Introduction
Osteoarthritis (OA), the most common disease of the
joint, is characterized by progressive degeneration of the
articular cartilage, resulting in narrowing of joint spaces,
osteophytosis and subchondral sclerosis (1). Although OA is a disease with multiple
genetic and environmental risk factors, genetics and epigenetics
contribute significantly to the pathogenesis and are currently an
important subject in the field of OA research (2). Articular chondrocytes are vital for
maintaining the homeostasis of the articular cartilaginous tissue
and numerous factors may impair their genetic transcriptional
function, leading to phenotypic changes and ultimately cartilage
degradation (3).
The study of epigenetics involves exploring changes
in gene expression in organisms capable of cellular differentiation
without changes in the DNA sequence (4). Previous studies have indicated that
various types of epigenetics have an impact on the pathogenesis of
OA, including DNA methylation, microRNA, long noncoding RNA and
histone modification (5). Among
these, DNA methylation is an intensively studied epigenetic
modifications. The dynamic DNA methylation process is potentiated
by DNA methyltransferase (DNMT) enzymes, including DNMT1, 3A and
3B, and the demethylation enzymes ten-eleven translocation
methylcytosine dioxygenases (TETs), which include TET1, 2 and 3.
DNMTs catalyze the addition of a methyl group to a
cytosine-phosphate-guanine dinucleotide (CpG) to form
5-methylcytosine (5mC) (6).
Studies have demonstrated that DNA methylation may
have a key role in regulating gene expression (7), whether at the promoter, enhancer or
the gene body (6). In chondrocytes
of individuals with OA, the deletion of DNA methylation forms the
basis of abnormal expression of certain important catabolic genes,
such as the matrix metalloproteinase (MMP), ADAMTS4, IL-1β and NOS2
(8-10).
Previous studies have primarily focused on the methylation level at
the gene promoters, while other regions of DNA methylation, such as
gene body and enhancers, warrant further study (11).
In the recent decade, a variety of genome-wide
methods for mapping 5mC have been developed, most commonly by
treating the DNA with bisulfite followed by analysis via Illumina
450K microarray (12). Through this
method, the acquisition of a complete methylated genome has
advanced the understanding of the role of DNA methylation in
epigenetic processes (11). One of
the important advancements in knowledge is that the distribution of
methylation across the genome dictates its subsequent expression.
Numerous studies have confirmed that methylation of the promoter
leads to gene silencing. Specifically, Imagawa et al
(13) indicated that a total of 6
CpG sites of the COL9A1 promoter in OA chondrocytes were
significantly hypermethylated, while the mRNA expression was
significantly decreased. The expression level of COL9A1 mRNA was
increased after the treatment with the DNA methylation inhibitor
5-aza-2'-deoxycytidine (5'Aza). On the other hand, the effect of
gene body methylation is different from that of promoter
methylation, which may be related to transcriptional elongation and
selective splicing (14,15). It has been confirmed that gene body
methylation is able to activate gene transcription on the X
chromosome (16).
In the present study, the data of DNA methylation
and mRNA expression of normal and OA cartilage from public datasets
were analyzed and compared to identify key genes that may
contribute to the pathogenesis of OA. To investigate the functional
changes of gene transcription caused by the differences in DNA
methylation, the study focused on those genes with differentially
methylated sites (DMS) located in the promoter together with their
methylation level that was negatively correlated with the mRNA
levels, or those genes with DMS localized in the gene body or the
3'UTR together with a positive relationship between methylation
levels and gene transcription. Cartilage samples from patients
undergoing knee replacement for primary OA were collected to verify
the mRNA expression levels of the differentially expressed genes
(DEGs), which were then assessed in the in vitro cell
experiments to validate the association between DNA methylation and
gene transcription.
Materials and methods
Human articular cartilage samples
Articular cartilage was obtained from 3 knee joint
of patients (patient 1: Male, 75 years; patient 2: Female, 69
years; patient 3: Female, 90 years) with OA undergoing knee
replacement surgery at Renmin Hospital of Wuhan University (Wuhan,
China) in July 2020. Previous studies have confirmed that the outer
lateral tibial plateau (oLT), the inner lateral tibial plateau
(iLT) and the inner medial tibial plateau (iMT) regions of the knee
represent the early, intermediate and late stages of OA (17). Normal and OA cartilage samples were
collected from the outer region of the lateral tibial plateau and
the inner medial tibial plateau. The present study was approved by
the Ethics Committee of Renmin Hospital of Wuhan University (Wuhan,
China).
Cell culture
The primary human chondrocytes (cat. no. CP-H096)
were purchased from Procell Life Science & Technology Co., Ltd.
5'Aza, a DNA methylation inhibitor, was purchased from Aladdin
(cat. no. A119533). The primary human chondrocytes were cultured in
90% DMEM/F12 medium (cat. no. SH30023.01; Hyclone; Cytiva) with
added 10% FBS (Gibco; Thermo Fisher Scientific, Inc.). To gain
further insight into the mechanisms of transcriptional regulation
by DNA demethylation, the cultured primary chondrocytes were
treated with 5'Aza and compared with those that were untreated. The
concentration of 5'Aza and cartilage culture time according to
those described in previous studies (18,19).
Total RNA was then extracted to detect the mRNA expression of the
target genes.
