1. Introduction
Ischemic stroke is a common clinical disease caused
by an insufficient blood supply to the brain. Characterized by high
morbidity, lethality and mortality, it is the third leading cause
of death in the world and the primary cause of death in China
(1,2). As such, it is a heavy burden on
families and society. The pathobiology of ischemic stroke is
complicated and involves energy metabolism disorders, peroxidation,
calcium ion (Ca2+) overload, neurotoxicity, as well as
other complications (3). Ion
homeostasis and osmotic pressure are abnormal in the early stage of
cerebral ischemia and destroy the dynamic balance of the
blood-brain barrier (BBB), which causes an overflow of plasma
components into the brain. Subsequently, endogenous edema of the
blood vessels and a series of cascade reactions occur, which
aggravate the neurons and any existing brain injury (4). Following cerebral ischemia, ATP
synthesis disorders further result in insufficient energy supply,
metabolism and homeostasis (5,6).
Additionally, blood flow to the brain is markedly reduced after a
few minutes of cerebral ischemia, which causes irreversible brain
damage to varying degrees, including cell necrosis and even
neurovascular unit injury (7,8).
Transient receptor potential (TRP) channels consist
of a group of 28-30 TRP proteins, which are closely related
structurally and form ion channels in the membranes of numerous
animals (9). A number of studies in
previous years have revealed that the expression and activity of
various TRP channel proteins are altered during the course of
cerebral ischemia-reperfusion (IR) injury (10,11)
and a large number of in vivo and in vitro
experiments have demonstrated that multiple pathways interfering
with the regulation of the TRP channel effectively prevent IR
injury (12,13). Studies have demonstrated that the
pathogenesis of cerebral ischemia is closely associated with
Ca2+ (14) and that
members of the TRP vanilloid (TRPV) family have high permeability
to Ca2+ (15). Moreover,
cerebral ischemic injury has been associated with TRPVs, which
indicates that the regulation of TRPVs could affect the repair of
damaged neurons (16).
TRPVs are divided into TRPV1, TRPV2, TRPV3, TRPV4,
TRPV5 and TRPV6. Table I displays
their functions and organization. TRPV1 to TRPV4 belong to the
non-selective ion channel group, with TRPV1, TRPV2 and TRPV4
distributed predominantly in the central nervous system (CNS) and
TRPV3 being distributed in the skin. TRPV5 and TRPV6 are selective
osmotic Ca2+ channels, and TRPV5 is important in
Ca2+ reabsorption due to its expression in renal
epithelial cells (17). TRPV6 is
predominantly expressed in the intestinal tract of animals and
exerts an effect on intestinal Ca2+ absorption (18). Evidence indicates that there is a
mutual relationship between ischemic injury and TRPVs (10-13).
Due to the distribution of TRPV3, TRPV5 and TRPV6, this paper
focuses on the function of TRPV1, TRPV2 and TRPV4, which are highly
expressed in the brain, and their role in cerebral ischemia or
under hypoxic conditions. Therefore, the aim of the present review
is to provide new strategies for research and to advance the
clinical treatment of strokes by reviewing the existing literature
on TRPVs and cerebral ischemia.
 | Table IFunctions and distributions of TRPV1
to TRPV6. |
Table I
Functions and distributions of TRPV1
to TRPV6.
| A, Non-selective
ion channels |
|---|
| Channel | Function | Distribution |
|---|
| TRPV1 | Vanilloid receptor
and noxious thermosensor (43˚C) | CNS, PNS |
| TRPV2 | Osmosis and noxious
heat thermosensor (52˚C) | CNS, spleen,
lung |
| TRPV3 | Warmth sensor
channel (33-39˚C) | Skin, CNS, PNS |
| TRPV4 | Osmosis and warmth
sensor channel (27-34˚C) | CNS, internal
organs |
| B, Selective
osmotic Ca2+ channels |
| Channel | Function | Distribution |
| TRPV5 | Calcium-selective
channel | Intestine, kidney,
placenta |
| TRPV6 | Calcium-selective
channel | Kidney,
intestine |
2. Methods
PubMed (https://pubmed.ncbi.nlm.nih.gov/) and China National
Knowledge Infrastructure databases (http://www.cnkiTabet) were
searched using the following search terms: TRPV OR transient
receptor potential vanilloid OR TRPV1 OR TRPV2 OR TRPV4 AND
cerebral ischemia OR cerebral ischemia-reperfusion injury from the
time of construction of the library to January 2020. Based on the
title, abstract and other information in the literature, such as
models and the relationship to ischemic injury, certain search
results were excluded. The included literature relating to TRPV1,
TRPV2 or TRPV4 were read carefully and individually. Finally, the
roles of TRPV1, TRPV2 and TRPV4 in cerebral ischemia were reviewed
and summarized.
