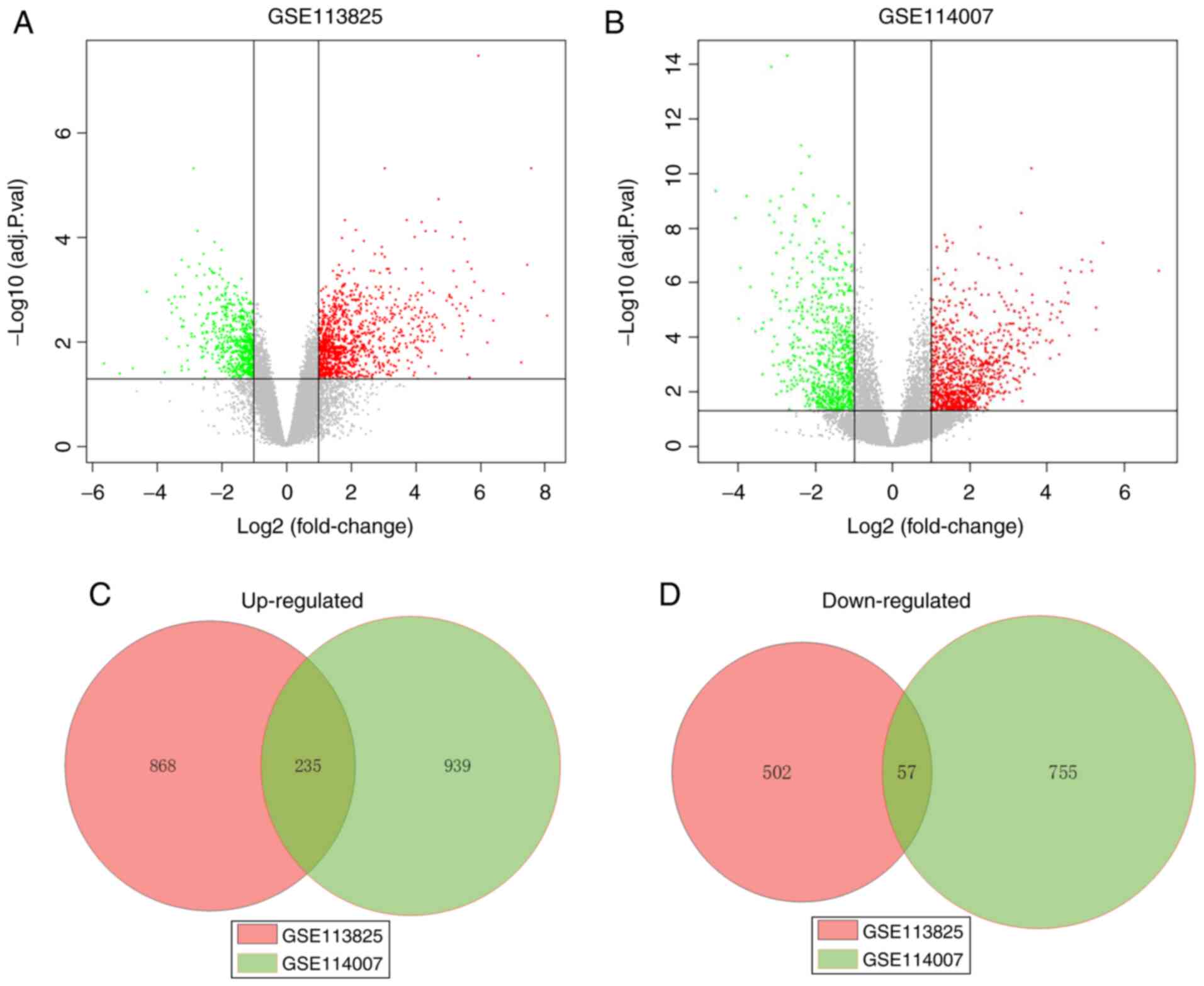
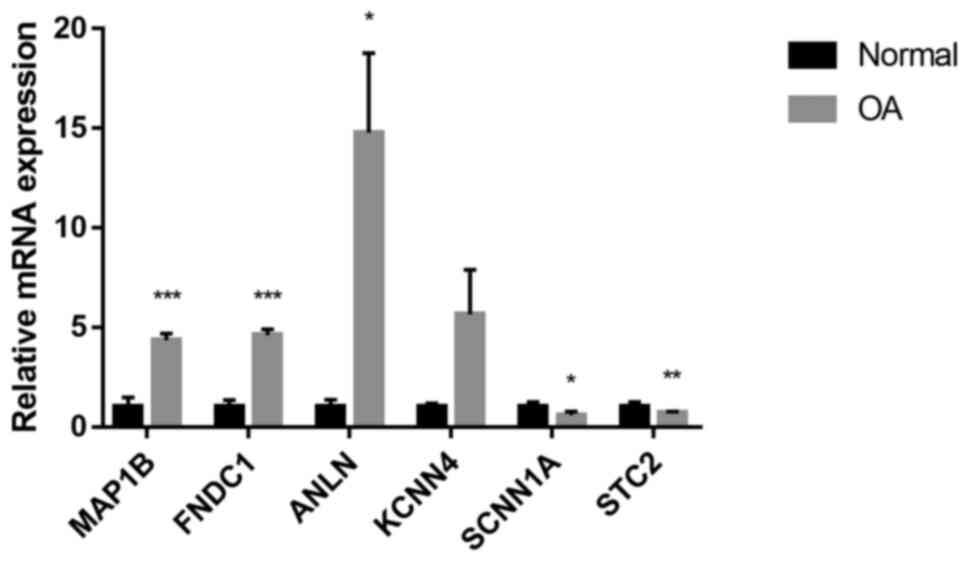
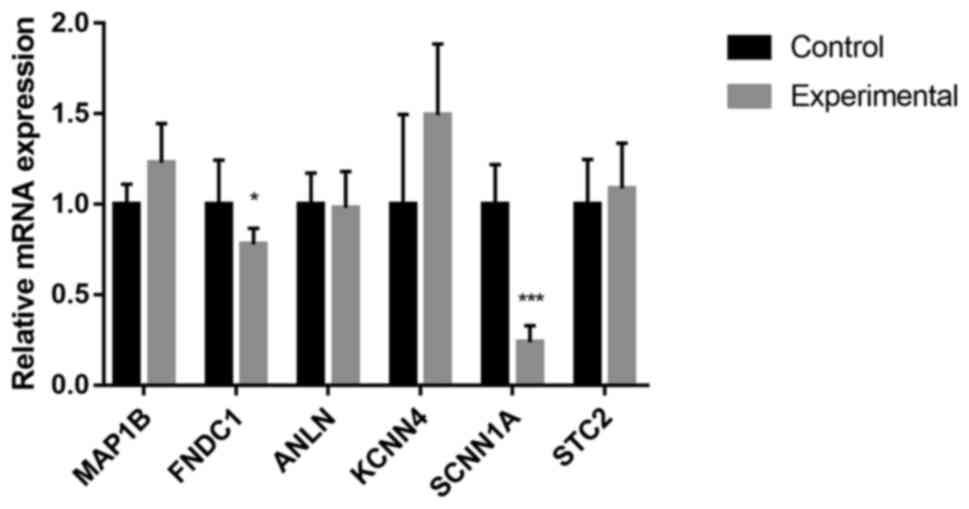
|
1
|
Miranda-Duarte A: DNA methylation in
osteoarthritis: Current status and therapeutic implications. Open
Rheumatol J. 12:37–49. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Jeffries MA: Osteoarthritis year in review
2018: Genetics and epigenetics. Osteoarthritis Cartilage.
27:371–377. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lotz M and Loeser RF: Effects of aging on
articular cartilage homeostasis. Bone. 51:241–248. 2012.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Goldberg AD, Allis CD and Bernstein E:
Epigenetics: A landscape takes shape. Cell. 128:635–638.
2007.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Barter MJ, Bui C and Young DA: Epigenetic
mechanisms in cartilage and osteoarthritis: DNA methylation,
histone modifications and microRNAs. Osteoarthritis Cartilage.
20:339–349. 2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Shen J, Abu-Amer Y, O'Keefe RJ and
McAlinden A: Inflammation and epigenetic regulation in
osteoarthritis. Connect Tissue Res. 58:49–63. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Im GI and Choi YJ: Epigenetics in
osteoarthritis and its implication for future therapeutics. Expert
Opin Biol Ther. 13:713–721. 2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Roach HI, Yamada N, Cheung KS, Tilley S,
Clarke NM, Oreffo RO, Kokubun S and Bronner F: Association between
the abnormal expression of matrix-degrading enzymes by human
osteoarthritic chondrocytes and demethylation of specific CpG sites
in the promoter regions. Arthritis Rheum. 52:3110–3124.
2005.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Imagawa K, de Andres MC, Hashimoto K, Pitt
D, Itoi E, Goldring MB, Roach HI and Oreffo RO: The epigenetic
effect of glucosamine and a nuclear factor-kappa B (NF-κB)
inhibitor on primary human chondrocytes-implications for
osteoarthritis. Biochem Biophys Res Commun. 405:362–367.
