Introduction
Atherosclerosis is a chronic, multistep inflammatory
disease characterized by cholesterol accumulation in the arterial
walls (1,2). The disease starts with the recruitment
of macrophages and the formation of foam cells, ending with
myocardial infarction, ischemic stroke or gangrene (3-10).
Currently, atherosclerosis, especially coronary artery disease,
remains to be a leading cause of morbidity and mortality in both
developed and some developing countries (3,4).
However, drugs for the treatment of atherosclerosis are still under
development though breakthroughs in atherosclerosis treatment could
be imminent (11).
Epigenetic regulation of transcription refers to
modifications in gene expression without changing the DNA sequence
(11,12). Additionally, epigenetic regulations
are increasingly being associated with atherosclerosis in human and
animal models, and are of interest from both therapeutic and
biomarker perspectives (11-14).
Previous studies have reported that apolipoprotein E-deficient
(ApoE-/-) mice fed with a high-fat diet have
aberrant epigenetic changes, including decreased global methylation
in aorta tissue samples and peripheral blood monocytes, as well as
special gene hypermethylation in atherosclerotic plaques and
lesion-predisposed regions (15-18).
Generally, histone modifications include methylation, acetylation,
ubiquitination, phosphorylation, glycosylation and sumoylation are
key epigenetic markers that play critical roles in the modification
of gene expression and have been identified to play an important
role in the development of atherosclerosis (19-21).
Histone methylation and acetylation in macrophages and vascular
endothelial cells have been reported to markedly influence the
progression of atherosclerosis (21).
Histone methyltransferase enhancer of zeste homolog
2 (EZH2) is a histone methyltransferase and is the catalytic
subunit of the polycomb repressive complex-2 (PRC2)/EED-EZH2
protein complex, which can catalyze trimethylation of histone H3 at
Lys 27 (H3K27me3) to dramatically regulate gene expression through
epigenetic machinery. Previous studies have found evidence that
EZH2-induced downregulation of ATP binding cassette subfamily A
member 1 (ABCA1) and insulin like growth factor binding protein 5
(IGFBP5) gene expression levels (22,23),
which promotes lipid accumulation in cells and the development of
atherosclerosis. Therefore, inhibition of the histone
methyltransferase EZH2 may have considerable potential for
suppressing atherogenesis. GSK126 is a small-molecule inhibitor of
both wild-type and mutant EZH2 methyltransferase, that has been
tested in a phase 1/2 dose escalation study to investigate its
safety, pharmacokinetics, pharmacodynamics and clinical activity in
patients with relapsed or refractory diffuse large B cell and
transformed follicular lymphoma (24). To determine the efficacy of GSK126
for suppressing atherogenesis, the effects and mechanisms of EZH2
inhibition were investigated in vitro and in vivo in
the present study.
Materials and methods
Chemicals and antibodies
The EZH2 methyltransferase inhibitor GSK126 (cat.
no. S7061) was purchased from Selleck Chemicals and was dissolved
in DMSO to obtain a stock solution (100 µg/ml) and stored at -80˚C.
The following antibodies were used in the present study: Mouse
monoclonal anti-H3K27me3 (cat. no. ab6147), rabbit polyclonal
anti-ABCA1 (cat. no. ab7360), rabbit monoclonal anti-vascular cell
adhesion molecule 1 (VCAM1; for immunohistochemistry; cat. no.
ab134047) and rabbit polyclonal anti-lectin-like oxidized
low-density lipoprotein (Ox-LDL) receptor 1 (LOX-1; cat. no.
ab60178) were purchased from Abcam. Rabbit monoclonal anti-VCAM1
(cat. no. PA5-102452; for immunohistochemistry) was purchased from
Invitrogen. Rabbit monoclonal anti-EZH2 (cat. no. 5246), rabbit
monoclonal anti-β-actin (cat. no. 8457), rabbit monoclonal
anti-VCAM1 (cat. no. 13662; for western blot), rabbit monoclonal
anti-intercellular adhesion molecule 1 (ICAM1; cat. no. 4915) and
rabbit monoclonal anti-histone H3 (cat. no. 4499) were purchased
from Cell Signaling Technology, Inc.
Cell culture
THP-1 and HUVEC cells were purchased from the
American type culture collection (ATCC). THP-1 and HUVEC cells were
cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 10% FBS (Gibco; Thermo Fisher Scientific,
Inc.) and 100 U/ml penicillin-streptomycin (Gibco; Thermo Fisher
Scientific, Inc.) at 37˚C in an atmosphere containing 5%
CO2. For foam cell transformation, THP-1 cells were
treated with phorbol-12-myristate-13-acetate (PMA, 100 nmol/l;
STEMCELL Technologies, Inc.; cat. no. 74044) for 48 h at 37˚C in an
atmosphere containing 5% CO2. The medium was then
replaced with a serum-free medium containing Ox-LDL (50 µg/ml;
Thermo Fisher Scientific, Inc.; cat. no. L34357) for 48 h at 37˚C
in an atmosphere containing 5% CO2 to transform THP-1
cells to foam cells.
Monocyte-endothelial cell adhesion
assay
Following stimulation by Ox-LDL for 24 h, THP-1
cells were collected by centrifugation and re-suspended in culture
medium plus 2',7'-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein
(BCECF-AM; Beyotime Institute of Biotechnology). After incubation
for an hour at 37˚C, fluorescently labeled THP-1 cells were
collected by centrifugation and plated in 96-well plates pre-seeded
with HUVECs. After incubation for an hour, the plates were washed
with PBS for 3 times. Images of fluorescently stained cells were
captured with a fluorescence microscope (Olympus Corporation).
CRISPR-Cas9 system for gene knockout
(KO)
Single guide RNAs (sgRNAs) targeting human EHZ2 or
control (AAVS1) were cloned into lentiCRISPRv2 (Addgene) as
previously described (25). HEK293T
(ATCC; cat. no. CRL-3216) cells were cultured in DMEM (Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS and 100 U/ml
penicillin-streptomycin at 37˚C in an atmosphere containing 5%
CO2. Cells were transfected with 6.7 mg psPAX2 (Addgene,
Inc.), 4.1 mg VSV-G (Addgene, Inc.) and 10 mg lentiviral vectors
using 60 mg of linear polyethylenimine (Polysciences, Inc.).
