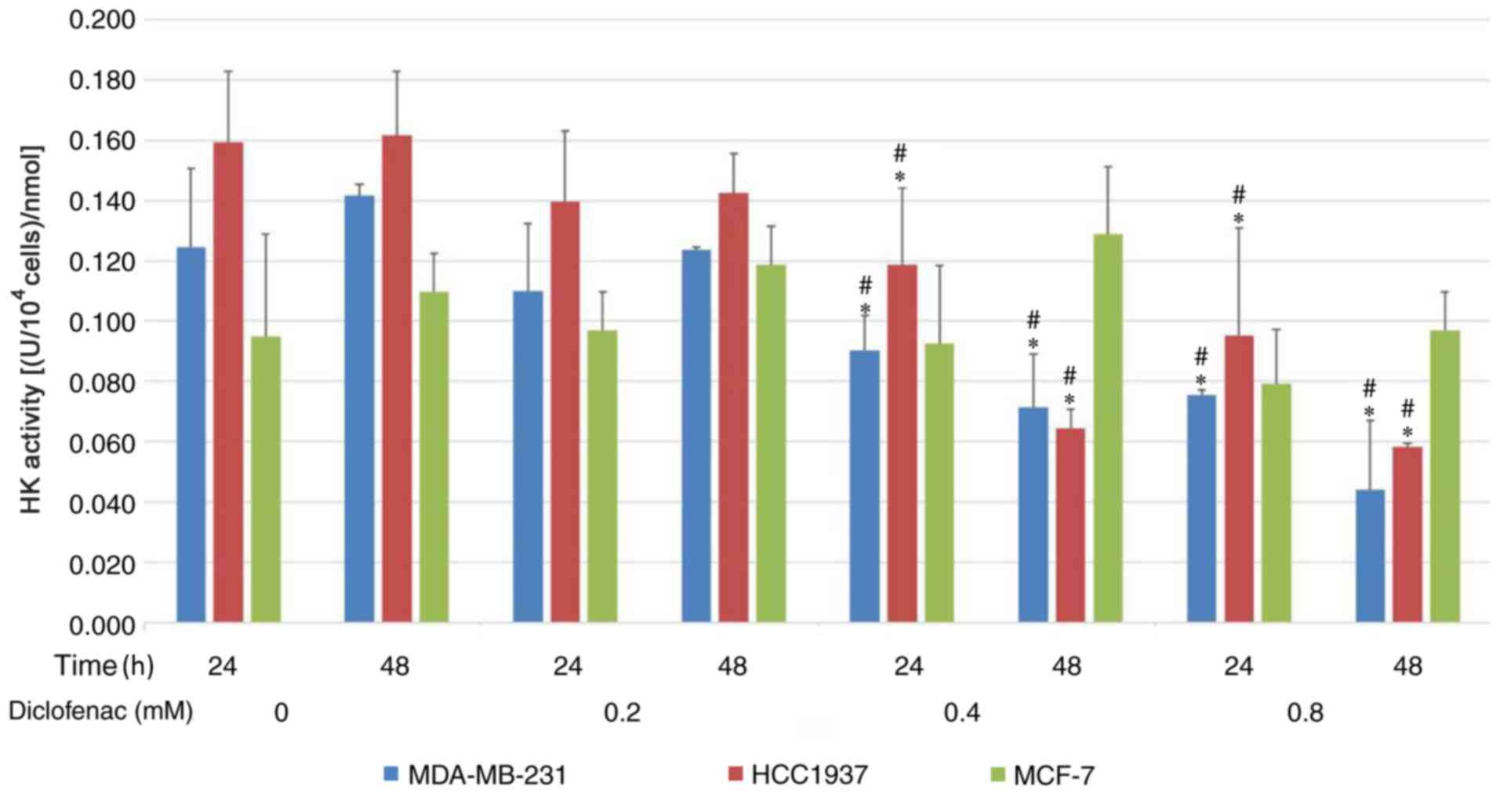
To investigate the effect of diclofenac on
glycolytic metabolism, the activity of HK was assessed. As shown in
Fig. 3, after incubation with
diclofenac for 24 and 48 h, the activity of HK was downregulated in
a dose-dependent manner in the TNBC cell lines (MDA-MB-231 and
HCC1937). HK activity was most significantly downregulated under
treatments with 0.4 and 0.8 mM diclofenac. However, no difference
was observed in HK activity in the non-TNBC cell line (MCF-7)
groups compared with the corresponding control groups.