2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Hashimoto K, Oreffo RO, Gibson MB,
Goldring MB and Roach HI: DNA demethylation at specific CpG sites
in the IL1β promoter in response to inflammatory cytokines in human
articular chondrocytes. Arthritis Rheum. 60:3303–3313.
2009.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Jones PA: Functions of DNA methylation:
Islands, start sites, gene bodies and beyond. Nat Rev Genet.
13:484–492. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Harris RA, Wang T, Coarfa C, Nagarajan RP,
Hong C, Downey SL, Johnson BE, Fouse SD, Delaney A, Zhao Y, et al:
Comparison of sequencing-based methods to profile DNA methylation
and identification of monoallelic epigenetic modifications. Nat
Biotechnol. 28:1097–1105. 2010.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Imagawa K, de Andres MC, Hashimoto K, Itoi
E, Otero M, Roach HI, Goldring MB and Oreffo RO: Association of
reduced type IX collagen gene expression in human osteoarthritic
chondrocytes with epigenetic silencing by DNA hypermethylation.
Arthritis Rheumatol. 66:3040–3051. 2014.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Hahn MA, Wu X, Li AX, Hahn T and Pfeifer
GP: Relationship between gene body DNA methylation and intragenic
H3K9me3 and H3K36me3 chromatin marks. PLoS One.
6(e18844)2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Laurent L, Wong E, Li G, Huynh T, Tsirigos
A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J and Wei CL:
Dynamic changes in the human methylome during differentiation.
Genome Res. 20:320–331. 2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hellman A and Chess A: Gene body-specific
methylation on the active X chromosome. Science. 315:1141–1143.
2007.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Chou CH, Lee CH, Lu LS, Song IW, Chuang
HP, Kuo SY, Wu JY, Chen YT, Kraus VB, Wu CC and Lee MT: Direct
assessment of articular cartilage and underlying subchondral bone
reveals a progressive gene expression change in human
osteoarthritic knees. Osteoarthritis Cartilage. 21:450–461.
2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Zhao L, Wang Q, Zhang C and Huang C:
Genome-wide DNA methylation analysis of articular chondrocytes
identifies TRAF1, CTGF, and CX3CL1 genes as hypomethylated in
osteoarthritis. Clin Rheumatol. 36:2335–2342. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Iliopoulos D, Malizos KN and Tsezou A:
Epigenetic regulation of leptin affects MMP-13 expression in
osteoarthritic chondrocytes: Possible molecular target for
osteoarthritis therapeutic intervention. Ann Rheum Dis.
66:1616–1621. 2007.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Morris TJ, Butcher LM, Feber A,
Teschendorff AE, Chakravarthy AR, Wojdacz TK and Beck S: ChAMP:
450k chip analysis methylation pipeline. Bioinformatics.
30:428–430. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Teschendorff AE, Marabita F, Lechner M,
Bartlett T, Tegner J, Gomez-Cabrero D and Beck S: A beta-mixture
quantile normalization method for correcting probe design bias in
Illumina Infinium 450 k DNA methylation data. Bioinformatics.
29:189–196. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Li Z, Zhang R, Yang X, Zhang D, Li B,
Zhang D, Li Q and Xiong Y: Analysis of gene expression and
methylation datasets identified ADAMTS9, FKBP5, and PFKBF3 as
biomarkers for osteoarthritis. J Cell Physiol. 234:8908–8917.