Subsequently, supernatants containing lentivirus were harvested at
48 h post-transfection and concentrated by ultracentrifugation
(26,000 x g for 2 h at 4˚C). THP-1 cells were plated into 12-well
plates (0.5-3 x106 cells per well) with medium
supplemented with 8 mg/ml polybrene (Sigma-Aldrich; Merck KGaA) and
spin-infected at 700 x g for 2 h at 37˚C (26). Cells were used for subsequent
experiments 2 days after the transfection procedure. CRISPR sgRNA
sequences were as follows: Control sgRNA (AAVS1), 5'-CACCGCTCCCTG
GCCACTTTGCAC-3'; EZH2 KO sgRNA#1, 5'-CACCGTGTT GGGGGTACATTCAGG-3';
EZH2 KO sgRNA#2, 5'-CAC CGTATCAGAAGGAAATTTCCG-3'; and EZH2 KO
sgRNA#3 5'-CACCGTATGATGGGAAAGTACACG-3'.
Cholesterol efflux assay
THP-1 or HUVEC cells were cultured at 60% confluence
and then incubated with 0.2 mCi/ml [3H] cholesterol (PerkinElmer,
Inc.). After incubation for 24 h, cells were washed with cold PBS
twice and equilibrated with fresh medium. Equilibrated [3H]
cholesterol-labeled cells were then washed with cold PBS and
incubated in efflux medium containing RPMI-1640 medium, 0.1% BSA
(EMD Millipore; cat. no. A7906) and human plasma apoA-I (25 µg/ml)
for 6 h. The medium was collected and centrifuged at 14,000 x g for
at least 10 min at 4˚C. Total cell-associated radioactivity was
determined by dissolving the cells in isopropanol (96.7%, 200 µl).
Medium and cell-associated [3H] cholesterol was then measured by
liquid scintillation counting (22). The % efflux was calculated using the
following equation: [total media count/(total cellular count +
total medium count)] x100% (22,27).
Oil Red O stain
THP-1 cells were cultured in RPMI-1640 medium and
treated with PMA (100 nmol/l) for 48 h. Subsequently, THP-1 cells
were washed with PBS and cultured in a serum free medium containing
Ox-LDL (50 µg/ml) and human apoA-I (25 µg/ml; Biodesign
International, Inc.) for a further 48 h. After fully
differentiating into foam cells, the cells were verified by fixing
with 4% paraformaldehyde at room temperature for 30 min, and
subsequently staining with 0.5% Oil Red O and hematoxylin
separately at room temperature for 10 min. Hematoxylin was used for
counterstaining cell nuclei and cells were photographed at x200
magnification under a light microscope (Olympus DP80 Microscope).
Cells stained with Oil Red O were quantified using ImageJ/FIJI
platform (National Institutes of Health).
In vivo protocol
Male ApoE-/- mice (n=18; age, 6-8
weeks; weight ~20 g) were purchased from Beijing HFK Bioscience
Co., Ltd. and maintained under a specific pathogen-free environment
in microisolator cages. Animals were kept at 18-23˚C at 40-60%
humidity under a 12 h light/dark cycle. The mice were separated
into the control group (chow diet + DMSO/PBS injection), vehicle
group (high-fat diet + DMSO/PBS injection) and treatment group
(high-fat diet + GSK126 treatment), with six mice in each group.
The mice in the control group were fed a chow diet, whereas the
mice in the vehicle and treatment groups were fed a high-fat diet
(21% fat, 0.15% cholesterol; Mediscience Diets Co., Ltd.). All
animals had free access to foods. In addition, the mice in the
treatment group received a daily intraperitoneal injection of
GSK126 at a dose of 50 mg/kg/day (mouse body weight) for 10 weeks.
The mice in the vehicle group were administered a daily
intraperitoneal injection of 20% DMSO/PBS (volume, 0.1 ml). Animals
were sacrificed at 16-18 weeks of age. CO2 was supplied
in a precisely regulated and purified form. Flow meter was used in
order to maintain a gradual fill/displacement rate of 10-30% of
chamber volume per min. CO2 flow was maintained for at
least 1 min after respiratory arrest. Following sacrifice by
inhalation of CO2, the animals' heart tissue samples
were collected, embedded in the OCT gel without fixation and stored
at -80˚C immediately. All animal procedures were approved by the
Animal Ethics Committee of Dalian Medical University (Dalian,
China). All in vivo experiments were performed in accordance
with the national legislation and institutional guidelines
(approval no. 17-007).
Atherosclerotic lesion analysis
Frozen sections (OCT gel frozen samples; 8 µm
sections) of the aortic root were stained with Oil Red O and
H&E as previously described. The plaque area in the aortic root
sections was then measured using computer-assisted image
quantification with Image Pro Plus 6.0 (Media Cybernetics, Inc.).
Images were captured using an Olympus fluorescent microscope (DP80;
Olympus Corp). Frozen sections were also stained with Masson's
trichrome at room temperature for 30 min according to
manufacturer's instructions to measure the collagen content in
plaques. All staining solutions were obtained from BASO Precision
Optics Ltd.
Immunohistochemistry
Frozen sections of the aortic root (8 µm) were first
fixed at room temperature for 20 min in methanol (98%,
Sigma-Aldrich; Merck KGaA), then were incubated with 3%
H2O2 for 15 min at room temperature (OriGene
Technologies, Inc.), air-dried for 15 min and incubated with 10%
goat serum (OriGene Technologies, Inc.) for ~30 min at room
temperature. The frozen sections were incubated with anti-LOX-1
(1:200 dilution) and anti-VCAM1 (1:400 dilution) antibodies
overnight at 4˚C overnight. Images were captured using an Olympus
fluorescent microscope (DP80; Olympus Corp). Five
immunohistochemistry sections were quantified using the ImageJ/FIJI
platform (National Institutes of Health).
Lipoprotein uptake assay
THP-1 cells were cultured in RPMI-1640 medium and
treated with PMA for 48 h. Subsequently, the cells were treated
with GSK126 (5 µM) for 36 h at 37˚C in a CO2 incubator.
THP-1 macrophages were then washed with PBS and cultured in
serum-free RPMI-1640 medium. Following overnight fasting, the
macrophages were washed with PBS again and cultured in medium with
or without Ox-LDL (100 µg/ml; Guangzhou Yiyuan Biotechnology Co.,
Ltd.) for 48 h. After incubation, macrophages were fixed in
methanol at room temperature (98%, Sigma-Aldrich; Merck KGaA) for
30 min and stained with Oil Red O for 20 min at room temperature
and observed under a DP80 fluorescent microscope at x200
magnification. Experiments were repeated in triplicate in each
group.
Western blot analysis
Cell lysates of THP-1 cells were isolated using a
Total Protein Extraction kit from Nanjing KeyGen Biotech. Co.,
Ltd., following the manual provided by the manufacturing company.
The protein concentration was determined using a PierceTM BCA
Protein Assay kit (Thermo Fisher Scientific, Inc.) following the
manufacturer's protocols. Denatured proteins (30-50 µg) were
separated by 12% SDS-polyacrylamide gel electrophoresis for 120 min
at 120 V in electrophoretic buffer solution. The proteins were then
transferred to PVDF membranes (EMD Millipore) for 90 min at 400 mA.