Analysis of mRNA expression profiling
data
Two mRNA expression profiling datasets, GSE114007
and GSE113825, were obtained from the Gene Expression Omnibus (GEO)
database (https://www.ncbi.nlm.nih.gov/gds/). GSE114007
contained 18 normal and 20 OA cartilage samples, while the
GSE113825 contained 5 normal and 5 OA cartilage samples from
patients without history of OA and patients with OA. The Limma
package in R (v3.6.0) was used to identify DEGs between the normal
and OA cartilage samples. The cutoff value for DEGs was abs(logFC)
>1 and adjusted P-value <0.05.
Data normalization and analysis of DNA
methylation profiling data
The methylation profiling dataset GSE63695 was
obtained from the GEO database (https://www.ncbi.nlm.nih.gov/gds/) and it contained 21
samples of normal cartilage and 23 samples of primary OA cartilage
from healthy subjects and patients with OA. Raw intensity data were
analyzed by using the ChAMP methylation analysis package in R
(v3.6.0) (20). After filtering the
unnecessary CpG-including probes with single nucleotide
polymorphisms or probes located on X and Y chromosomes, the
remaining probes were normalized to perform a type-II probe
normalization by using Beta Mixture Quantile dilation (21). DMS were defined as CpG sites with
absolute (abs)[log fold change (FC)] >0.1 and adjusted P-value
<0.05 (22,23).
Enrichment analysis of CpG sites
The distribution of DMS across the genome may affect
their regulation of gene expression. The percentages of DMS and
array sites (after filtering) in different subregions and their
positions relative to a CpG island (CGI) were analyzed separately.
The seven different subregions included transcription start site
(TSS)200, TSS1500, 1st exon, 3'UTR, 5'UTR, intergenic regions and
gene body. The positions relative to a CGI included the shore,
shelf and opensea regions (24).
Reverse transcription-quantitative PCR
(RT-qPCR)
RNA was isolated from cartilage tissue using a
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.).
Subsequently, RNA was transcribed into complementary (c)DNA using
the RevertAid First Strand cDNA Synthesis Kit (cat. no. K1622;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. Quantitative analysis of mRNA levels was performed using
the FastStart Universal SYBR Green Master Mix (Rox; Roche
Diagnostics GmbH) according to the manufacturer's protocol. The
primer sequences were presented in Table I. GAPDH was used as the internal
reference gene. The thermocycling conditions were as follows:
Initial denaturation at 95˚C for 30 sec, followed by 40 cycles of
95˚C for 15 sec and 60˚C for 60 sec. Relative mRNA expression
levels were calculated using the 2-ΔΔCq method (25).
 | Table IPrimer sequences of the genes used
for PCR. |
Table I
Primer sequences of the genes used
for PCR.
| Gene name | Forward
(5'-3') | Reverse
(5'-3') |
|---|
| GAPDH |
CAATGACCCCTTCATTGACC |
GACAAGCTTCCCGTTCTCAG |
| MAP1B |
GGAGCGAGACACTTCGCC |
TGATCATTAAGCGCACCTCG |
| FNDC1 |
CCACCCAAAGATGCTACCAGT |
TTGGGCACTTCCTTTTCTGTG |
| ANLN |
GGTGTGGTAAGTCCAGAGAGTT |
CACCAGATTCAGCTCGAGGG |
| KCNN4 |
ATCGGCGCTCTCAATCAAGT |
ATTAACAGCCTGCCTCTCGG |
| SCNN1A |
CATGAGCAGTATCAAGGGGAA |
ACGAGCTTGTCCGAGTTGAG |
| STC2 |
CTGTGGAGGTCAGTGGGTGTC |
AGCCCAGACAGTACAATGGA |
Statistical analysis
Values are expressed as the mean ± standard error of
the mean. Comparisons between the two groups were performed using
the unpaired Student's t-test. GraphPad Prism v.7 (GraphPad
Software, Inc.) was used to draw graphs. P<0.05 was considered
to indicate statistical significance.
