Introduction
Coffin-Siris syndrome1 [CSS1; Online Mendelian
Inheritance in Man (OMIM) no. 135900] was first described as a
multiple malformation syndrome in 1970 and is characterized by
intellectual and/or developmental delay and hypoplastic or absent
fifth fingernails and/or toenails (1-3).
Haploinsufficiency of the AT-rich interaction domain-containing
protein 1B (ARID1B) has been reported to cause CSS1(4). The ARID1B variant is reported to be
the most frequently mutated gene in CSS (3,5-7)
and the majority of reported cases are sporadic pathogenic variants
(5,8).
ARID1B encodes a component of the Brahma-associated
factor complex, also known as the mammalian SWItch/sucrose
non-fermentable (SWI/SNF) complex (9,10). The
SWI/SNF complex is an ATP-dependent chromatin remodeling complex,
which modifies chromatin structure and facilitates transcription
factor access to DNA (9-11).
Forms of CSS have been demonstrated to be caused by pathogenic
variants in numerous genes encoding subunits of the SWI/SNF
complex, including CSS2-8 (OMIM nos. 614607, 614608, 614609,
616938, 617808, 618027 and 618362) which are caused by variants in
the AT-rich interaction domain-containing protein 1A (ARID1A),
SWI/SNF related matrix-associated actin-dependent regulator of
chromatic subfamily (SMARC) B member 1 (SMARCB1), SMARCA4, SMARCE1,
AT-rich interaction domain-containing protein 2 (ARID2), double
plant homeodomain finger 2 (DPF2) and SMARCC2 genes (OMIM nos.
603024, 601607, 603254, 603111, 609539, 601671 and 601734,
respectively) (2,3,5,6). A
similar phenotype, Nicolaides-Baraitser syndrome (OMIM no. 601358)
is caused by a pathogenic variant in a subunit of the SMARCA2
complex (OMIM no. 600014) (3).
Mosaicism is a biological phenomenon that describes
an individual who has developed from a single fertilized egg and
has two or more populations of cells with distinct genotypes
(12). Specific types of mosaicism
describe the parts of the body that harbor the variant cells and
the potential for transmission to offspring, including germline
mosaicism (also known as gonadal mosaicism), somatic mosaicism and
somatogonadal mosaicism (a combination of germline and somatic
mosaicism) (12-16).
Postzygotic mosaicism refers to mutations that result in distinct
cell populations within the same individual when somatogonadal
mosaicism cannot be fully excluded (14-16).
The developmental timing and cell lineage are affected and,
combined with the phenotypic consequences of the pathogenic
variant, ultimately determine the tissue distribution of mosaicism
(somatic, germline or somatogonadal), as well as the patterns of
disease reoccurrence within families (12,14).
Somatogonadal mosaicism that arises at an early embryonic stage can
involve both somatic and germ cells, and individuals with
somatogonadal mosaicism are at risk of having affected children
(12,14).
The present study reported two siblings with CSS1
and demonstrated rare genetic inheritance from their mother with
somatogonadal mosaicism.
Materials and methods
Ethical statement
Ethical approval for the present study was obtained
from the Institutional Review Board of the Children's Hospital of
Chongqing Medical University, Chongqing, China (ethical approval
no. 2018-64). Written informed consent was obtained from the
parents of the siblings.
Clinical characterizations
The patient in the present study was a girl aged 6
years and 3 months, born full-term to non-consanguineous Chinese
parents. She was delivered normally (gravida 3; para 3) with a
birth weight of 4,150 g. The length of the baby and head
circumference at birth were unknown. Feeding problems were observed
during the infant period. The first baby of the parents had died
shortly following birth.
The patient came to hospital and her mother reported
that the patient had intellectual and linguistic developmental
delays. The patient spoke her first words ‘mum mum’ at the age of 4
years and had slightly improved; however, she only said ‘mum mum’
during the clinical evaluation. She began to walk by herself at 2
years old. She has a severe intellectual delay and an IQ<32
according to the Wechsler Preschool and Primacy Scale of
Intelligence test (17). According
to the operation manual, the average score is 100 and the 95%
confidence interval is 70-130.