Following blocking with 5% milk/Tris-buffered saline (w/v;
Sigma-Aldrich; Merck KGaA) at 25˚C for 1 h, the membranes were
incubated with primary antibodies at 4˚C overnight, including
anti-LOX-1 (dilution, 1:1,000), anti-β-actin (dilution, 1:1,000),
anti-histone H3 (dilution, 1:500), anti-H3K27me3 (dilution,
1:1,000), anti-ABCA1 (dilution, 1:1,000) and anti-EZH2 (dilution,
1:1,000). The membranes were then washed with TBST (1% Tween-20)
and incubated with horseradish peroxidase-conjugated goat
anti-rabbit IgG (cat. no. sc-2004; dilution, 1:10,000) or
horseradish peroxidase-conjugated goat anti-mouse IgM (cat. no.
sc-2064; dilution, 10,000) secondary antibodies obtained from Santa
Cruz Biotechnology, Inc., and Texas Red-X conjugated goat anti-rat
IgG secondary antibody (dilution, 1:200; Thermo Fisher Scientific,
Inc.; cat. no. T-6392) for 1 h at room temperature. The blots were
then treated with the WesternBright ECL kit (Advansta Inc.) and
visualized using the Bio-Rad imaging system (Bio-Rad Laboratories,
Inc.) and analyzed using Image Pro Plus 6.0 (Media Cybernetics,
Inc.).
Statistical analysis
Statistical analysis was performed using SPSS
version 19.0 statistical software (IBM Corp.) and GraphPad Prism 7
(GraphPad Software). All data are presented as the mean ± SD. A
one-way ANOVA was performed to analyze multiple group comparisons
of quantitative data. Bonferroni post hoc-tests were used after
one-way ANOVAs. P<0.05 was considered to indicate a
statistically significant difference.
Results
EZH2 inhibition markedly attenuated
lipoprotein uptake and reduced foam cell formation in human THP-1
monocytes
To gain molecular insights into the therapeutic
effect of EZH2 inhibition on the development of atherosclerosis,
the role of EZH2 was examined using an in vitro model of
atherosclerosis, specifically using THP-1 human monocytes, which
are commonly used for in vitro studies into atherosclerosis
(10). The present study performed
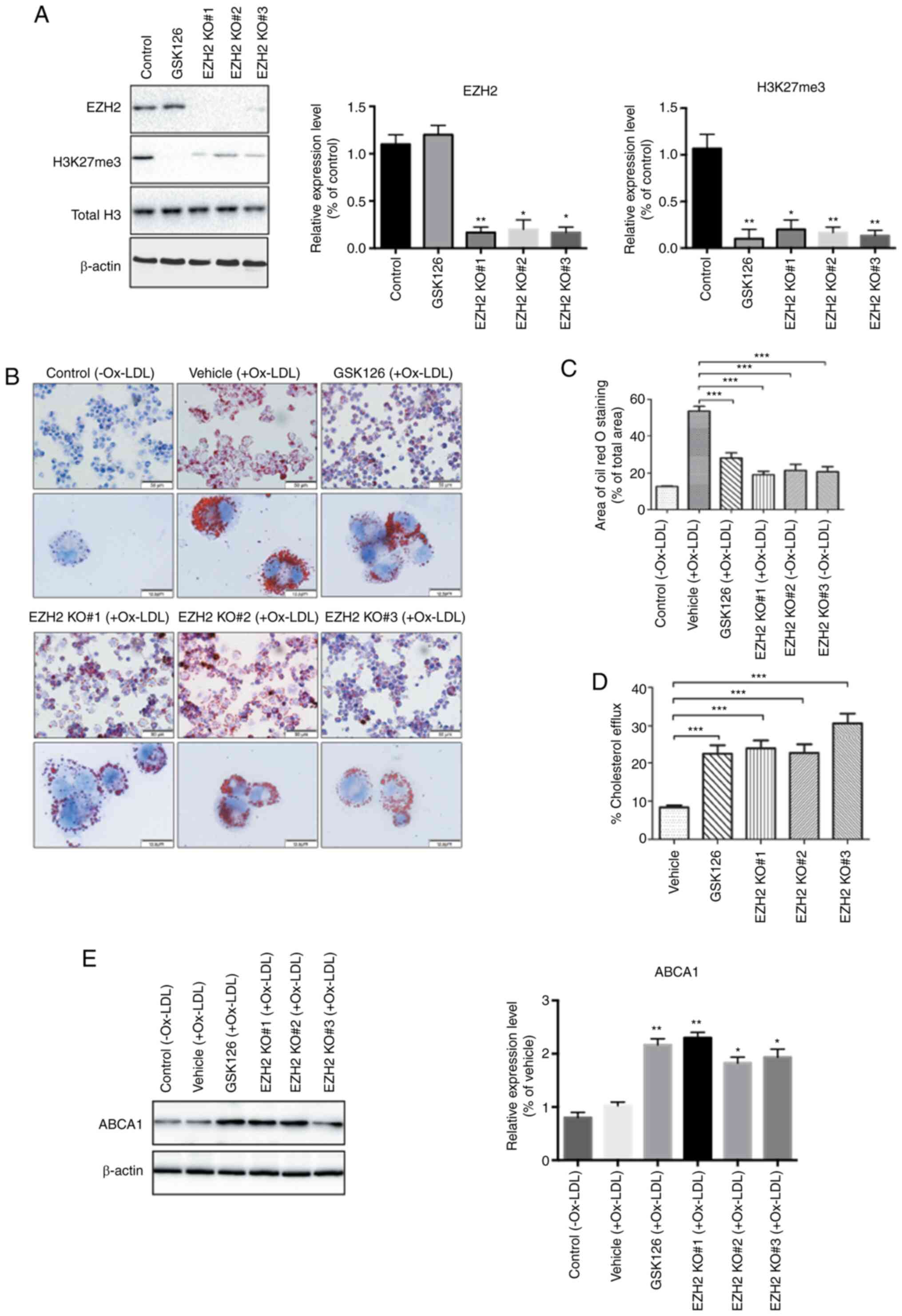
KO of the EZH2 gene in THP-1 monocytes and it was observed that
EZH2 KO significantly decreased the abundance of H3K27me3 in THP-1
cells compared to control cells (Fig.
1A). Meanwhile, administration of GSK126, a specific EZH2
inhibitor, completely abolished H3K27 methylation in THP-1 cells
but did not affect the protein expression levels of EZH2 (Fig. 1A).