Results
Screening of DEGs
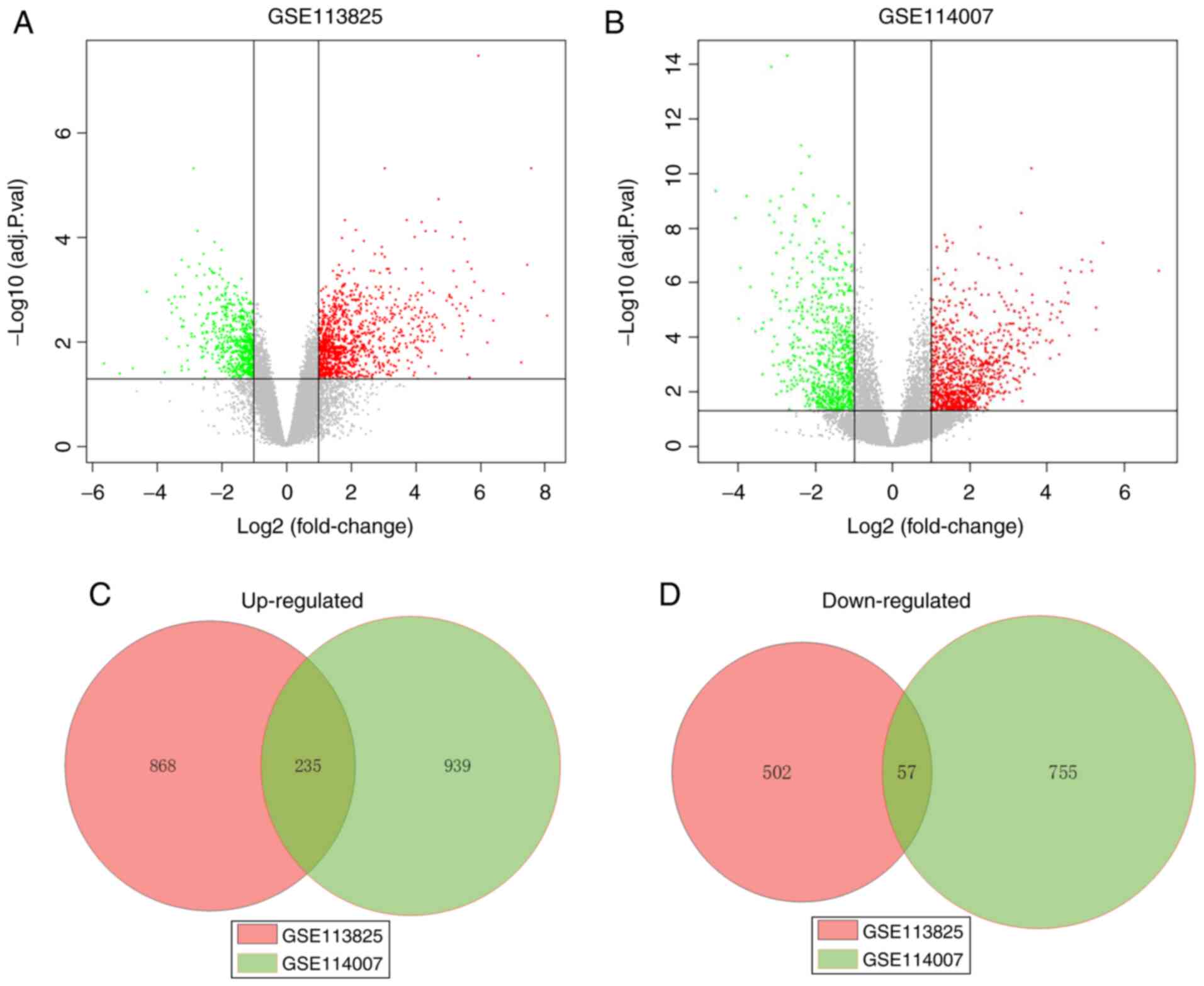
A total of 1,662 and 1,986 DEGs were identified
between the expression profiles of OA and normal human cartilage
from the GSE113825 and GSE114007 dataset, respectively. In the
GSE113825 dataset, 1,103 DEGs were upregulated and 559 were
downregulated in the OA samples, and in the GSE114007 dataset,
1,174 DEGs were upregulated and 812 were downregulated in the OA
samples. Of these, further screening revealed that 292 genes shared
common differences between the two sets of data, among which 235
were upregulated and 57 were downregulated in OA. The full list of
the common DEGs is provided in Table
SI. The volcano plots and Venn diagrams of DEGs among GSE113825
and GSE114007 are presented in Fig.
1.
Screening of DMGs
A total of 574 DMS that contained 394 DMGs were
identified between the normal and OA cartilage from GSE63695. A
complete list of the DMS is provided in Table SII. Among these DMS in the OA
cartilage, 220 sites were hypomethylated, while 355 were
hypermethylated. Of those 394 DMGs, certain genes that were related
to the generation and degradation of extracellular matrix,
including ADAMTS17, COL9A1, COL11A2 and COL28A1, and members of the
TGF signaling pathway, including BMP6 and SMAD6(26).
Enrichment analysis of CpG sites
Enrichment analysis was performed for DMS and total
array sites (after filtering) across the genome or locations
relative to CGIs and the results are provided in Table II. The percentages of DMS and total
array sites in the gene body were 45 and 34%, respectively, while
the percentages in the ‘promoter’ regions (including TSS1500,
TSS200, 5'UTR and the 1st Exon) were 23 and 40%, respectively. In
addition, 9% of DMS in comparison with 33% of total array sites
were located on CGIs. These results indicated that the occurrence
of differential methylation was related to the genomic subregions,
which appeared more prominent in the regions that had a low CpG
density. A previous study has also demonstrated that CGIs were
preferentially observed in the promoter region of genes (27).
| Table IIDistribution of DMS and array sites
(after filtering) in different subregions. |
Table II
Distribution of DMS and array sites
(after filtering) in different subregions.
| CpG subregion | DMS | DMS rate (%) | Array sites (after
filtering) | Array rate (after
filtering; %) |
|---|
| 3'UTR | 44 | 3 | 13,781 | 3 |
| 5'UTR | 117 | 9 | 35,183 | 9 |
| 1st exon | 20 | 2 | 19,213 | 5 |
| Gene body | 580 | 45 | 130,554 | 34 |
| TSS200 | 41 | 3 | 44,891 | 12 |
| TSS1500 | 109 | 9 | 55,991 | 14 |
| IGR | 365 | 29 | 87,573 | 23 |
| Island | 121 | 9 | 129,101 | 33 |
| Opensea | 761 | 60 | 131,751 | 34 |
| Shelf | 128 | 10 | 35,245 | 9 |
| Shore | 266 | 21 | 91,089 | 24 |
Comprehensive analysis of DEGs and
DMGs
When comparing DEGs and DMGs, 15 common genes were
identified as presented in Table
III. Of these, six genes (MAP1B, FNDC1, ANLN, KCNN4, SCNN1A and
STC2) were further investigated. The DMS of STC2 and SCNN1A were
located in the promoter, with the level of methylation negatively
associated with gene transcription. On the other hand, the DMS of
MAP1B, FNDC1, ANLN and KCNN4 were located in the gene body, with
the level of methylation positively associated with gene
transcription.