3. Role of TRPV1 in cerebral ischemic
injury
TRPV1 is highly expressed in the CNS and is
activated by high temperatures (>43˚C), acidic conditions
(pH<6.0), exogenous stimuli compounds (capsaicin and allyl
isothiocyanate) (19) and
endogenous stimuli (cannabinoid anandamide, N-oleoyldopamine and
N-arachidonic acid-dopamine) (20,21).
Neuronal damage is the main cause of brain dysfunction, and a
previous study has revealed that activation of TRPV1 confers
neuroprotection by enhancing axonal signaling in neurons (22). Autophagy, which promotes neuronal
survival and induces neuronal apoptosis or cell death, is activated
during cerebral ischemia. This process occurs several hours after
stroke and is key in determining the fate of neurons (23). Apoptosis also requires activation of
apoptotic genes, involvement of the mitochondria, release of
cytochrome c and activation of caspase family cascades
(24). The opposing roles of
autophagy may be related to the degree of autophagy activation
occurring at different stages of a stroke. Furthermore, moderate
autophagy removes damaged organelles and inhibits neuronal
apoptosis to exert a protective effect (25). Therefore, inhibition of the
development of cell apoptosis appears to be an effective way of
reducing the degree of cerebral ischemia or cerebral IR injury
(26). Notably, a previous study
revealed that, following activation of TRPV1, nerve cell apoptosis
under hypoxia/re-oxygenation was clearly improved following an
influx of Ca2+ (27).
TRPV1 receptors are present in cerebrovascular
endothelial cells and are activated by capsaicin, resulting in an
influx of Ca2+ (28).
Notably, the TRPV1 channel, which is activated by capsaicin,
reduces the infarct volume in IR rats, improving their motor
coordination and neurobehavioral scores (29). Evodiamine induces protective
autophagy via the Ca2+/c-JNK pathway (30); however, capsicum also reduces
Ca2+ influx and increases apoptosis (31). In addition, autophagy is activated
by various stress factors associated with mediated cytotoxic damage
processes (32). Notably, contrary
evidence also exists demonstrating that a TRPV1 antagonist
(capsazepine) reduces infarct size without influencing cerebral
blood flow in a middle cerebral artery occlusion (MCAO) model
(33). Wild-type and TRPV1-knockout
mice with cerebral ischemia exhibited no differences in the
increased number of ionized calcium binding adaptor molecule
1-positive microglia/macrophages, glial fibrillary acidic
protein-positive astrocytes or granulocyte receptor-1-positive
neutrophils (33). Together, these
outcomes suggest that TRPV1 activated by ischemic injury could
cause neurological and motor deficits, as well as brain infarction,
but the exact cause remains unclear. Therefore, the relationship
between TRPV1 and injuries has not yet been definitively
determined. Based on the existing literature, it is apparent that
the varying outcomes cannot prove that TRPV1 has biphasic
directional regulation. Studies to date have demonstrated that
regulation of the TRPV1 channel alters neurological function scores
and relieves ischemic injury by regulating cerebral blood flow,
inhibiting nerve excitation, promoting anti-inflammation and
inducing hypothermia (34-55).
TRPV1 in blood vessels
A previous study established that TRPV1 mediates
endothelium-dependent and -independent vasodilation, and
participates in the regulation of cerebral blood flow (34), thereby reducing ischemia and hypoxia
neuronal injury. A study revealed that cannabinoids could activate
the TRPV1 receptor at the neuron ends, which resulted in the
release of calcitonin gene-related peptide (CGRP), the
hyperpolarization of smooth muscle cells to activate K+
channels and vasodilation (35). A
similar study has also demonstrated that activation of TRPV1 in
perivascular sensory nerve endings promotes the release of CGRP and
vasodilation; however, the vasodilation was significantly reduced
in knockout TRPV1 mice (36).
Further studies revealed that activation of TRPV1 induced
extracellular Ca2+ influxes with nitric oxide (NO)
production or vasodilation (37-39).
TRPV1 in neurotoxicity
Following cerebral ischemia, the release of
glutamates into the presynaptic membranes is increased. Glutamate
aggregation in the synaptic cleft and activation of glutamate
receptors results in an excessive Ca2+ influx, which
causes cell death or excitotoxicity (40). Previous in vitro studies have
revealed that capsaicin reduces glutamate-induced Ca2+
influx resulting from cortical neuron excitotoxicity and levels of
phospho-NMDA receptor 1 (GluN) 1, GluN2B and the
N-methyl-D-aspartate receptor (NMDAR). Furthermore, capsaicin can
reduce infarct volume, and improve neurobehavioral scores and motor
coordination in IR rats (29,38).
These studies suggested that capsaicin exerted a neuroprotective
effect in cortical neurons via TRPV1.
TRPV1 in inflammation
Inflammation is important in the process of cerebral
ischemic injury. Following cerebral ischemia, leukocyte aggregation
and microglia activation leads to the production of a variety of
pro-inflammatory cytokines. Moreover, microglia contribute to
inflammation of the brain, particularly in the ischemic penumbra.