Effect of diclofenac on the protein
expression levels of c-Myc and GLUT1
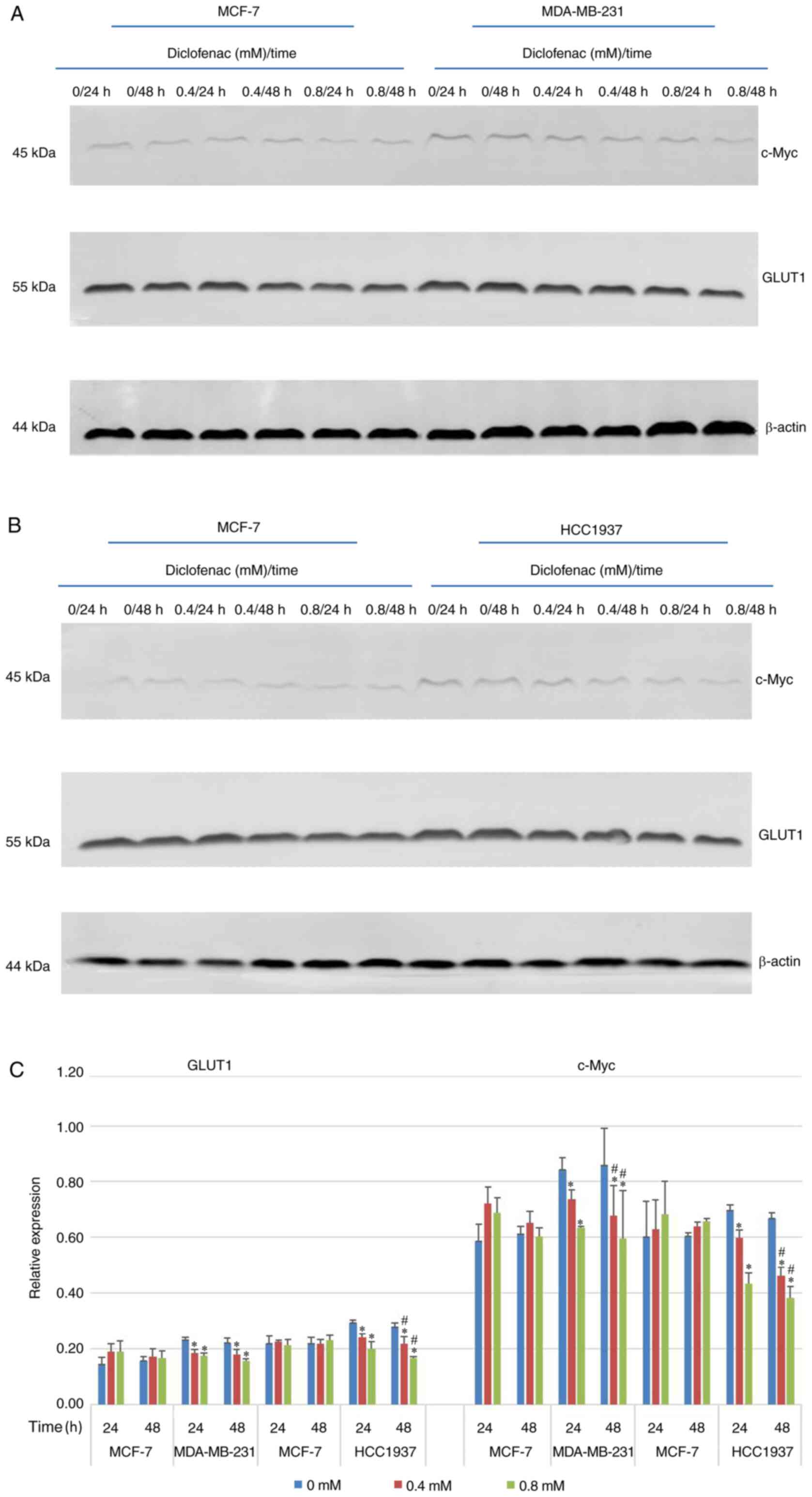
To study the effect of diclofenac on the relative
expression levels of GLUT1 and c-Myc, TNBC cells (MDA-MB-231 and
HCC1937) and non-TNBC cells (MCF-7) were exposed to diclofenac (0,
0.4 and 0.8 mM) for 24 and 48 h, and the protein expression levels
of GLUT1 and c-Myc were measured by western blotting. As shown in
Fig. 4, after incubation with
diclofenac for 24 and 48 h, the protein expression levels of both
c-Myc and GLUT1 were decreased in a dose- and time-dependent manner
in the TNBC cell lines (MDA-MB-231 and HCC1937). The protein
expression levels of c-Myc and GLUT1 were most significantly
decreased under treatment with 0.4 and 0.8 mM diclofenac. In the
non-TNBC cell line (MCF-7) groups, although the protein expression
levels of c-Myc and GLUT1 were decreased in a time-dependent
manner, the differences were not significant.
Discussion
TNBC is a unique subtype of breast cancer with a
5-year survival rate of <80%, and most commonly occurs in young
premenopausal patients. TNBC is highly invasive and has a higher
incidence of distant metastases compared with non-TNBC subtypes
(19); thus, its prognosis is poor,
but targeted therapies are lacking. Compared with ER-positive
cells, TNBC cells possess unique metabolic characteristics: High
glucose uptake, increased lactate production, and low mitochondrial
respiration levels (20). These
characteristics suggest that suppressing breast cancer cell
proliferation by inhibiting the glycolytic pathway may be a novel