| Table IIIDifferentially expressed genes
harboring differentially methylated sites between OA and normal
knee articular cartilage. |
Table III
Differentially expressed genes
harboring differentially methylated sites between OA and normal
knee articular cartilage.
| A, Hypermethylated
in OA |
|---|
| Gene symbol | Gene expression
status in OA | CpG genomic
feature |
|---|
| ANPEP | Upregulated | 5'UTR |
| MAP1B | Upregulated | Body |
| FNDC1 | Upregulated | Body |
| TPPP3 | Upregulated | TSS1500 |
| ANLN | Upregulated | Body |
| BMPR1B | Upregulated | 5'UTR |
| KCNN4 | Upregulated | Body |
| SCNN1A | Downregulated | 5'UTR |
| STC2 | Downregulated | TSS1500 |
| B, Hypomethylated
in OA |
| Gene symbol | Gene expression
status in OA | CpG genomic
feature |
| TRPV2 | Upregulated | Body |
| LHFPL2 | Upregulated | Body |
| IGFBP4 | Upregulated | Body |
| BTBD11 | Upregulated | Body |
| CAPZB | Upregulated | Body |
| AQP1 | Upregulated | 1st exon, body |
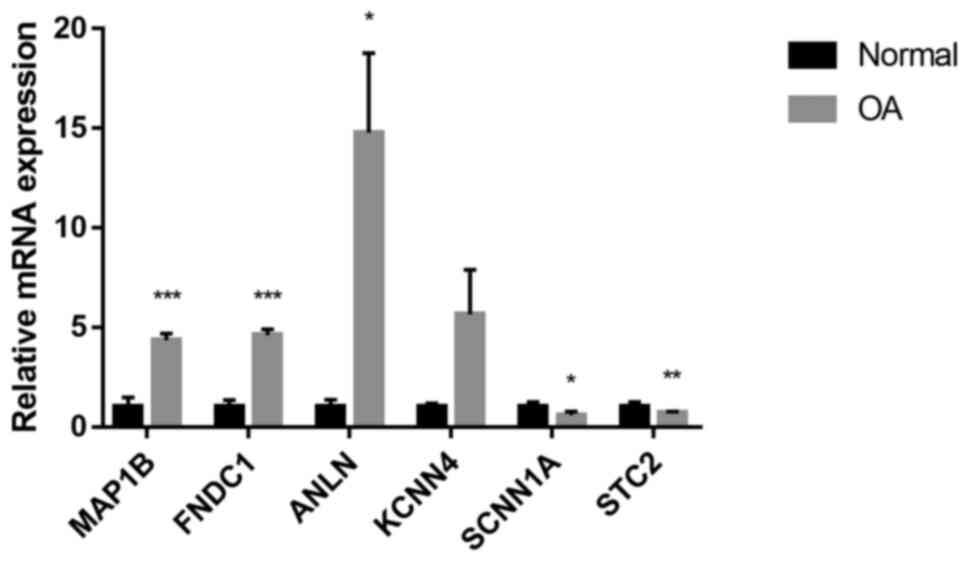
Validation of screened genes
The mRNA expression of those selected screened genes
was verified by RT-qPCR, which indicated that in the OA cartilage,
MAP1B, FNDC1 and ANLN were upregulated, while SCNN1A and STC2 were
downregulated (P<0.05, P<0.01 or P<0.001; Fig. 2). No significant alteration in the
mRNA levels of ANLN was observed. This was consistent with the
results obtained from the mRNA expression datasets.
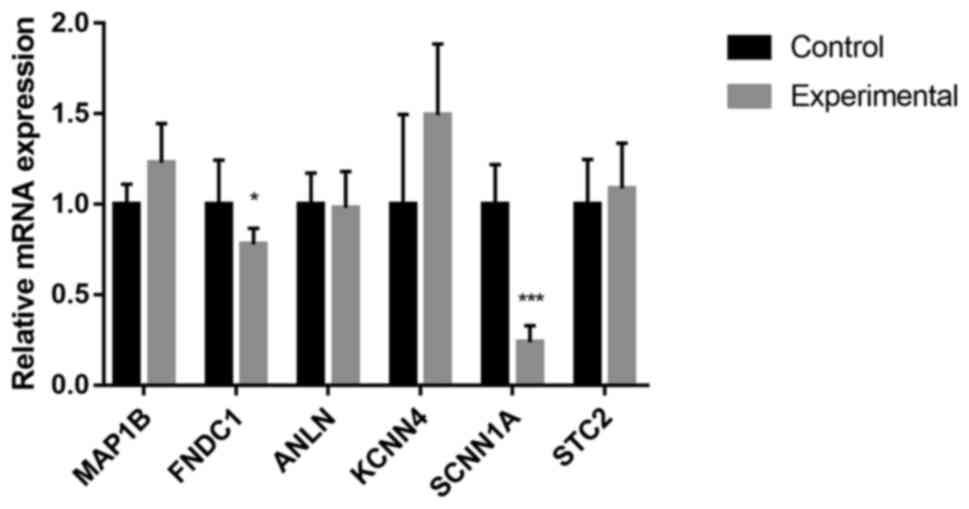
Changes in gene expression in cultured
chondrocytes after DNA demethylation
To investigate whether hypomethylation affected the
transcription of those genes, 5'Aza, a DNA methylation inhibitor
(28), was applied as a useful tool
in a further experiment. The effects of 5'Aza on the expression of
the MAP1B, FNDC1, ANLN, KCNN4, SCNN1A and STC2 genes in cultured
human articular chondrocytes were assessed. As indicated in
Fig. 3, the expression of FNDC1 and
SCNN1A was downregulated after 5'Aza treatment (P<0.05 and
P<0.001, respectively). No significant alterations in the mRNA
levels of MAP1B, ANLN, KCNN4 and STC2 were observed.