The uptake of Ox-LDL by macrophages and subsequent
formation of foam cells is initiated by the development of
atherosclerosis (28). To evaluate
whether EZH2 regulates lipid uptake and foam cell formation, THP-1
cells were incubated with Ox-LDL or vehicle for 48 h after KO or
pharmacological inhibition of EZH2. Compared with the vehicle
treatment, the accumulation of cytoplasmic lipid droplets, as
determined by Oil Red O staining, was visibly reduced in the GSK126
treated and EZH2-null THP-1 cells, implying that foam cell
formation is dependent of EZH2 (Fig.
1B and C). Furthermore, the
present study determined whether the inhibition of EZH2 with sgRNA
or treatment with GSK126 altered the cholesterol efflux in THP-1
macrophage-derived foam cells. Both GSK126 treatment and EZH2-gRNA
lentiviral infection significantly increased cellular cholesterol
efflux (Fig. 1D). To further
understand the inhibitory effect of EZH2 inhibition on foam cell
formation, the protein expression levels of ATP Binding Cassette
Subfamily A Member 1 (ABCA1) were examined, a major regulator of
cellular cholesterol and phospholipid homeostasis in human beings
(22). It was found that the
protein expression levels of ABCA1 were significantly increased
following EZH2 inhibition by EZH2 KO and GSK126 in THP-1 cells as
evidenced by western blotting (Fig.
1E). mRNA expression levels of other important regulators of
cellular cholesterol homeostasis were also measured, including CD36
and ATP Binding Cassette Subfamily G Member 1 (ABCG1) in THP-1
cells treated by GSK126 or vehicle. No significant differences were
observed to the expression levels of CD36 and ABCG1 (data not
shown). Taken together, these data suggested that an EZH2-dependent
epigenetic regulation is involved in foam cell formation, likely
through the upregulation of ABCA1 at the protein level in THP-1
cells.
Inhibition of EZH2 significantly
blocks Ox-LDL induced monocyte adhesion
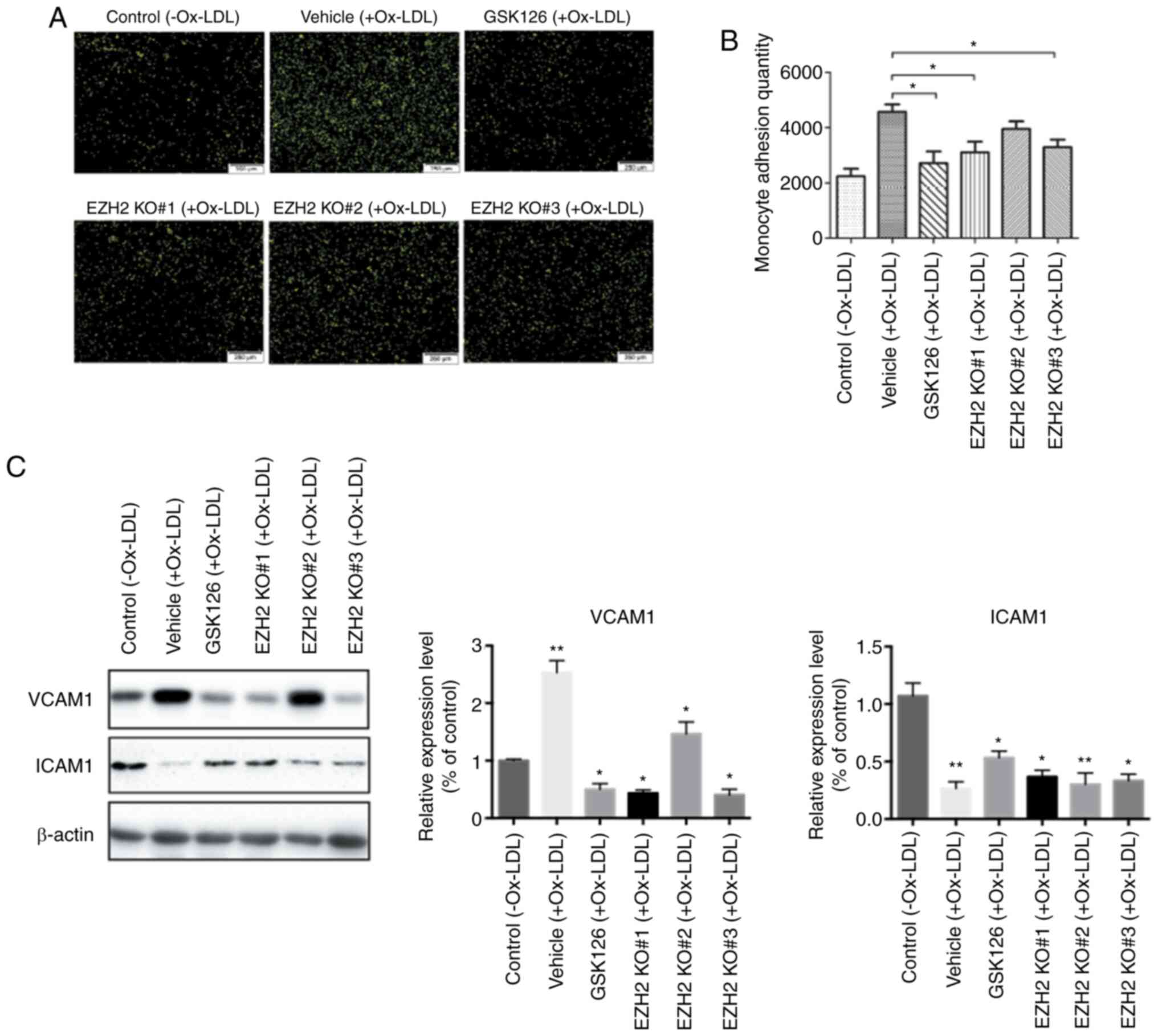
Monocyte adhesion to the vascular wall is another
crucial step in the development of atherosclerosis (29). The present study assessed whether
EZH2 KO or GSK126 could decrease the extent of monocyte adhesion to
HUVECs. THP-1 and HUVECs were co-cultured, then the number of
BCECF-AM-labeled THP-1 monocytes bound to HUVECs following Ox-LDL
stimulation was counted. Following treatment with GSK126 and EZH2
KO, the number of THP-1 monocytes adhered to HUVECs was
significantly reduced when compared to those in vehicle-treated
wells (Fig. 2A and B). Furthermore western blotting analysis
revealed that inhibition of EZH2 activity significantly
downregulated the protein expression levels of VCAM1 and
upregulated ICAM1 levels (Fig. 2C),
both of which have been previously implicated to affect the
recruitment of monocytes to endothelial cells (30). However, VCAM1 is unique in that its
expression is largely restricted to lesions and lesion-predisposed
regions, whereas ICAM1 expression extends into uninvolved healthy
aorta and lesion-protected regions (31). The difference in expression patterns
suggest different functions for VCAM1 and ICAM1 in lesion
initiation. In addition, the expression of VCAM1 was significantly
reduced in THP-1 cells treated with GSK126, providing a molecular
basis for the effects of EZH2 on Ox-LDL stimulated monocyte
adhesion. Taken together, these findings provide direct evidence
that methylation of H3K27 contributes to monocyte adhesion in an
EZH2-dependent manner.