Upon examination, the patient weighed 14 kg
(z-score, -2.91), her height was 105.2 cm (z-score, -2.23), head
circumference was 46.5 cm (~20-month average) and BMI was 12.7
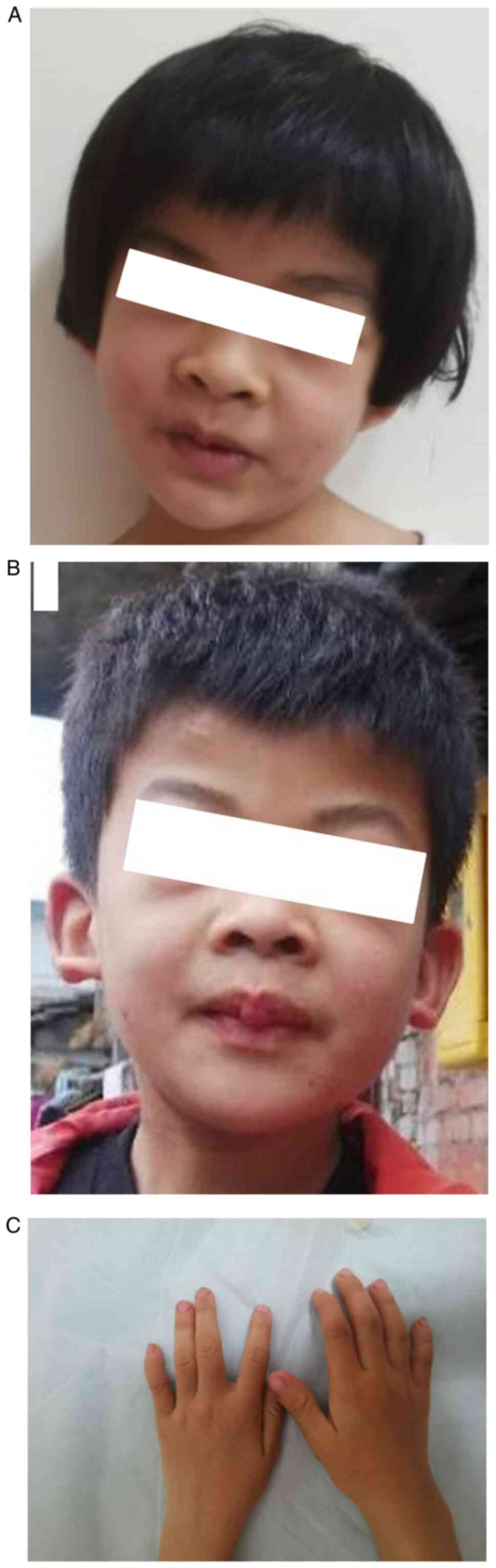
(z-score, -2.05), all of which are abnormal. She displayed
dysmorphic features, including brachytelephalangy and hypoplastic
nails of the fifth digit phalanges and fingers, coarse facial
features, low hairline, short philtrum, thick lips, mild scoliosis
(Fig. 1A-C) and body
hypertrichosis. Brain MRI and metabolic screening were normal.
The patient's older brother exhibited similar
disease onset and progression. He was 10 years old at the time of
the current study and had learning difficulties. Physical
examination demonstrated similar dysmorphic features tothe patient,
except for moderate scoliosis (Fig.
1B). The parents were clinically healthy and exhibited no
dysmorphic features.
Whole-exome sequencing (WES)
Peripheral blood samples were collected from the
patient and her family in EDTA tubes. The genomic DNA of the
patient, her brother and their parents were screened for genetic
variations using trio WES. Briefly, the DNA was sheared using an
ultrasonic processor (version no. KQ218; Kunshan Ultrasonic
Instruments Co., Ltd.) and hybridized with the xGen Exome Research
Panel probe sequence capture array (version no. 1.0; Integrated
Device Technology, Inc.) to enrich the exonic region, according to
the manufacturer's protocol. Quantitative PCR was performed for
effective molecular concentration detection of the exome libraries
using a KAPA Library Quantification kit (cat. no. KR0405-v8.17; the
kit included SYBR-Green; Kapa Biosystems, Roche Diagnostics),
according to the manufacturer's protocol. The primers and
thermocycling conditions are presented in Table I. The size distribution and
concentration of the exome libraries was tested using Bioanalyzer
2100 (Agilent Technologies, Inc.), according to the manufacturer's
protocol. A standard curve was generated and was used to convert
the average Cq score for each library. The average size-adjusted
concentration for each library that was assayed was calculated by
multiplying the calculated average concentration using the
following formula: Size of DNA standard in bp (452)/average
fragment length of library in bp.