Discussion
In the present study, both the DNA methylation data
and mRNA expression data of OA and normal cartilage were analyzed
to identify genes that may have a role in the pathogenesis of the
disease. A total of two mRNA expression datasets were used to
accurately identify DEGs. The methylation profiling dataset
GSE63695 obtained from GEO database used for the Illumina Human
Methylation 450 BeadChip microarray. The array typically measures a
total of 485,512 CpG sites and its probes have a wide range of
genomic characteristics, including enhancers, promoters, UTR and
gene body. This may provide a broader understanding of the
epigenetic changes that occur in the pathogenesis of OA when
compared with the Illumina 27K methylation microarray (29).
Previous studies have indicated that differential
mRNA expression of certain key genes in the pathogenesis of OA is
associated with changes in their epigenetic status, such as COL9A1.
COL9A1 encodes type IX collagen, which is paramount in the
formation of a collagen network that maintains the integrity of
cartilage tissue while protecting cartilage tissue from mechanical
damage (30,31). The study by Imagawa et al
(13) indicated that
hypermethylation of the COL9A1 promoter resulted in its
downregulation in OA cartilage tissue. Indeed, studies have
revealed numerous distinct DMS between OA and normal tissue, but
only a small fraction of these sites have been indicated to affect
gene expression. To date, in OA, the impact of DMS on changes to
articular cartilage-related gene expression and the specific
mechanisms have remained to be fully elucidated.
DMS between normal and OA cartilage may be
distinguished by their genomic features such as promoters,
enhancers, UTR and gene bodies. In the present study, an enrichment
analysis was performed for DMS and total array sites (after
filtering) across the genome. Most of the DMS were enriched in the
intergenic region and gene body, while only 23% were enriched in
the promoter (32). Furthermore,
when the positions of the DMS relative to a CGI were analyzed, the
percentage of DMS on CGIs was small, indicating that regions
lacking CGIs, such as the gene body and enhancer subregions, were
more likely to undergo changes in methylation levels than promoters
that were rich in CGIs.
Methylation of the promoter primarily imposes a
negative regulatory effect on transcription when compared with
methylation of the gene body (24,33).
The 1st exon has been considered as an important negative factor in
the regulation of transcription (34). In the present study, 15 common key
genes were identified and six of these were verified, namely MAP1B,
FNDC1, ANLN, KCNN4, SCNN1A and STC2. First, mRNAs were extracted
from the knee cartilage tissues of patients with OA. RT-qPCR was
then performed to validate the expression levels of mRNA of these
six genes in OA cartilage. Subsequently, validation of these genes
was performed in vitro using primary chondrocytes cultured
with 5'Aza or vehicle, revealing that only STC2 and FNDC1 had met
our expectations; DMS located in the promoter was negatively
associated with mRNA levels, while DMS located in the gene body or
the 3'UTR was positively associated with gene transcription.
The present study suggested a relationship between
methylation levels and gene expression, which appears more complex
than initially postulated. Although methylation of promoter regions
has been associated with silencing of the downstream gene
expression, it has long been debated whether it is silencing or
methylation that comes first (11).
Of note, the study by Keller et al (35) has indicated that the role of DNA
methylation at the promoter regions of C. intestinalis was
related to the methylation states of the nearby gene body regions.
Furthermore, promoter methylation adjacent to the methylated gene
body was negatively correlated with gene expression levels, but
this phenomenon was not observed when not in the direct vicinity to
the methylated gene body (35).
The role of gene body methylation has remained to be
fully elucidated (36,37). It has long been postulated that gene
body DNA methylation suppresses spurious transcription in the
coding region, given that DNA methylation is usually repressive
(38). Through this, gene body
methylation may effectively inhibit ‘transcriptional noise’,
meaning that gene expression levels between cells may vary despite
the same genetic materials and biological conditions (36). This hypothesis, if true, would
account for the consistent positive correlation between the
methylation of genes and their transcriptional levels. In addition,
other studies have suggested that gene body DNA methylation may
function in conjunction with other epigenetic modifications. For
instance, the initiation and elongation of forged transcripts are
inhibited by excluding the occupation of RNA polymerase II in
addition to recruiting multiple inhibitory histone markers
(39). The study by Jjingo et
al (37) also has uncovered the
relationship between gene body methylation and expression levels,
which appeared bell-shaped rather than monotonic. Hence, a model
has been proposed that may explain the relationship as a result of
interactions between chromatin openness and RNA polymerase II
density, in which genes with low or high expression had the lowest
methylation level, while genes with medium expression had the
highest methylation level (37).