therapeutic avenue for antitumor drugs.
NSAIDs have potent anticancer effects. Recent
studies have attributed this effect of NSAIDs mainly to their role
as COX inhibitors. Diclofenac is an old classic NSAID, which is
mainly used for antipyretic, analgesic and anti-inflammatory
purposes in the clinic. The main side effects include damage of
gastrointestinal mucosa, myocardial infarction ad hypertension
(21,22). Recent studies showed that diclofenac
can inhibit the cell proliferation by targeting Myc and lactate
transport, but the non-selective COX inhibitor aspirin did not have
this effect (16).
In the present study, TNBC cell lines (MDA-MB-231
and HCC1937) and a non-TNBC cell line (MCF-7) were treated with
different concentrations of diclofenac and assessed their
proliferation and apoptosis. Diclofenac significantly inhibited the
proliferation of TNBC cells (Fig.
1A and B) and promoted
apoptosis. Interestingly, the effect of diclofenac on the
proliferation and apoptosis of non-TNBC cells was not obvious,
suggesting that diclofenac may inhibit the proliferation and
promote the apoptosis of TNBC cells through a unique mechanism.
Studies have demonstrated that glucose transport
requires the participation of GLUT1 and that glucose molecules rely
mainly on GLUT1 to cross the lipid bilayer of the cell membrane
along a concentration gradient (8,23).
GLUT1 is very important in cancer-specific metabolism (24). GLUT1 is highly expressed in several
malignancies and plays an important role in mediating the Warburg
effect in these malignancies (25,26).