 | Table IPrimers and thermocycling conditions
of quantitative-PCR for effective molecular concentration of the
exome librarie. |
Table I
Primers and thermocycling conditions
of quantitative-PCR for effective molecular concentration of the
exome librarie.
| Primer | Sequence | Step 1 | Step 2 | Step 3 |
|---|
| Forward |
5'-AATGATACGGCGACCACCGA-3' | Pre-denaturation | Denaturation at 95˚C
for | Melt curve |
| Reverse |
5'-CAAGCAGAAGACGGCATACGA-3' | at 95˚C for 5
min | 30 sec, annealing at
60˚C for 45 sec, 35 cycles | analysis at
65-95˚C |
Sanger sequencing
In order to validate the pathogenic variants
detected by WES, the genomic DNA of the patient, her brother and
their parents was evaluated using PCR and the products were
analyzed using Sanger sequencing. PCR amplification was performed
using a KAPA2G Robust HotStart PCR kit (Kapa Biosystems) on a Hema
9600 PCR Thermo Cycler (Zhuhai Hema Medical Instrument Co., Ltd.),
according to the manufacturer's protocol. The primers and
thermocycling conditions are presented in Table II.
| Table IIPrimers and thermocycling conditions
used for Sanger sequencing of the AT-rich interaction
domain-containing protein 1B gene. |
Table II
Primers and thermocycling conditions
used for Sanger sequencing of the AT-rich interaction
domain-containing protein 1B gene.
| Primer | Sequence | Step 1 | Step 2 | Step 3 |
|---|
| Forward |
5'-GCCATCAGCAGGTTCCCTAA-3' | Predenaturation | Denaturation at 95˚C
for | Final extension
at |
| Reverse |
5'-CGCCACTTACCAGGAGATGG-3' | at 95˚C for 5
min | 30 sec, annealing at
60˚C for 30 sec, chain extension at 72˚C for 30 sec, 30 cycles | 72˚C for 10 min |
Sequences were assembled and analyzed using the
DNASTAR Laser gene software package (version no. 7.1; DNASTAR,
Inc.; www.dnastar.com/documentation).
Amplicon-based deep sequencing
A two-stage PCR was performed. For the first stage
of PCR, the PCR reaction system was prepared using sample DNA. Each
step of the two-stage PCR was identical to that described
previously, except for the specific primers (Chigene; not
commercially available). The product directly served as the
template for the second stage of PCR with the same procedure,
except for the second pair specific primers. The primers used in
each stage were specifically designed by Chigene (Chigene
Translational Medical Research Center Co. Ltd.; chigene.org) and are not commercially available. The
purified DNA products underwent Next Generation Sequencing
(NGS)-based ultra-deep sequencing (sequencing depth, >500,000X).
The sequencing data were then used for bioinformatics analysis.
Bioinformatics analysis
Raw image files were processed using a BCL2FASTQ
software package (version no. 1.8.4; Illumina, Inc.; support.illumina.com/content/dam/illumina-support/documents/documentation/software_documentation/bcl2fastq/bcl2fastq_letterbooklet_15038058brpmi.pdf)
to generate fastq data. Low-quality variations of the quality score
<20 were filtered out. The sequencing reads were aligned to the
National Center for Biotechnology Information (www.ncbi.nlm.nih.gov/) human reference genome using
Burrows-Wheeler Aligner (version no. 0.7.11-r1034; https://github.com/lh3/bwa). SAMtools (version no.
0.1.19; github.com/samtools/samtools) and Pindel (version no.
0.2.4; github.com/genome/pindel) software were used to call
single-nucleotide variants and indels of the reads. The minor
allele frequency (MAF) was annotated using the databases dbSNP
(version no. 151; http://www.ncbi.nlm.nih.gov/projects/SNP/), 1,000
Genomes MAF (Chinese; https://www.internationalgenome.org/), Genome
Aggregation Database (gnomAD r2.0.2; https://gnomad.broadinstitute.org/), ExAC (merged with
the gnomAD database) and an in-house MAF (Chigene; not commercially
available) that includes >100,000 Chinese exomes. Synonymous
substitutions or single-nucleotide variants with MAF >5% were
filtered out. Transcriptions and translations of non-synonymous
variants were predicted using Sorting Intolerant from Tolerant and
Protein Variation Effect Analyzer (version no. 1.1.3; http://provean.jcvi.org), Polymorphism Phenotyping
(version no. 2; http://genetics.bwh.harvard.edu/pph2/). The
pathogenicity of variants was annotated according to the American
College of Medical Genetics and Genomics (ACMG) standards and
guidelines (18).