The present results may also be explained by other
studies. For instance, Den Hollander et al (40) postulated a novel theory that
epigenetic regulation only affects the transcription of certain
genes, such as genes involved in maintaining the phenotype of
chondrocytes. In addition, Imagawa et al (13) suggested that the mRNA levels of
COL2A1 in OA chondrocytes were >10-fold higher than those in the
control group, but this was not mediated by changes in the
methylation status of gene promoters or enhancers. However, it may
be inferred that the fold changes of mRNA expression does not
necessarily contribute to higher significance. Furthermore, setting
a threshold of fold change to screen for differential genes may
exclude certain genes with small differential multiples but have an
important functional role.
The present results concerning FNDC1, which may
represent a bone metabolism-related factor (41), were novel. It was previously
reported that inhibition of FNDC1 expression in bone marrow
mesenchymal stem cells resulted in the impediment of postmenopausal
osteoporosis (41). However, to
date, there has been no clear evidence to suggest the role of FNDC1
in cartilage tissue in the pathogenesis of OA. Based on several
previous studies, FNDC1 may share similar functions to fibronectin
(FN), which is a high-molecular-weight glycoprotein comprised of
three types of repeating amino acid units, type I, II and III
repeats (42). The function of FN
varies with the tissue type and fibronectin serves as a scaffold
for cell adhesion and migration that contributes to the healing of
skin wounds (43). However, in the
cartilage tissues of OA, if the removal of fibronectin is not
complete, the persistence of fibronectin fragments or aggregates
impairs tissue regeneration and remodeling. When degraded into
multiple fragments by proteases, FN may bind to C1q of the
complement system, which may lead to chronic leukocyte stimulation
(44,45). In addition, FN fragments may inhibit
the synthesis of cartilage matrix such as sulfated proteoglycans
and stimulate the release of proinflammatory cytokines and MMP,
thus mediating cartilage injury in OA (42). Thus, FNDC1 may potentially be a
valuable biomarker for OA.
In the present study, certain key genes associated
with the distribution of DMS across the genome were identified and
further validated via in vitro experiments. The present
results suggested that the regulation of gene transcription by DNA
methylation may not have a relatively fixed rule. However, compared
with the conventional genome-wide DNA methylation analysis, this
may improve the current understanding of the function of DNA
methylation and its regulation of transcription. Nevertheless, the
establishment of a causal relationship remains a great challenge in
epigenetics research (23). Further
mechanistic studies, such as large-scale longitudinal interference
in animal experiments, are therefore warranted. In addition, for
the key genes identified in the present study, further analysis and
functional verification will be performed in the future. For
instance, alterations in the binding status of transcription
factors to genes following changes in DNA methylation levels may be
detected by a chromatin immunoprecipitation assay. Alternatively,
by knocking out or overexpressing these genes, the effect of these
changes on chondrocyte or tissue phenotype and the specific
regulatory mechanisms may be observed.
There were certain limitations to the present study.
First, the methylation level of target genes prior to and after
5'Aza treatment was not investigated. Undoubtedly, the assessment
of changes in methylation levels may assist in determining the role
of 5'Aza in affecting the methylation levels more accurately and
may provide a clearer presumption that it was the changes in DNA
methylation levels that regulated the transcription of genes.
However, according to previous studies, this part is non-essential
(18,46). Furthermore, most previous studies
have indicated that 5'Aza accurately demethylates the DNA of cells
(28,47). Therefore, the measurement of the
methylation level of target genes concerning 5'Aza treatment is
considered non-essential. As another limitation, the analysis of
two mRNA expression data using the intersection method to obtain
common differential genes lacks novelty (22). Finally, the sample size of patients
from whom articular cartilage tissue was obtained and the sample
size of the DNA methylation and mRNA expression data obtained from
the GEO database were small.
In the in vitro experiments involving culture
of human chondrocytes, glucosamine has been indicated to prevent
cytokine-induced demethylation of the promoter of IL-1β, resulting
in decreased IL-1β expression. This suggests that modification of
DNA methylation may be a potential therapeutic strategy to
intervene in the OA process (9).
In conclusion, in the present study, a group of key
genes in OA regulated by DNA methylation was identified and several
of them were validated. The present results provide an enhanced
understanding of the regulation of gene transcription of these key
genes by epigenetics in OA, which may serve as potential biomarkers
in the pathogenesis of OA and will provide avenues for targeted
therapeutic intervention of OA pathogenesis through modification of
DNA methylation.
Supplementary Material
The full list of the common DEGs
between OA and normal cartilage from the datasets GSE113825 and
GSE114007.
A complete list of the DMS between OA
and normal knee articular cartilage from the dataset GSE63695.
Acknowledgements
Not applicable.
Funding
Funding: The authors are grateful for the financial support from
the National Natural Science Foundation of China (grant no.
81071494) and Hubei Provincial Science and Technology Support
Program (grant no. 2015BCA316).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
PY and BQ designed the study. PY, XX and JY acquired
and interpreted the data. PY and XX analyzed the data and were
major contributors in writing the manuscript. BQ prepared the
manuscript and supervised the study. BQ and XX approve the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Renmin Hospital of Wuhan University (Wuhan,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Miranda-Duarte A: DNA methylation in
osteoarthritis: Current status and therapeutic implications. Open
Rheumatol J. 12:37–49. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Jeffries MA: Osteoarthritis year in review
2018: Genetics and epigenetics. Osteoarthritis Cartilage.