2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Rushton MD, Reynard LN, Barter MJ, Refaie
R, Rankin KS, Young DA and Loughlin J: Characterization of the
cartilage DNA methylome in knee and hip osteoarthritis. Arthritis
Rheumatol. 66:2450–2460. 2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Mishra NK and Guda C: Genome-wide DNA
methylation analysis reveals molecular subtypes of pancreatic
cancer. Oncotarget. 8:28990–29012. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Jung SM, Lee JH, Park J, Oh YS, Lee SK,
Park JS, Lee YS, Kim JH, Lee JY, Bae YS, et al: Smad6 inhibits
non-canonical TGF-β1 signalling by recruiting the deubiquitinase
A20 to TRAF6. Nat Commun. 4(2562)2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Brena RM, Huang TH and Plass C: Toward a
human epigenome. Nat Genet. 38:1359–1360. 2006.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kim KI, Park YS and Im GI: Changes in the
epigenetic status of the SOX-9 promoter in human osteoarthritic
cartilage. J Bone Miner Res. 28:1050–1060. 2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wright ML, Dozmorov MG, Wolen AR,
Jackson-Cook C, Starkweather AR, Lyon DE and York TP: Establishing
an analytic pipeline for genome-wide DNA methylation. Clin
Epigenetics. 8(45)2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Mustafa Z, Chapman K, Irven C, Carr AJ,
Clipsham K, Chitnavis J, Sinsheimer JS, Bloomfield VA, McCartney M,
Cox O, et al: Linkage analysis of candidate genes as susceptibility
loci for osteoarthritis-suggestive linkage of COL9A1 to female hip
osteoarthritis. Rheumatology (Oxford). 39:299–306. 2000.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Loughlin J, Mustafa Z, Dowling B, Southam
L, Marcelline L, Raina SS, Ala-Kokko L and Chapman K: Finer linkage
mapping of a primary hip osteoarthritis susceptibility locus on
chromosome 6. Eur J Hum Genet. 10:562–568. 2002.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Li K, Qin L, Jiang S, Li A, Zhang C, Liu
G, Sun J, Sun H, Zhao Y, Li N and Zhang Y: The signature of
HBV-related liver disease in peripheral blood mononuclear cell DNA
methylation. Clin Epigenetics. 12(81)2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Gutierrez-Arcelus M, Lappalainen T,
Montgomery SB, Buil A, Ongen H, Yurovsky A, Bryois J, Giger T,
Romano L, Planchon A, et al: Passive and active DNA methylation and
the interplay with genetic variation in gene regulation. Elife.
2(e00523)2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Brenet F, Moh M, Funk P, Feierstein E,
Viale AJ, Socci ND and Scandura JM: DNA methylation of the first
exon is tightly linked to transcriptional silencing. PLoS One.
6(e14524)2011.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Keller TE, Han P and Yi SV: Evolutionary
transition of promoter and gene body DNA methylation across
invertebrate-vertebrate boundary. Mol Biol Evol. 33:1019–1028.
2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Huh I, Zeng J, Park T and Yi SV: DNA
methylation and transcriptional noise. Epigenetics Chromatin.
6(9)2013.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Jjingo D, Conley AB, Yi SV, Lunyak VV and
Jordan IK: On the presence and role of human gene-body DNA
methylation. Oncotarget. 3:462–474. 2012.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Bird AP and Wolffe AP: Methylation-induced
repression-belts, braces, and chromatin. Cell. 99:451–454.
1999.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Lorincz MC, Dickerson DR, Schmitt M and
Groudine M: Intragenic DNA methylation alters chromatin structure
and elongation efficiency in mammalian cells. Nat Struct Mol Biol.
11:1068–1075. 2004.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Den Hollander W, Ramos YF, Bomer N,
Elzinga S, Van der Breggen R, Lakenberg N, De Dijcker WJ, Suchiman
HE, Duijnisveld BJ, Houwing-Duistermaat JJ, et al: Transcriptional
associations of osteoarthritis-mediated loss of epigenetic control
in articular cartilage. Arthritis Rheumatol. 67:2108–2116.
2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Xiao Y, Wei R, Yuan Z, Lan X, Kuang J, Hu
D, Song Y and Luo J: Rutin suppresses FNDC1 expression in bone
marrow mesenchymal stem cells to inhibit postmenopausal
osteoporosis. Am J Transl Res. 11:6680–6690. 2019.PubMed/NCBI
|
|
42
|
Stoffels JM, Zhao C and Baron W:
Fibronectin in tissue regeneration: Timely disassembly of the
scaffold is necessary to complete the build. Cell Mol Life Sci.
70:4243–4253. 2013.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Pankov R and Yamada KM: Fibronectin at a
glance. J Cell Sci. 115:3861–3863. 2002.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Bing DH, Almeda S, Isliker H, Lahav J and
Hynes RO: Fibronectin binds to the C1q component of complement.
Proc Natl Acad Sci USA. 79:4198–4201. 1982.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Carsons SE, Schwartzman S, Diamond HS and
Berkowitz E: Interaction between fibronectin and C1q in rheumatoid
synovial fluid and normal plasma. Clin Exp Immunol. 72:37–42.
1988.PubMed/NCBI
|
|
46
|
Alvarez-Garcia O, Fisch KM, Wineinger NE,
Akagi R, Saito M, Sasho T, Su AI and Lotz MK: Increased DNA
methylation and reduced expression of transcription factors in
human osteoarthritis cartilage. Arthritis Rheumatol. 68:1876–1886.
2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Christman JK: 5-Azacytidine and
5-aza-2'-deoxycytidine as inhibitors of DNA methylation:
Mechanistic studies and their implications for cancer therapy.
Oncogene. 21:5483–5495. 2002.PubMed/NCBI View Article : Google Scholar
|