Results
WES
Due to the absence of phenotypes in the parents and
the presence of similar phenotypes in the patient and her older
brother, the condition was assumed to exhibit an autosomal
recessive mode of inheritance. However, evaluation according to
ACMG guidelines and clinical features did not present a homozygous
or compound heterozygous variant that would have been expected in a
recessive inheritance model. Therefore, the condition was
hypothesized to exhibit ta dominant inheritance model. The present
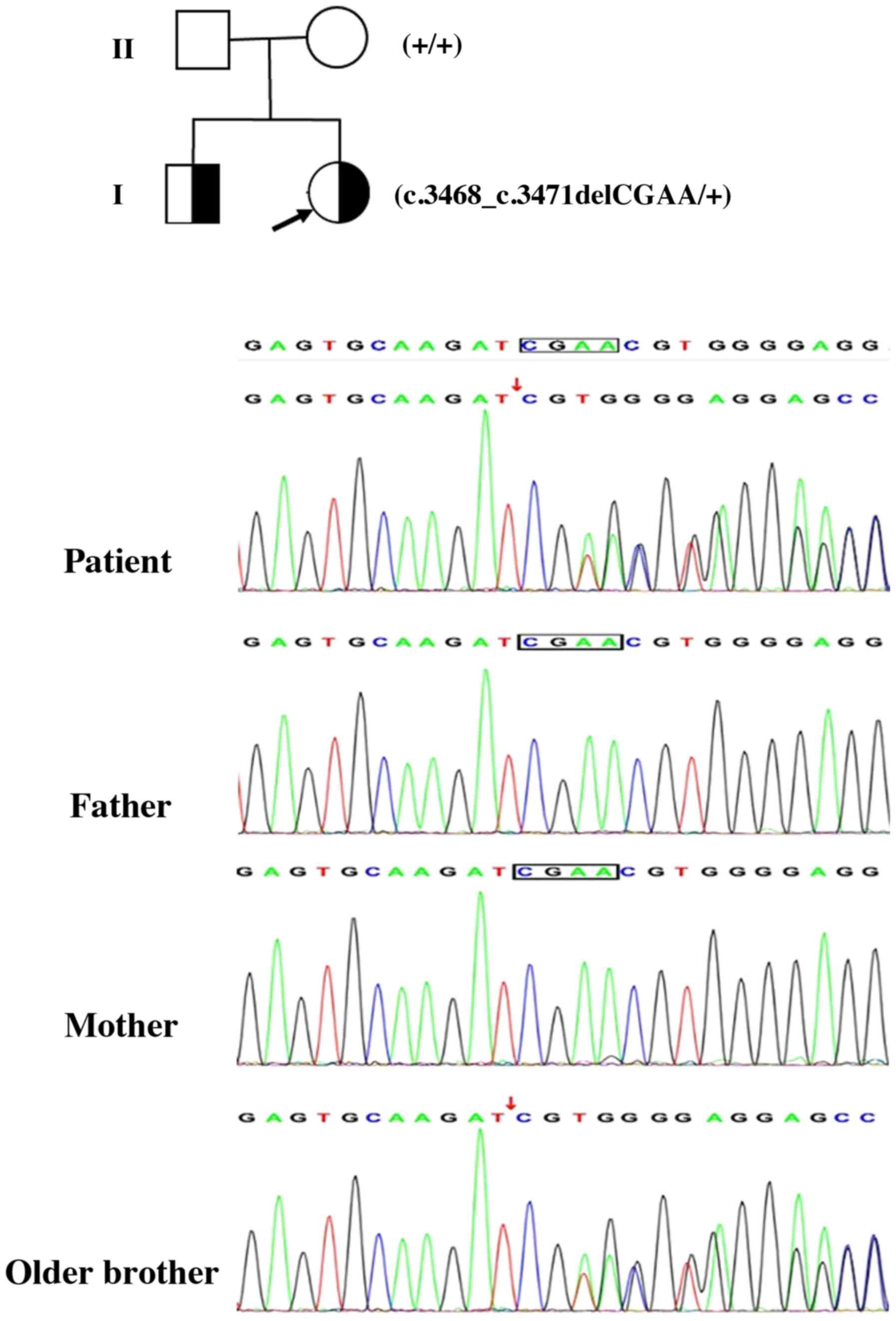
study therefore identified a de novo heterozygous 4-bp
deletion (c.3468_3471del) in the 13th exon of the ARID1B gene
(NM_020732; Fig. 2). This deletion
is predicted to result in a frame shift and premature termination.