GSK126 attenuates the development of
atherosclerosis in ApoE-/- mouse models fed a high-fat
diet
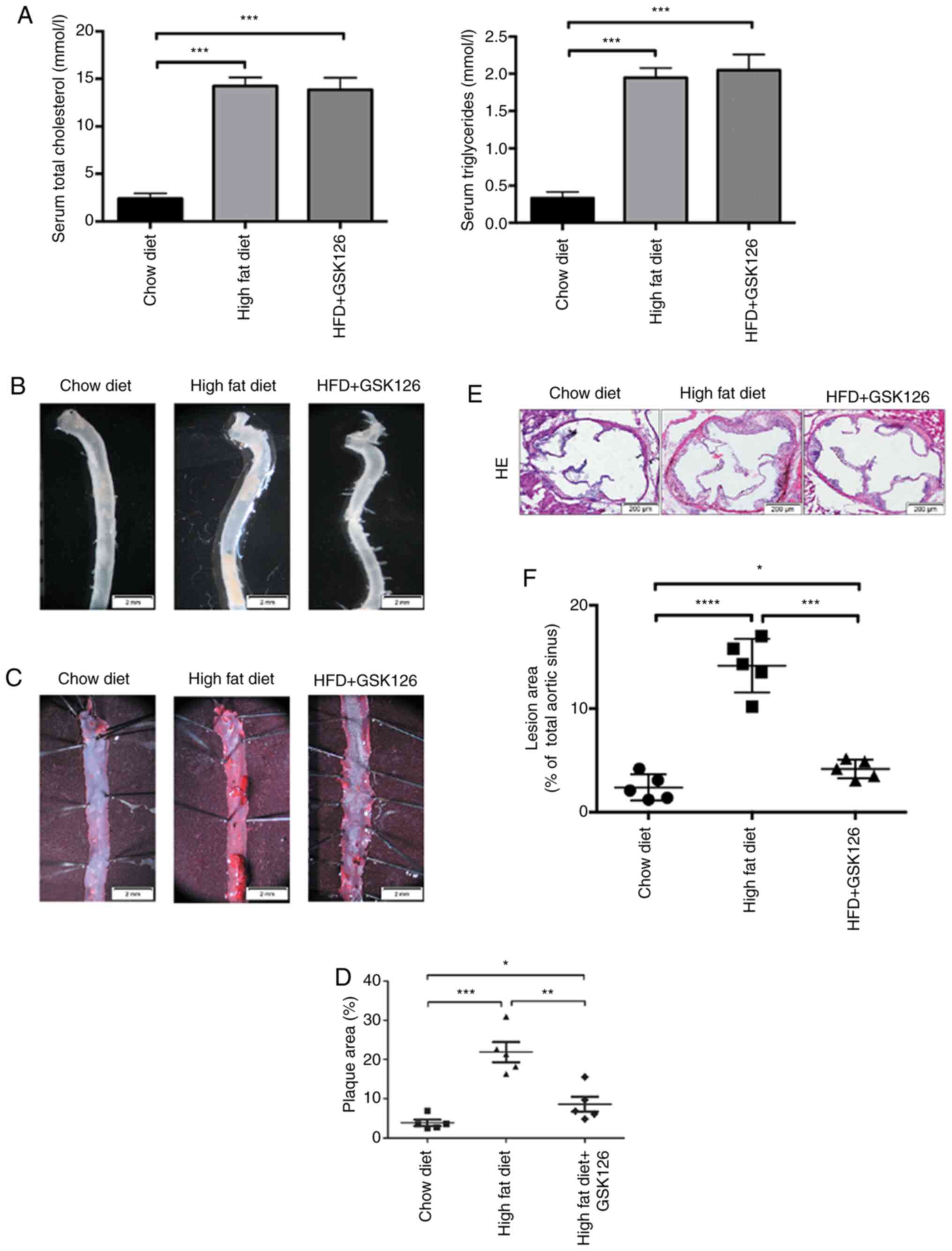
To address the role of EZH2 inhibition on the
development of atherosclerosis, male ApoE-/-
mice, which were fed an atherogenic high-fat diet, were treated
with GSK126 (50 mg/kg/day) or vehicle (20% DMSO/PBS, 0.1 ml) via a
daily intraperitoneal injection for a period of 10 weeks.
Peripheral blood and aortas from animals were collected for
cholesterol testing and Oil Red O staining, respectively.
Consistent with the aforementioned in vitro findings,
ApoE-/- mice fed with a high-fat diet exhibited a
significant increase in cholesterol and triglyceride levels, but a
a significant decrease with GSK126 treatment, as compared with the
animals fed a chow diet (Fig. 3A).
Subsequently, whole mouse aortas were harvested (Fig. 3B). En face Oil Red O staining was
performed on the thoracic aortas isolated from
ApoE-/- mice fed an atherogenic high-fat diet and
treated with GSK126 or vehicle Mice treated with GSK126 exhibited
significantly reduced atherosclerotic plaque size (Fig. 3C and D), indicating that GSK126 exerts a
protective effect, suppressing atherosclerotic plaque development
in mouse models. H&E staining of serial cross sections of
aortic roots also revealed reduced atherosclerotic lesion sizes in
the GSK126-treated animals, as compared with the vehicle-treated
mice (Fig. 3E and F). These results indicated that GSK126 has
the ability to attenuate the progression of atherosclerosis in
atherosclerosis-prone models.