As the most aggressive breast cancer, TNBC exhibits a higher GLUT1
expression level compared with other subtypes (13). The present study demonstrated that
the protein expression levels of GLUT1 in TNBC cells were
significantly higher compared with those in non-TNBC cells and that
diclofenac downregulated GLUT1 protein expression. Moreover, this
downregulating effect was stronger in TNBC cells compared with
non-TNBC cells. HK is a key rate-limiting enzyme in the first step
of glycolysis in tumor tissues, and its expression and activity
increase significantly in tumor tissues to ensure a sufficient
energy supply, even under anaerobic conditions. The expression of
HK is upregulated in most malignant tumors because most malignant
tumors prefer aerobic glycolysis. A recent study demonstrated that
the expression of HK was higher in MDA-MB-231 cells compared with
MCF-7 cells (27). The present
study demonstrated that diclofenac can decrease the activity of HK
and showed that the activity of HK in tumor cells was decreased in
a dose-dependent manner with increasing doses of diclofenac.
However, this effect was not seen in non-TNBC cells. Therefore, it
was concluded that diclofenac inhibits the proliferation of TNBC
cells by inhibiting glycolytic enzymes such as GLUT1 and HK.
c-Myc is an important member of the Myc family. The
c-Myc gene can promote cell division and acts as a ‘switch’ that
determines the entry of cells into S phase from
G0/G1 phase. The protein encoded by c-Myc is
closely associated with cell proliferation (28). The c-Myc oncogene is deregulated in
>50% of human cancer types, and this deregulation is frequently
associated with poor prognosis and unfavourable patient survival
outcomes (29). c-Myc has a central
role in almost every aspect of the oncogenic process, orchestrating
proliferation, apoptosis, differentiation and metabolism (29). As the core of the glycolytic
metabolism of cancer cells, c-Myc controls the metabolism of cancer
cells through a variety of ways. For example, in recent reports,
AMPK factor can regulate c-Myc bidirectionally, AMPK pathway
positively regulates the expression of oncogene c-Myc to promote
cancer cell apoptosis (30), and
AMPK can also reversely regulate c-Myc to promote cancer autophagy
(31). Furthermore, AMPK is
considered to be a factor associated with glucose-mediated cancer
progression (32-34).
Recent studies have shown that the molecular mechanism of TNBC
cells is unique compared with that of other types of breast cancer
cells: TNBC cells have higher expression of c-Myc and lower
expression of TXNIP, and c-Myc can promote glucose uptake and its
use in tumor cells, which in turn accelerates cancer cell
proliferation (2). In addition,
other studies have shown that several pathways of cell metabolism
are regulated by c-Myc and that the key enzymes in glucose
metabolism, such as GLUT1 and HK, are targets of c-Myc (35-38).
In the present study, TNBC cell lines (MDA-MB-231 and HCC1937) and
a non-TNBC cell line (MCF-7) were treated with diclofenac at
different concentrations and measured c-Myc protein expression in
these cells by western blotting. The protein expression level of
c-Myc in TNBC cells was significantly higher compared with that in
non-TNBC cells. Moreover, diclofenac decreased the protein
expression level of c-Myc, and this effect was stronger in TNBC
cells compared with non-TNBC cells.
Glycolysis plays a central role in tumor metabolism
and growth, and this is reflected in a high rate of glucose uptake.
But in fact, in addition to c-Myc, GLUT1, and HK in the present
study, there are various other factors that play a role in tumour
metabolism and growth, which is reflected by a high rate of glucose
uptake effect. For example, SGLT2, which is a sodium-glucose
cotransporter, is involved in glucose metabolism. In recent years,
it has been proposed as a target for diabetes and cancer (39). Its inhibitors have been proved to
decrease the proliferation of breast cancer cells (40). Drugs have always been
multi-targeted. Diclofenac in the present study has an impact on
the glucose uptake and proliferation of cancer cells. It also has
the possibility of targeting other glycolysis-associated genes
including SGLT2, but this requires more extensive research.