27:371–377. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lotz M and Loeser RF: Effects of aging on
articular cartilage homeostasis. Bone. 51:241–248. 2012.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Goldberg AD, Allis CD and Bernstein E:
Epigenetics: A landscape takes shape. Cell. 128:635–638.
2007.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Barter MJ, Bui C and Young DA: Epigenetic
mechanisms in cartilage and osteoarthritis: DNA methylation,
histone modifications and microRNAs. Osteoarthritis Cartilage.
20:339–349. 2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Shen J, Abu-Amer Y, O'Keefe RJ and
McAlinden A: Inflammation and epigenetic regulation in
osteoarthritis. Connect Tissue Res. 58:49–63. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Im GI and Choi YJ: Epigenetics in
osteoarthritis and its implication for future therapeutics. Expert
Opin Biol Ther. 13:713–721. 2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Roach HI, Yamada N, Cheung KS, Tilley S,
Clarke NM, Oreffo RO, Kokubun S and Bronner F: Association between
the abnormal expression of matrix-degrading enzymes by human
osteoarthritic chondrocytes and demethylation of specific CpG sites
in the promoter regions. Arthritis Rheum. 52:3110–3124.
2005.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Imagawa K, de Andres MC, Hashimoto K, Pitt
D, Itoi E, Goldring MB, Roach HI and Oreffo RO: The epigenetic
effect of glucosamine and a nuclear factor-kappa B (NF-κB)
inhibitor on primary human chondrocytes-implications for
osteoarthritis. Biochem Biophys Res Commun. 405:362–367.
2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Hashimoto K, Oreffo RO, Gibson MB,
Goldring MB and Roach HI: DNA demethylation at specific CpG sites
in the IL1β promoter in response to inflammatory cytokines in human
articular chondrocytes. Arthritis Rheum. 60:3303–3313.
2009.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Jones PA: Functions of DNA methylation:
Islands, start sites, gene bodies and beyond. Nat Rev Genet.
13:484–492. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Harris RA, Wang T, Coarfa C, Nagarajan RP,
Hong C, Downey SL, Johnson BE, Fouse SD, Delaney A, Zhao Y, et al:
Comparison of sequencing-based methods to profile DNA methylation
and identification of monoallelic epigenetic modifications. Nat
Biotechnol. 28:1097–1105. 2010.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Imagawa K, de Andres MC, Hashimoto K, Itoi
E, Otero M, Roach HI, Goldring MB and Oreffo RO: Association of
reduced type IX collagen gene expression in human osteoarthritic
chondrocytes with epigenetic silencing by DNA hypermethylation.
Arthritis Rheumatol. 66:3040–3051. 2014.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Hahn MA, Wu X, Li AX, Hahn T and Pfeifer
GP: Relationship between gene body DNA methylation and intragenic
H3K9me3 and H3K36me3 chromatin marks. PLoS One.
6(e18844)2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Laurent L, Wong E, Li G, Huynh T, Tsirigos
A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J and Wei CL:
Dynamic changes in the human methylome during differentiation.
Genome Res. 20:320–331. 2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hellman A and Chess A: Gene body-specific
methylation on the active X chromosome. Science. 315:1141–1143.
2007.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Chou CH, Lee CH, Lu LS, Song IW, Chuang
HP, Kuo SY, Wu JY, Chen YT, Kraus VB, Wu CC and Lee MT: Direct
assessment of articular cartilage and underlying subchondral bone
reveals a progressive gene expression change in human
osteoarthritic knees. Osteoarthritis Cartilage. 21:450–461.
2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Zhao L, Wang Q, Zhang C and Huang C:
Genome-wide DNA methylation analysis of articular chondrocytes
identifies TRAF1, CTGF, and CX3CL1 genes as hypomethylated in
osteoarthritis. Clin Rheumatol. 36:2335–2342. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Iliopoulos D, Malizos KN and Tsezou A:
Epigenetic regulation of leptin affects MMP-13 expression in
osteoarthritic chondrocytes: Possible molecular target for
osteoarthritis therapeutic intervention. Ann Rheum Dis.
66:1616–1621. 2007.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Morris TJ, Butcher LM, Feber A,
Teschendorff AE, Chakravarthy AR, Wojdacz TK and Beck S: ChAMP:
450k chip analysis methylation pipeline. Bioinformatics.
30:428–430. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Teschendorff AE, Marabita F, Lechner M,
Bartlett T, Tegner J, Gomez-Cabrero D and Beck S: A beta-mixture
quantile normalization method for correcting probe design bias in
Illumina Infinium 450 k DNA methylation data. Bioinformatics.
29:189–196. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Li Z, Zhang R, Yang X, Zhang D, Li B,
Zhang D, Li Q and Xiong Y: Analysis of gene expression and
methylation datasets identified ADAMTS9, FKBP5, and PFKBF3 as
biomarkers for osteoarthritis. J Cell Physiol. 234:8908–8917.