GSK126 reduces macrophage recruitment
to atherosclerotic plaques
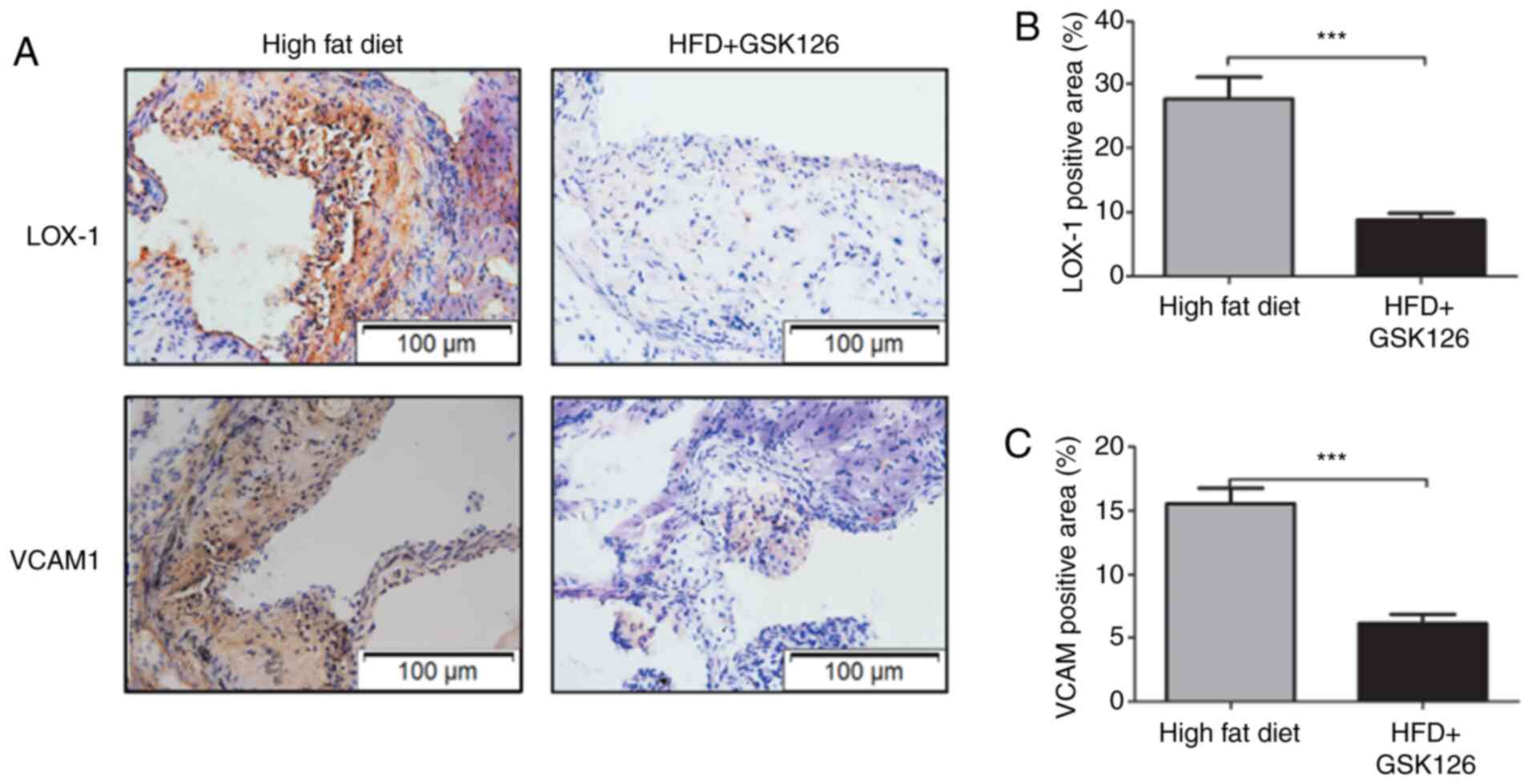
Previous studies have demonstrated that
overexpression of lectin-type oxidized LOX-1, a macrophage and
endothelial cell receptor for Ox-LDL (32,33),
in animal models of atherosclerosis, enhances the levels of
valvular accumulation of Ox-LDL, thereby favouring endothelial
dysfunction, vascular inflammation and plaque formation (34). According to our previous studies
(10), chow fat diet does not
induce atherosclerosis and Lox-1 expression in
ApoE-/- mice. In order to investigate whether
GSK126 modulates the expression levels of Lox-1 during plaque
formation in mice, the protein expression levels of Lox-1 in
plaques were examined from mice fed a high-fat diet.
Immunohistochemical staining of cross sections of the mouse aorta
revealed a decreased protein expression of Lox-1 in GSK126-treated
animals, as compared with the vehicle-treated control mice
(Fig. 4A and B). Upon initiation of plaque formation,
Ox-LDL stimulates inflammatory pathways in monocytes and
endothelial cells, inducing monocyte adhesion to endothelial cells
(35). In line with this concept,
the present study observed an increase in VCAM1 expression in
GSK126-treated mice compared with mice treated with vehicle, as
shown by immunohistochemical staining of aortas (Fig. 4A). The relative VCAM1-positive area
in the GSK126-treated mice was significantly decreased compared
with that of the vehicle-treated animals (Fig. 4C). These results suggested that
GSK126 is sufficient to suppress atherosclerotic plaque formation
in mouse models, possibly through the modulation of LOX-1 induced
lipid uptake and VCAM1 induced monocyte adhesion to endothelial
cells.
Discussion
Histone modification controls gene expression by
changing DNA accessibility or chromatin structure, and plays
important roles in a wide range of pathological processes, such as
tumorigenesis, atherosclerosis and inflammation (36-38).
Recent studies have highlighted the importance of EZH2 histone
methyltransferase on the development and progression of
atherosclerosis (21,22,34,37).
Interestingly, contradictory results have been observed in studies
from different groups. For example, Greißel et al (21) observed significantly increased H3K27
methylation levels in endothelial cells from human early and
advanced atherosclerotic plaques. However, other studies have
reported a global decrease in H3K27 methylation levels during
progression of atherosclerosis (21,36,39).
Meanwhile, EZH2 has also been reported to recruit DNA
methyltransferase 1 to the promoter region of the ABCA1 gene, which
in turn increases methylation of CpG dinucleotides at the ABCA1
promoter and consequently suppresses its transcriptional activity
(22).
Additionally, EZH2 has been shown to play a crucial
role in macrophage/microglial activation, mediating the expression
of toll-like receptor-induced proinflammatory genes, such as IL-6,
IL-12b, TNF-α, CeC motif chemokine ligand 2 and C-X-C motif
chemokine 10. EZH2 ChIP-seq analysis has identified that the
expression of suppressor of cytokine signaling 3, an
anti-inflammatory gene, is regulated by EZH2 mediated H3K27me3.
Furthermore, EZH2 deficiency significantly attenuates the
expression of proinflammatory genes at both the mRNA and protein
levels, and therefore diminishes macrophage/microglial activation
and attenuates the autoimmune inflammation (40). These results suggest that EZH2 may
be a potential candidate for anti-atherosclerosis therapy.