To the best of our knowledge, this pathogenic variant in the ARID1B
gene has not previously been reported. De novo occurrence
was confirmed using Sanger sequencing of samples from the patient
and her parents (Fig. 2). No other
pathogenic variants were identified in screening of all associated
CSS genes (ARID1B, SMARCB1, SMARCA4, SMARCE1, ARID2, DPF2, SMARCC2
and SMARCA2).
Amplicon-based deep sequencing
After excluding the possibility of incomplete
penetrance of CSS1, one of the parents was suspected to be a
gonadal mosaic. In order to evaluate this hypothesis,
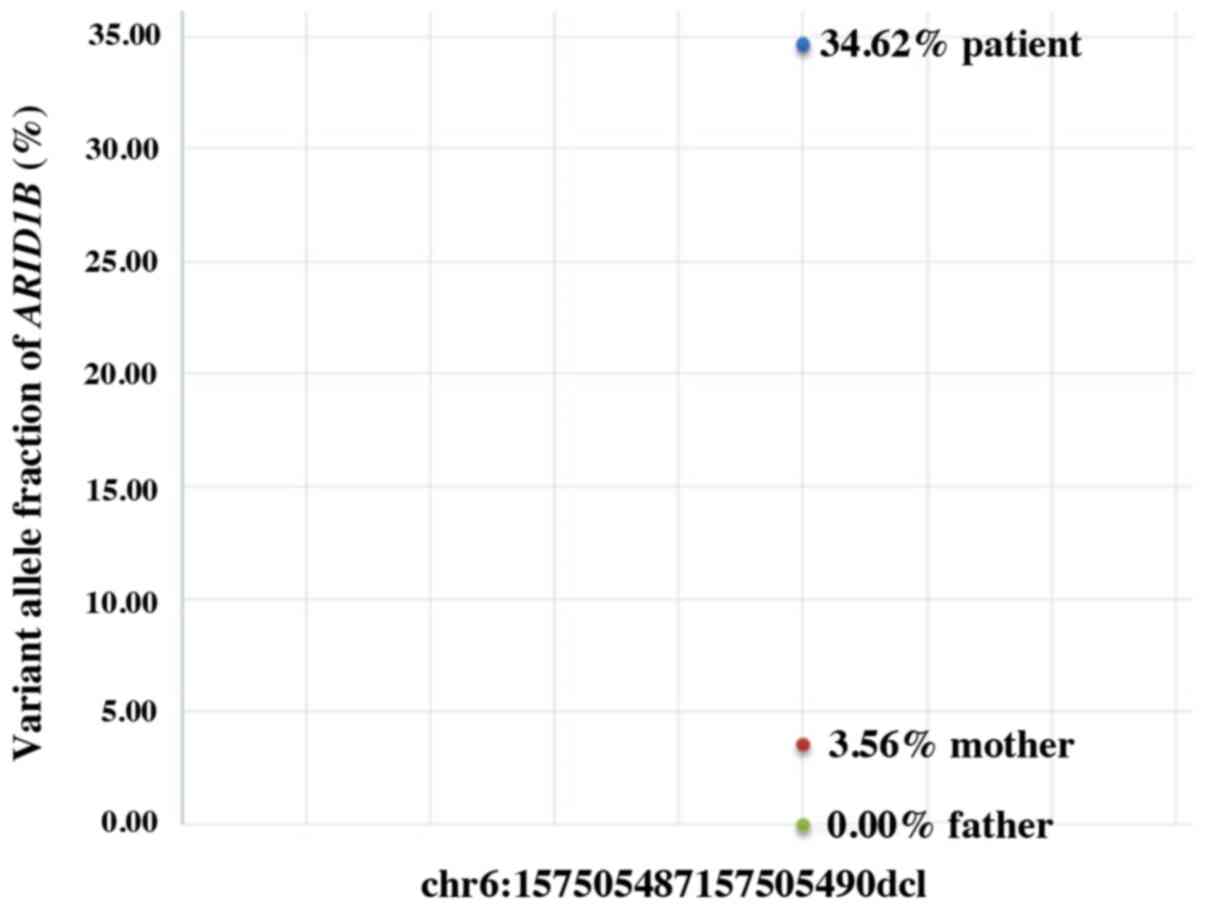
amplicon-based deep sequencing was performed using DNA from the
peripheral blood of the patient's parents, which confirmed a 4%
somatic ARID1B mosaicism in the patient's mother (Fig. 3). The previous detection of the
ARID1B pathogenic variant in the patient's brother indicated the
presence of somatogonadal ARID1B mosaicism in the mother.