In summary, the present study results provide
evidence that diclofenac can decrease the protein expression of
GLUT1 and HK activity in cancer cells by downregulating the
expression of c-Myc, lowering glucose uptake, preventing the supply
of energy and inhibiting glycolysis, eventually inhibiting the
growth of TNBC cells. Changes in energy metabolism in tumours
significantly affect tumour proliferation and metastasis. For the
first time, the effect and potential mechanism of diclofenac in
TNBC cells was explored, and the potential value of glycolysis
inhibitors in the treatment of TNBC was proposed. The use of
glycolysis inhibitors alone or in combination with chemotherapeutic
drugs has been currently proposed in the clinic. Further studies on
the effects of drugs targeting c-Myc can provide basic data on
abnormal energy metabolism and new therapeutic targets for drugs
inhibiting energy metabolism in TNBC.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by the National Natural
Science Foundation of China (grant no. 8176100111) and Basic
Ability Enhancement Project of Young and Middle-aged Teachers in
Guangxi Universities (grant no. 2019KY0145).
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LY and JiaL participated in the preliminary
experimental design, preliminary experiment, main experiment
operation including cell culture, western blotting and flow
cytometry detection, data analysis, manuscript writing and
revision. YL, YZ, ZW and DZ participated in data analysis,
manuscript writing and revision. JinL and XZ participated in early
experimental design including selecting drugs and designing
possible signal pathways. LY and JiaL confirm the authenticity of
all the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Foulkes WD, Smith IE and Reis-Filho JS:
Triple-negative breast cancer. N Engl J Med. 363:1938–1948.
2010.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Shen L, O'Shea JM, Kaadige MR, Cunha S,
Wilde BR, Cohen AL, Welm AL and Ayer DE: Metabolic reprogramming in
triple-negative breast cancer through Myc suppression of TXNIP.
Proc Natl Acad Sci USA. 112:5425–5430. 2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Luo C, Wang Y, Wei C, Chen Y and Ji Z: The
anti-migration and anti-invasion effects of Bruceine D in human
triple-negative breast cancer MDA-MB-231 cells. Exp Ther Med.
19:273–279. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Yin L, Qi XW, Liu XZ, Yang ZY, Cai RL, Cui
HJ, Chen L and Yu SC: Icaritin enhances the efficacy of cetuximab
against triple-negative breast cancer cells. Oncol Lett.
19:3950–3958. 2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Li C, Li X, Li G, Sun L, Zhang W, Jiang J
and Ge Q: Identification of a prognosis-associated signature
associated with energy metabolism in triple-negative breast cancer.
Oncol Rep. 44:819–837. 2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Long JP, Li XN and Zhang F: Targeting
metabolism in breast cancer: How far we can go? World J Clin Oncol.
7:122–130. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Deng D and Yan N: GLUT, SGLT, and SWEET:
Structural and mechanistic investigations of the glucose
transporters. Protein Sci. 25:546–558. 2016.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Godoy A, Ulloa V, Rodríguez F, Reinicke K,
Yañez AJ, García Mde L, Medina RA, Carrasco M, Barberis S, Castro
T, et al: Differential subcellular distribution of glucose
transporters GLUT1-6 and GLUT9 in human cancer: Ultrastructural
localization of GLUT1 and GLUT5 in breast tumor tissues. J Cell
Physiol. 207:614–627. 2006.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Rogers S, Docherty SE, Slavin JL,
Henderson MA and Best JD: Differential expression of GLUT12 in
breast cancer and normal breast tissue. Cancer Lett. 193:225–233.
2003.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Garrido P, Morán J, Alonso A, González S
and González C: 17β-estradiol activates glucose uptake via GLUT4
translocation and PI3K/Akt signaling pathway in MCF-7 cells.