Development of EZH2 small molecule inhibitors has
been an active area of investigation (41). Pharmaceutical companies have been
increasingly developing compounds and promising preclinical results
have been obtained for the treatment of breast cancer, with early
results suggesting a potential treatment utility (42). GSK126 is a potent and specific
methyltransferase inhibitor which is highly selective for the
histone methyltransferase EZH2, with a very low IC50 of
9.9 nM (40). GSK126 has been shown
to have a 1,000-fold selectivity for EZH2 over other human
methyltransferases (43,44). However, it has not yet been
clarified whether inhibition of EZH2 by a small-molecule inhibitor
has anti-atherosclerotic effects. The present study provided
evidence that EZH2 plays a critical role in the initiation of
atherosclerosis, specifically in foam cell formation and monocyte
adhesion. Pharmacological inhibition of EZH2 appears to be
antiatherogenic in ApoE-/- mice fed a high-fat diet. Additionally,
pharmacological inhibition of EZH2 by GSK126, as shown in the
present study, or by GSK343 as previously shown (42), significantly decreases the
expression levels of proatherogenic genes, including VCAM1 and
LOX-1 in macrophages or atherosclerotic plaques, thereby
attenuating the ability of lipid uptake and monocyte adhesion to
arterial walls (45). These results
suggested that GSK126 is a potential drug candidate for the
treatment of atherosclerosis.
In conclusion, the present study identified that
EZH2, as a key epigenetic regulator, promotes the development of
atherosclerotic lesions and further demonstrated that the EZH2
inhibitor GSK126 is able to effectively reduce atherosclerosis in
ApoE-/- mice by suppressing foam cell formation
and monocyte adhesion. The present study also provided novel
insights into the protective effects of EZH2 inhibition on blocking
lipid uptake by upregulating the expression of ABCA1 and reducing
monocyte adhesion by downregulating the expression of VCAM1. These
results suggested that EZH2 may be considered a potential
therapeutic target for the prevention of atherosclerotic disease
progression.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by funds from the Affiliated
Zhongshan Hospital of Dalian University and the National Natural
Science Foundation of China (grant no. 81372858).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XWa, XWe, and LX contributed in the conception and
design of the study. XWe, YZ and KW contributed in data
acquisition, analysis and interpretation of the data, statistical
analysis, manuscript drafting, and critical revision of the
manuscript for important intellectual content. XWa and XWe are
responsible for the authenticity of all data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Animal procedures were approved (approval no.
17-007) by the Animal Ethics Committee of the Affiliated Zhongshan
Hospital of Dalian University (Dalian, China). All in vivo
experiments were performed in accordance with national legislation
and institutional guidelines.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Libby P, Ridker PM and Hansson GK:
Progress and challenges in translating the biology of
atherosclerosis. Nature. 473:317–325. 2011.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Libby P: Inflammation in atherosclerosis.
Nature. 420:868–874. 2002.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Hansson GK and Libby P: The immune
response in atherosclerosis: A double-edged sword. Nat Rev Immunol.
6:508–519. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Hansson GK: Inflammation, atherosclerosis,
and coronary artery disease. N Engl J Med. 352:1685–1695.
2005.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Jonasson L, Holm J, Skalli O, Bondjers G
and Hansson GK: Regional accumulations of T cells, macrophages, and
smooth muscle cells in the human atherosclerotic plaque.
Arteriosclerosis. 6:131–138. 1986.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Stary HC, Chandler AB, Dinsmore RE, Fuster
V, Glagov S, Insull W Jr, Rosenfeld ME, Schwartz CJ, Wagner WD and
Wissler RW: A definition of advanced types of atherosclerotic
lesions and a histological classification of atherosclerosis. A
report from the Committee on Vascular Lesions of the Council on
Arteriosclerosis, American Heart Association. Arterioscler Thromb
Vasc Biol. 15:1512–1531. 1995.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Ross R: Atherosclerosis is an inflammatory
disease. Am Heart J. 138:S419–S420. 1999.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kolodgie FD, Gold HK, Burke AP, Fowler DR,
Kruth HS, Weber DK, Farb A, Guerrero LJ, Hayase M, Kutys R, et al:
Intraplaque hemorrhage and progression of coronary atheroma. N Engl
J Med. 349:2316–2325. 2003.PubMed/NCBI View Article : Google Scholar
|
|
9
|
van der Wal AC, Das PK, Bentz van de Berg
D, van der Loos CM and Becker AE: Atherosclerotic lesions in
humans. In situ immunophenotypic analysis suggesting an immune
mediated response. Lab Invest. 61:166–170. 1989.PubMed/NCBI
|
|
10
|
Wang XQ, Wan HQ, Wei XJ, Zhang Y and Qu P:
CLI-095 decreases atherosclerosis by modulating foam cell formation
in apolipoprotein E-deficient mice. Mol Med Rep. 14:49–56.
2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Opar A: Where now for new drugs for
atherosclerosis? Nat Rev Drug Discov. 6:334–335. 2007.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Wierda RJ, Geutskens SB, Jukema JW, Quax
PHA and van den Elsen PJ: Epigenetics in atherosclerosis and
inflammation. J Cell Mol Med. 14 (6A):1225–1240. 2010.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sharma P, Kumar J, Garg G, Kumar A,
Patowary A, Karthikeyan G, Ramakrishnan L, Brahmachari V and
Sengupta S: Detection of altered global DNA methylation in coronary
artery disease patients. DNA Cell Biol. 27:357–365. 2008.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Yi-Deng J, Tao S, Hui-Ping Z, Jian-Tuan X,
Jun C, Gui-Zhong L and Shu-Ren W: Folate and ApoE DNA methylation
induced by homocysteine in human monocytes. DNA Cell Biol.
26:737–744. 2007.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Delaney C, Garg SK, Fernandes C, Hoeltzel
M, Allen RH, Stabler S and Yung R: Maternal diet supplemented with
methyl-donors protects against atherosclerosis in F1 ApoE(-/-)
mice. PLoS One. 8(e56253)2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Castro R, Rivera I, Struys EA, Jansen EE,
Ravasco P, Camilo ME, Blom HJ, Jakobs C and Tavares de Almeida I:
Increased homocysteine and S-adenosylhomocysteine concentrations
and DNA hypomethylation in vascular disease. Clin Chem.
49:1292–1296. 2003.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kim M, Long TI, Arakawa K, Wang R, Yu MC
and Laird PW: DNA methylation as a biomarker for cardiovascular
disease risk. PLoS One. 5(e9692)2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kim J, Kim JY, Song KS, Lee YH, Seo JS,
Jelinek J, Goldschmidt-Clermont PJ and Issa JP: Epigenetic changes
in estrogen receptor beta gene in atherosclerotic cardiovascular
tissues and in-vitro vascular senescence. Biochim Biophys Acta.
1772:72–80. 2007.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Jiang W, Agrawal DK and Boosani CS: Cell
specific histone modifications in atherosclerosis (Review). Mol Med
Rep. 18:1215–1224. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Khyzha N, Alizada A, Wilson MD and Fish
JE: Epigenetics of Atherosclerosis: Emerging Mechanisms and
Methods. Trends Mol Med. 23:332–347. 2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Greißel A, Culmes M, Burgkart R,
Zimmermann A, Eckstein HH, Zernecke A and Pelisek J: Histone
acetylation and methylation significantly change with severity of
atherosclerosis in human carotid plaques. Cardiovasc Pathol.