Discussion
CSS was first reported by Coffin and Siris (1) and is characterized by notable
hypoplasia of the fifth digit phalanges and/or nails, coarse facial
features and sparse scalp hair (1).
Worldwide, >100 CSS cases with ARID1B pathogenic variants have
been reported in the last two years and the clinical features have
been summarized (7,8). Previous evidence has indicated that
the majority of CSS cases exhibit a certain degree of intellectual
and/or developmental delay (4,5,8,19).
There are two facial features that have been categorized (4,20).
Patients with classical CSS have coarse facial features, bushy
eyebrows and thick vermilion of the lips, while patients with
variant CSS collectively display a less coarse facial features,
thinner eyebrows and thin vermillion border of the lips. However,
both CSS and ARID1B-associated disorders have a phenotypic spectrum
and cannot always be distinguished as two separate (classical or
variant) categories. As a result of this phenotypic heterogeneity,
no widely convincing clinical criteria and key diagnostic features
have yet been outlined (1,3,5,7,19,20).
Thus, molecular testing currently has an important role in the
diagnosis of CSS.
In 2012, CSS was reported to be caused by pathogenic
variants in genes encoding subunits of the SWI/SNF complex, which
function as chromatin remodeling factors (21-23).
These include SMARCB1, SMARCA2, SMARCA4, SMARCE1, ARID1A and ARID1B
at 22q11.23, 9p24.3, 19p13.2, 17q21.2, 1p36.11 and 6q25.3 (21-23).
Additional genes (including SOX11, ADNP, PHF6 and TBC1D24 at
2p25.2, 20q13.13, Xq26.2 and 16p13.3,) have alsobeen identified
(24). Evidence has indicated that
pathogenic variants in ARID1B are the primary genetic cause of CSS,
accounting for 51-75% of cases (3,5-7)
in European, Japan, Canada, USA, between 2013-2019. However, ARID1B
is the most frequently mutated gene in unspecified intellectual
disability cohorts (~1%) from the United Kingdom and Germany in the
past 8 years (7,25,26).
ARID1B pathogenic variants are associated with phenotypes that vary
from non-syndromic intellectual disability to CSS (7).
The patient and her brother presented with
intellectual disabilities, speech problems, developmental delay,
hypoplastic fifth fingernails and/or toenails and
brachytelephalangy. Additionally, the patient and her brother
exhibited dysmorphic features, including coarse facial features,
low hairline, bushy eyebrows, thick lips, scoliosis, body
hypertrichosis and dental anomalies. The two siblings were
clinically diagnosed with CSS1; this was confirmed using WES, which
demonstrated heterozygous ARID1B variants [c.3468 (exon13)_c.3471
(exon13)_delCGAA] present in both siblings.
The majority of reported CSS1 cases are caused by
heterozygous pathogenic variants in the ARID1B gene are sporadic
(5,8). However, exceptions have been noted,
including a pathogenic variant of ARID1B that was passed down from
an affected mother to her son (19). In a consanguineous Emirati family, a
heterozygous variant of ARID1B was present in three affected
siblings; however, this was absent in their parents and unaffected
siblings (8). This indicated that
one of the parents was a gonadal mosaic.