Endocrinology. 154:1979–1989. 2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Krzeslak A, Wojcik-Krowiranda K, Forma E,
Jozwiak P, Romanowicz H, Bienkiewicz A and Brys M: Expression of
GLUT1 and GLUT3 glucose transporters in endometrial and breast
cancers. Pathol Oncol Res. 18:721–728. 2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Choi J, Jung WH and Koo JS:
Metabolism-related proteins are differentially expressed according
to the molecular subtype of invasive breast cancer defined by
surrogate immunohistochemistry. Pathobiology. 80:41–52.
2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Hsieh AL, Walton ZE, Altman BJ, Stine ZE
and Dang CV: MYC and metabolism on the path to cancer. Semin Cell
Dev Biol. 43:11–21. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Palaskas N, Larson SM, Schultz N,
Komisopoulou E, Wong J, Rohle D, Campos C, Yannuzzi N, Osborne JR,
Linkov I, et al: 18F-fluorodeoxy-glucose positron emission
tomography marks MYC-overexpressing human basal-like breast
cancers. Cancer Res. 71:5164–5174. 2011.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Gottfried E, Lang SA, Renner K, Bosserhoff
A, Gronwald W, Rehli M, Einhell S, Gedig I, Singer K, Seilbeck A,
et al: New aspects of an old drug-diclofenac targets MYC and
glucose metabolism in tumor cells. PLoS One.
8(e66987)2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Penny HL, Sieow JL, Adriani G, Yeap WH,
See Chi Ee P, San Luis B, Lee B, Lee T, Mak SY, Ho YS, et al:
Warburg metabolism in tumor-conditioned macrophages promotes
metastasis in human pancreatic ductal adenocarcinoma.
Oncoimmunology. 5(e1191731)2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Guo X, Zhang X, Wang T, Xian S and Lu Y:
3-Bromopyruvate and sodium citrate induce apoptosis in human
gastric cancer cell line MGC-803 by inhibiting glycolysis and
promoting mitochondria-regulated apoptosis pathway. Biochem Biophys
Res Commun. 475:37–43. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Johnson J, Rychahou P, Sviripa VM, Weiss
HL, Liu C, Watt DS and Evers BM: Induction of AMPK activation by
N,N'-diarylurea FND-4b decreases growth and increases apoptosis in
triple negative and estrogen-receptor positive breast cancers. PLoS
One. 14(e0209392)2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Pelicano H, Zhang W, Liu J, Hammoudi N,
Dai J, Xu RH, Pusztai L and Huang P: Mitochondrial dysfunction in
some triple-negative breast cancer cell lines: Role of mTOR pathway
and therapeutic potential. Breast Cancer Res.
16(434)2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Butt JH, Barthel JS, Hosokawa MC and Moore
RA: NSAIDs: A clinical approach to the problems of gastrointestinal
side-effects. Aliment Pharmacol Ther. 2 (Suppl 1):S121–S129.
1988.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Grosser T, Ricciotti E and FitzGerald GA:
The cardiovascular pharmacology of nonsteroidal anti-inflammatory
drugs. Trends Pharmacol Sci. 38:733–748. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Barron CC, Bilan PJ, Tsakiridis T and
Tsiani E: Facilitative glucose transporters: Implications for
cancer detection, prognosis and treatment. Metabolism. 65:124–139.
2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhou YX, Zhou KM, Liu Q, Wang H, Wang W,
Shi Y and Ma YQ: The effect of Glut1 and c-myc on prognosis in
esophageal squamous cell carcinoma of Kazakh and Han patients.
Future Oncol. 14:1801–1815. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Carvalho KC, Cunha IW, Rocha RM, Ayala FR,
Cajaíba MM, Begnami MD, Vilela RS, Paiva GR, Andrade RG and Soares
FA: GLUT1 expression in malignant tumors and its use as an
immunodiagnostic marker. Clinics (Sao Paulo). 66:965–972.