25:79–86. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Lv YC, Tang YY, Zhang P, Wan W, Yao F, He
PP, Xie W, Mo ZC, Shi JF, Wu JF, et al: Histone methyltransferase
enhancer of Zeste Homolog 2-mediated ABCA1 promoter DNA methylation
contributes to the progression of atherosclerosis. PLoS One.
11(e0157265)2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Xu S, Xu Y, Yin M, Zhang S, Liu P,
Koroleva M, Si S, Little PJ, Pelisek J and Jin ZG: Flow-dependent
epigenetic regulation of IGFBP5 expression by H3K27me3 contributes
to endothelial anti-inflammatory effects. Theranostics.
8:3007–3021. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Yap TA, Winter JN, Giulino-Roth L, Longley
J, Lopez J, Michot JM, Leonard JP, Ribrag V, McCabe MT, Creasy CL,
et al: Phase I study of the novel enhancer of Zeste Homolog 2
(EZH2) inhibitor GSK2816126 in patients with advanced hematologic
and solid tumors. Clin Cancer Res. 25:7331–7339. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Shalem O, Sanjana NE, Hartenian E, Shi X,
Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, et
al: Genome-scale CRISPR-Cas9 knockout screening in human cells.
Science. 343:84–87. 2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yamauchi T, Masuda T, Canver MC, Seiler M,
Semba Y, Shboul M, Al-Raqad M, Maeda M, Schoonenberg VAC, Cole MA,
et al: Genome-wide CRISPR-Cas9 Screen Identifies Leukemia-Specific
Dependence on a Pre-mRNA Metabolic Pathway Regulated by DCPS.
Cancer Cell. 33:386–400.e5. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Low H, Hoang A and Sviridov D: Cholesterol
efflux assay. J Vis Exp. 61(e3810)2012.PubMed/NCBI View
Article : Google Scholar
|
|
28
|
Zimetti F, Favari E, Cagliero P, Adorni
MP, Ronda N, Bonardi R, Gomaraschi M, Calabresi L, Bernini F and
Guardamagna O: Cholesterol trafficking-related serum lipoprotein
functions in children with cholesteryl ester storage disease.
Atherosclerosis. 242:443–449. 2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Mestas J and Ley K: Monocyte-endothelial
cell interactions in the development of atherosclerosis. Trends
Cardiovasc Med. 18:228–232. 2008.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Ley K and Huo Y: VCAM-1 is critical in
atherosclerosis. J Clin Invest. 107:1209–1210. 2001.PubMed/NCBI View
Article : Google Scholar
|
|
31
|
Iiyama K, Hajra L, Iiyama M, Li H,
DiChiara M, Medoff BD and Cybulsky MI: Patterns of vascular cell
adhesion molecule-1 and intercellular adhesion molecule-1
expression in rabbit and mouse atherosclerotic lesions and at sites
predisposed to lesion formation. Circ Res. 85:199–207.
1999.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Moriwaki H, Kume N, Sawamura T, Aoyama T,
Hoshikawa H, Ochi H, Nishi E, Masaki T and Kita T: Ligand
specificity of LOX-1, a novel endothelial receptor for oxidized low
density lipoprotein. Arterioscler Thromb Vasc Biol. 18:1541–1547.
1998.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Sawamura T, Kume N, Aoyama T, Moriwaki H,
Hoshikawa H, Aiba Y, Tanaka T, Miwa S, Katsura Y, Kita T, et al: An
endothelial receptor for oxidized low-density lipoprotein. Nature.
386:73–77. 1997.PubMed/NCBI View
Article : Google Scholar
|
|
34
|
Akhmedov A, Rozenberg I, Paneni F, Camici
GG, Shi Y, Doerries C, Sledzinska A, Mocharla P, Breitenstein A,
Lohmann C, et al: Endothelial overexpression of LOX-1 increases
plaque formation and promotes atherosclerosis in vivo. Eur Heart J.
35:2839–2848. 2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Frostegård J, Nilsson J, Haegerstrand A,
Hamsten A, Wigzell H and Gidlund M: Oxidized low density
lipoprotein induces differentiation and adhesion of human monocytes
and the monocytic cell line U937. Proc Natl Acad Sci USA.
87:904–908. 1990.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Greißel A, Culmes M, Napieralski R, Wagner
E, Gebhard H, Schmitt M, Zimmermann A, Eckstein HH, Zernecke A and
Pelisek J: Alternation of histone and DNA methylation in human
atherosclerotic carotid plaques. Thromb Haemost. 114:390–402.
2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis
RT, Wu X, Stack EC, Loda M, Liu T, et al: EZH2 oncogenic activity
in castration-resistant prostate cancer cells is
Polycomb-independent. Science. 338:1465–1469. 2012.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Liu Y, Peng J, Sun T, Li N, Zhang L, Ren
J, Yuan H, Kan S, Pan Q, Li X, et al: Epithelial EZH2 serves as an
epigenetic determinant in experimental colitis by inhibiting
TNFα-mediated inflammation and apoptosis. Proc Natl Acad Sci USA.
114:E3796–E3805. 2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Wierda RJ, Rietveld IM, van Eggermond
MCJA, Belien JAM, van Zwet EW, Lindeman JHN and van den Elsen PJ:
Global histone H3 lysine 27 triple methylation levels are reduced
in vessels with advanced atherosclerotic plaques. Life Sci.
129:3–9. 2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Zhang X, Wang Y, Yuan J, Li N, Pei S, Xu
J, Luo X, Mao C, Liu J, Yu T, et al: Macrophage/microglial Ezh2
facilitates autoimmune inflammation through inhibition of Socs3. J
Exp Med. 215:1365–1382. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Villanueva MT: Anticancer drugs: All roads
lead to EZH2 inhibition. Nat Rev Drug Discov.
16(239)2017.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Kim KH and Roberts CWM: Targeting EZH2 in
cancer. Nat Med. 22:128–134. 2016.PubMed/NCBI View Article : Google Scholar
|
|
43
|
McCabe MT, Ott HM, Ganji G, Korenchuk S,
Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A III,
Diaz E, et al: EZH2 inhibition as a therapeutic strategy for
lymphoma with EZH2-activating mutations. Nature. 492:108–112.
2012.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Kaniskan HÜ and Jin J: Chemical probes of
histone lysine methyltransferases. ACS Chem Biol. 10:40–50.
2015.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Zhou J, Huang S, Wang Z, Huang J, Xu L,
Tang X, Wan YY, Li QJ, Symonds ALJ, Long H, et al: Targeting EZH2
histone methyltransferase activity alleviates experimental
intestinal inflammation. Nat Commun. 10(2427)2019.PubMed/NCBI View Article : Google Scholar
|