In the present study, the heterozygous pathogenic
variant [c.3468 (exon13)_c.3471 (exon13) delCGAA] was identified
using WES in two siblings; however, this was not initially observed
in the parents. Therefore, it was hypothesized that this was a
de novo pathogenic variant. Additionally, we hypothesized
that one of the patient's parents exhibited gonadal mosaicism for
the pathogenic variant, which has previously been reported
(8); however, this had not been
verified by laboratory tests. In order to test this hypothesis,
amplicon-based deep sequencing was performed using samples from the
patient's parents. An ARID1B pathogenic variant (4%;
chr6:157505487-157505490) was observed in the mother; however, this
was absent in the father. This indicated that the mother exhibited
the somatogonadal mosaic ARID1B variant. To the best of our
knowledge, this is the first report based on experimental evidence
of an ARID1B pathogenic variant in CSS1 that was inherited from the
clinically healthy somatogonadal mosaic mother.
In summary, a heterozygous ARID1B pathogenic variant
[c.3468 (exon13)_c.3471 (exon13)_delCGAA] was identified in the
patient and her brother, both of whom exhibited the classical
phenotype of CSS1. The results of the present study expand the
spectrum of known ARID1B pathogenic variants, as well as the ethnic
backgrounds of reported cases. To the best of our knowledge, the
present study is the first to report two siblings with a pathogenic
ARID1B variant inherited from their mother who exhibited
somatogonadal mosaicism.
Acknowledgements
The authors would like to thank Miss Wenna Lin from
Beijing Chigene Translational Medical Research Center Co. Ltd.,
Beijing, China. She provided the NGS test, genetic disease analyses
and helpful discussion.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZM, CQ and DY conceived and designed the current
study and analyzed data. ZM and DY prepared the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Ethical approval for the present study was obtained
from the Institutional Review Board, Children's Hospital of
Chongqing Medical University, Chongqing, China (approval no.
2018-64).
Patient consent for publication
Consent for publication of the patient's data/images
in this paper was obtained from the parents.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Coffin GS and Siris E: Mental retardation
with absent fifth fingernail and terminal phalanx. Am J Dis Child.
119:433–439. 1970.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Vergano SS and Deardorff MA: Clinical
features, diagnostic criteria, and management of Coffin-Siris
syndrome. Am J Med Genet C Semin Med Genet. 166C:252–256.
2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wieczorek D, Bögershausen N, Beleggia F,
Steiner-Haldenstätt S, Pohl E, Li Y, Milz E, Martin M, Thiele H,
Altmüller J, et al: A comprehensive molecular study on Coffin-Siris
and Nicolaides-Baraitser syndromes identifies a broad molecular and
clinical spectrum converging on altered chromatin remodeling. Hum
Mol Genet. 22:5121–5135. 2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Santen GW and Clayton-Smith J: ARID1B-CSS
consortium: The ARID1B phenotype: What we have learned so far. Am J
Med Genet C Semin Med Genet. 166C:276–289. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Santen GW, Aten E, Vulto-van Silfhout AT,
Pottinger C, van Bon BW, van Minderhout IJ, Snowdowne R, van der
Lans CA, Boogaard M, Linssen MM, et al: Coffin-Siris syndrome and
the BAF complex: Genotype-phenotype study in 63 patients. Hum
Mutat. 34:1519–1528. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Tsurusaki Y, Okamoto N, Ohashi H, Mizuno
S, Matsumoto N, Makita Y, Fukuda M, Isidor B, Perrier J, Aggarwal
S, et al: Coffin-Siris syndrome is a SWI/SNF complex disorder. Clin
Genet. 85:548–554. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
van der Sluijs PJ, Jansen S, Vergano SA,
Adachi-Fukuda M, Alanay Y, AlKindy A, Baban A, Bayat A, Beck-Wödl
S, Berry K, et al: The ARID1B spectrum in 143 patients: From
nonsyndromic intellectual disability to Coffin-Siris syndrome.
Genet Med. 21:1295–1307. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ben-Salem S, Sobreira N, Akawi NA,
Al-Shamsi AM, John A, Pramathan T, Valle D, Ali BR and Al-Gazali L:
Gonadal mosaicism in ARID1B gene causes intellectual disability and
dysmorphic features in three siblings. Am J Med Genet A.