2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Rushton MD, Reynard LN, Barter MJ, Refaie
R, Rankin KS, Young DA and Loughlin J: Characterization of the
cartilage DNA methylome in knee and hip osteoarthritis. Arthritis
Rheumatol. 66:2450–2460. 2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Mishra NK and Guda C: Genome-wide DNA
methylation analysis reveals molecular subtypes of pancreatic
cancer. Oncotarget. 8:28990–29012. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Jung SM, Lee JH, Park J, Oh YS, Lee SK,
Park JS, Lee YS, Kim JH, Lee JY, Bae YS, et al: Smad6 inhibits
non-canonical TGF-β1 signalling by recruiting the deubiquitinase
A20 to TRAF6. Nat Commun. 4(2562)2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Brena RM, Huang TH and Plass C: Toward a
human epigenome. Nat Genet. 38:1359–1360. 2006.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kim KI, Park YS and Im GI: Changes in the
epigenetic status of the SOX-9 promoter in human osteoarthritic
cartilage. J Bone Miner Res. 28:1050–1060. 2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wright ML, Dozmorov MG, Wolen AR,
Jackson-Cook C, Starkweather AR, Lyon DE and York TP: Establishing
an analytic pipeline for genome-wide DNA methylation. Clin
Epigenetics. 8(45)2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Mustafa Z, Chapman K, Irven C, Carr AJ,
Clipsham K, Chitnavis J, Sinsheimer JS, Bloomfield VA, McCartney M,
Cox O, et al: Linkage analysis of candidate genes as susceptibility
loci for osteoarthritis-suggestive linkage of COL9A1 to female hip
osteoarthritis. Rheumatology (Oxford). 39:299–306. 2000.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Loughlin J, Mustafa Z, Dowling B, Southam
L, Marcelline L, Raina SS, Ala-Kokko L and Chapman K: Finer linkage
mapping of a primary hip osteoarthritis susceptibility locus on
chromosome 6. Eur J Hum Genet. 10:562–568. 2002.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Li K, Qin L, Jiang S, Li A, Zhang C, Liu
G, Sun J, Sun H, Zhao Y, Li N and Zhang Y: The signature of
HBV-related liver disease in peripheral blood mononuclear cell DNA
methylation. Clin Epigenetics. 12(81)2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Gutierrez-Arcelus M, Lappalainen T,
Montgomery SB, Buil A, Ongen H, Yurovsky A, Bryois J, Giger T,
Romano L, Planchon A, et al: Passive and active DNA methylation and
the interplay with genetic variation in gene regulation. Elife.
2(e00523)2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Brenet F, Moh M, Funk P, Feierstein E,
Viale AJ, Socci ND and Scandura JM: DNA methylation of the first
exon is tightly linked to transcriptional silencing. PLoS One.
6(e14524)2011.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Keller TE, Han P and Yi SV: Evolutionary
transition of promoter and gene body DNA methylation across
invertebrate-vertebrate boundary. Mol Biol Evol. 33:1019–1028.
2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Huh I, Zeng J, Park T and Yi SV: DNA
methylation and transcriptional noise. Epigenetics Chromatin.
6(9)2013.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Jjingo D, Conley AB, Yi SV, Lunyak VV and
Jordan IK: On the presence and role of human gene-body DNA
methylation. Oncotarget. 3:462–474. 2012.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Bird AP and Wolffe AP: Methylation-induced
repression-belts, braces, and chromatin. Cell. 99:451–454.
1999.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Lorincz MC, Dickerson DR, Schmitt M and
Groudine M: Intragenic DNA methylation alters chromatin structure
and elongation efficiency in mammalian cells. Nat Struct Mol Biol.
11:1068–1075. 2004.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Den Hollander W, Ramos YF, Bomer N,
Elzinga S, Van der Breggen R, Lakenberg N, De Dijcker WJ, Suchiman
HE, Duijnisveld BJ, Houwing-Duistermaat JJ, et al: Transcriptional
associations of osteoarthritis-mediated loss of epigenetic control
in articular cartilage. Arthritis Rheumatol. 67:2108–2116.
2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Xiao Y, Wei R, Yuan Z, Lan X, Kuang J, Hu
D, Song Y and Luo J: Rutin suppresses FNDC1 expression in bone
marrow mesenchymal stem cells to inhibit postmenopausal
osteoporosis. Am J Transl Res. 11:6680–6690. 2019.PubMed/NCBI
|
|
42
|
Stoffels JM, Zhao C and Baron W:
Fibronectin in tissue regeneration: Timely disassembly of the
scaffold is necessary to complete the build. Cell Mol Life Sci.
70:4243–4253. 2013.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Pankov R and Yamada KM: Fibronectin at a
glance. J Cell Sci. 115:3861–3863. 2002.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Bing DH, Almeda S, Isliker H, Lahav J and
Hynes RO: Fibronectin binds to the C1q component of complement.
Proc Natl Acad Sci USA. 79:4198–4201. 1982.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Carsons SE, Schwartzman S, Diamond HS and
Berkowitz E: Interaction between fibronectin and C1q in rheumatoid
synovial fluid and normal plasma. Clin Exp Immunol. 72:37–42.
1988.PubMed/NCBI
|
|
46
|
Alvarez-Garcia O, Fisch KM, Wineinger NE,
Akagi R, Saito M, Sasho T, Su AI and Lotz MK: Increased DNA
methylation and reduced expression of transcription factors in
human osteoarthritis cartilage. Arthritis Rheumatol. 68:1876–1886.
2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Christman JK: 5-Azacytidine and
5-aza-2'-deoxycytidine as inhibitors of DNA methylation:
Mechanistic studies and their implications for cancer therapy.
Oncogene. 21:5483–5495. 2002.PubMed/NCBI View Article : Google Scholar
|