2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Geng C, Li J, Ding F, Wu G, Yang Q, Sun Y,
Zhang Z, Dong T and Tian X: Curcumin suppresses 4-hydroxytamoxifen
resistance in breast cancer cells by targeting SLUG/Hexokinase 2
pathway. Biochem Biophys Res Commun. 473:147–153. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Chauhan A, Paul R, Debnath M, Bessi I,
Mandal S, Schwalbe H and Dash J: Synthesis of fluorescent
binaphthyl amines that bind c-MYC G-quadruplex DNA and repress
c-MYC expression. J Med Chem. 59:7275–7281. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Chen H, Liu H and Qing G: Targeting
oncogenic Myc as a strategy for cancer treatment. Signal Transduct
Target Ther. 3(5)2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Kfoury A, Armaro M, Collodet C,
Sordet-Dessimoz J, Giner MP, Christen S, Moco S, Leleu M, de Leval
L, Koch U, et al: AMPK promotes survival of c-Myc-positive melanoma
cells by suppressing oxidative stress. EMBO J.
37(e97673)2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Jayasooriya RGPT, Dilshara MG,
Karunarathne WAHM, Molagoda IMN, Choi YH and Kim GY: Camptothecin
enhances c-Myc-mediated endoplasmic reticulum stress and leads to
autophagy by activating Ca2+-mediated AMPK. Food Chem
Toxicol. 121:648–656. 2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Duan Q, Li H, Gao C, Zhao H, Wu S, Wu H,
Wang C, Shen Q and Yin T: High glucose promotes pancreatic cancer
cells to escape from immune surveillance via AMPK-Bmi1-GATA2-MICA/B
pathway. J Exp Clin Cancer Res. 38(192)2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Gutiérrez-Salmerón M, García-Martínez JM,
Martínez-Useros J, Fernández-Aceñero MJ, Viollet B, Olivier S,
Chauhan J, Lucena SR, De la Vieja A, Goding CR, et al: Paradoxical
activation of AMPK by glucose drives selective EP300 activity in
colorectal cancer. PLoS Biol. 18(e3000732)2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Li Y, Liang R, Sun M, Li Z, Sheng H, Wang
J, Xu P, Liu S, Yang W, Lu B, et al: AMPK-dependent phosphorylation
of HDAC8 triggers PGM1 expression to promote lung cancer cell
survival under glucose starvation. Cancer Lett. 478:82–92.
2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Osthus RC, Shim H, Kim S, Li Q, Reddy R,
Mukherjee M, Xu Y, Wonsey D, Lee LA and Dang CV: Deregulation of
glucose transporter 1 and glycolytic gene expression by c-Myc. J
Biol Chem. 275:21797–21800. 2000.PubMed/NCBI View Article : Google Scholar
|
|
36
|
O'Connell BC, Cheung AF, Simkevich CP, Tam
W, Ren X, Mateyak MK and Sedivy JM: A large scale genetic analysis
of c-Myc-regulated gene expression patterns. J Biol Chem.
278:12563–12573. 2003.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Dang CV, O'Donnell KA, Zeller KI, Nguyen
T, Osthus RC and Li F: The c-Myc target gene network. Semin Cancer
Biol. 16:253–264. 2006.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Liu Y, Xiang F, Huang Y, Shi L, Hu C, Yang
Y, Wang D, He N, Tao K, Wu K and Wang G: Interleukin-22 promotes
aerobic glycolysis associated with tumor progression via targeting
hexokinase-2 in human colon cancer cells. Oncotarget.
8:25372–25383. 2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Koepsell H: The Na+-D-glucose
cotransporters SGLT1 and SGLT2 are targets for the treatment of
diabetes and cancer. Pharmacol Ther. 170:148–165. 2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Komatsu S, Nomiyama T, Numata T, Kawanami
T, Hamaguchi Y, Iwaya C, Horikawa T, Fujimura-Tanaka Y, Hamanoue N,
Motonaga R, et al: SGLT2 inhibitor ipragliflozin attenuates breast
cancer cell proliferation. Endocr J. 67:99–106. 2020.PubMed/NCBI View Article : Google Scholar
|