170A:156–161. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Alfert A, Moreno N and Kerl K: The BAF
complex in development and disease. Epigenetics Chromatin.
12(19)2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Hargreaves DC and Crabtree GR:
ATP-dependent chromatin remodeling: Genetics, genomics and
mechanisms. Cell Res. 21:396–420. 2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Nie Z, Yan Z, Chen EH, Sechi S, Ling C,
Zhou S, Xue Y, Yang D, Murray D, Kanakubo E, et al: Novel SWI/SNF
chromatin-remodeling complexes contain a mixed-lineage leukemia
chromosomal translocation partner. Mol Cell Biol. 23:2942–2952.
2003.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Biesecker LG and Spinner NB: A genomic
view of mosaicism and human disease. Nat Rev Genet. 14:307–320.
2013.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
D'Gama AM and Walsh CA: Somatic mosaicism
and neurodevelopmental disease. Nat Neurosci. 21:1504–1514.
2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Happle R: Gonosomal versus somatogonadal
mosaicism: What is in a name. Am J Med Genet A.
179(1678)2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Lim ET, Uddin M, De Rubeis S, Chan Y,
Kamumbu AS, Zhang X, D'Gama AM, Kim SN, Hill RS, Goldberg AP, et
al: Rates, distribution and implications of postzygotic mosaic
mutations in autism spectrum disorder. Nat Neurosci. 20:1217–1224.
2017.PubMed/NCBI View
Article : Google Scholar
|
|
16
|
Wright CF, Prigmore E, Rajan D, Handsaker
J, McRae J, Kaplanis J, Fitzgerald TW, FitzPatrick DR, Firth HV and
Hurles ME: Clinically-relevant postzygotic mosaicism in parents and
children with developmental disorders in trio exome sequencing
data. Nat Commun. 10(2985)2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Watkins MW and Beaujean AA: Bifactor
structure of the wechsler preschool and primary scale of
intelligence-fourth edition. Sch Psychol Q. 29:52–63.
2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Mannino EA, Miyawaki H, Santen G and
Schrier Vergano SA: First data from a parent-reported registry of
81 individuals with Coffin-Siris syndrome: Natural history and
management recommendations. Am J Med Genet A. 176:2250–2258.
2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Vergano SA, van der Sluijs PJ and Santen
G: ARID1B-Related Disorder (ARID1B-RD). GeneReviews® [Internet],
University of Washington, Seattle, WA, 1993–2021.
|
|
21
|
Santen GW, Aten E, Sun Y, Almomani R,
Gilissen C, Nielsen M, Kant SG, Snoeck IN, Peeters EA,
Hilhorst-Hofstee Y, et al: Mutations in SWI/SNF chromatin
remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat
Genet. 44:379–380. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Smith JA, Holden KR, Friez MJ, Jones JR
and Lyons MJ: A novel familial autosomal dominant mutation in
ARID1B causing neurodevelopmental delays, short stature, and
dysmorphic features. Am J Med Genet A. 170:3313–3318.
2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Tsurusaki Y, Okamoto N, Ohashi H, Kosho T,
Imai Y, Hibi-Ko Y, Kaname T, Naritomi K, Kawame H, Wakui K, et al:
Mutations affecting components of the SWI/SNF complex cause
Coffin-Siris syndrome. Nat Genet. 44:376–378. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
24
|
Kosho T, Miyake N and Carey JC:
Coffin-Siris syndrome and related disorders involving components of
the BAF (mSWI/SNF) complex: Historical review and recent advances
using next generation sequencing. Am J Med Genet C Semin Med Genet.
166C:241–251. 2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wright CF, Fitzgerald TW, Jones WD,
Clayton S, McRae JF, van Kogelenberg M, King DA, Ambridge K,
Barrett DM, Bayzetinova T, et al: Genetic diagnosis of
developmental disorders in the DDD study: A scalable analysis of
genome-wide research data. Lancet. 385:1305–1314. 2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Hoyer J, Ekici AB, Endele S, Popp B,
Zweier C, Wiesener A, Wohlleber E, Dufke A, Rossier E, Petsch C, et
al: Haploinsufficiency of ARID1B, a member of the SWI/SNF-a
chromatin-remodeling complex, is a frequent cause of intellectual
disability. Am J Hum Genet. 90:565–572. 2012.PubMed/NCBI View Article : Google Scholar
|