In addition, endothelial cells, astrocytes and neurons secrete
pro-inflammatory cytokines following ischemic injury. The
combination of these inflammatory cells and pro-inflammatory
cytokines results in further damage to the neurons (41). Preventing the inflammatory response
is therefore another important approach that is essential for
protecting the brain. A previous study demonstrated that TRPV1
inhibition decreased levels of TNF-α and IL-10 in plasma, which
reduced infarction size, thus conferring a neuroprotective effect
(42). Another study revealed that
inhibiting TRPV1 exerted neuroprotection in rats with cerebral IR
injury, which was partially associated with TRPV1-mediated
antioxidant stress and anti-inflammation due to inhibiting p38 MAPK
activation (43). TRPV1 promotes
activation of astrocytes and the release of astrocyte-derived
IL-1β, predominantly via the Janus kinase 2-signal transducer and
activator of transcription 3 signaling pathway, and activation of
the nucleotide-binding oligomerization domain-like receptor protein
3 inflammasome in hypoxic-ischemic encephalopathy (44). Overall, these comprehensive analyses
suggest that the expression of TRPV1 is closely associated with the
inflammatory response and that regulation of TRPV1 may reduce the
inflammation induced by ischemic injury (42).
TRPV1 in hypothermia
A role for TRPV1 in the regulation of body
temperature has also previously been described (45). TRPV1 is tonically active in
vivo. Numerous TRPV1 antagonists are used as analgesics, but
are accompanied by hyperthermia, which indicates that TRPV1
activation could regulate body temperature (45). This explains the propensity of the
TRPV1 agonist capsaicin to cause sweating in order to reduce body
temperature. Without such signals, the body overheats. In a
previous study, it was found that tonically active TRPV1 channels
are presented in the viscera and confer a suppressive effect on
body temperature (46). Body
temperature maintenance was also recently proposed to be the
predominant function of TRPV1(47)
and additional studies have determined that hypothermia inhibits
neuronal cell apoptosis during ischemic injury (48-50).
The activation of TRPV1 triggers an autonomic nerve reaction that
promotes heat loss in the hypothalamus; body temperature and oxygen
metabolism are reduced, and blood oxygen saturation is increased,
which alleviates nerve injury (51-53).
Additionally, the activation of TRPV1 blocks or alleviates cell
excitotoxicity and reduces inflammation factors and free radical
levels. Studies have determined that TRPV1 agonists induce
hypothermia, thus reducing infarct size and improving neurological
function scores in IR animals (54,55).
These observations suggest that activation or inhibition of TRPV1
promotes a neuroprotective effect via both positive and negative
feedback regulation.
4. Role of TRPV2 in cerebral ischemic
injury
Structurally, TRPV2 is 50% homologous to TRPV1;
however, the activation temperature for TRPV2 channel thermal
stimulation is higher (≥52˚C) compared with TRPV1(56). In addition, TRPV2 is activated by
various mechanical and chemical stimuli, such as osmotic pressure.
Studies have revealed that TRPV2, which is associated with
Ca2+ transport, is abundantly expressed in the cell
membranes of astrocytes (57,58).
TRPV2 is also present in neurons and other non-neuronal tissues,
such as the heart and lungs, and it serves an important role in
basic cellular functions, such as cell contraction, proliferation
and death (59). The main
expression sites, however, are neurons. For neurons, neurite
outgrowth is the key to the formation of functional circuits during
neuronal development (60). A
previous study demonstrated that TRPV2 increases the expression
levels of nerve growth factor (NGF) and upregulates Ca2+
permeability via an MAPK pathway, thereby activating the
extracellular regulated kinase (ERK) signaling pathway to enhance
the outgrowth of neurites (61).
Ca2+ is also increased in astrocytes following
oxygen-glucose deprivation and re-oxygenation (OGD/R) treatment
(62). Moreover, blocking TRPV2
increases the synthesis and secretion of NGF, and promotes
astrocyte proliferation via the MAPK-JNK signaling pathway.
Notably, activation of TRPV2 induces the release of NGF, and
protects neurons and blood vessels via a JNK-dependent pathway
(63). Furthermore, a TRPV2 agonist
induced proliferation, migration and tubulogenesis, as well as
increased transendothelial electrical resistance in human brain
endothelial cells, which could regulate the function of the BBB
(64). These contradictory
experimental results support the conclusion that TRPV2 serves an
important role in promoting NGF synthesis during ischemic
stroke.
5. Role of TRPV4 in cerebral ischemic
injury
TRPV4 is another non-selective Ca2+
channel that is expressed in various tissues, such as the CNS,
heart, liver, kidney and lungs. In the CNS, TRPV4 is activated by
multiple stimuli and is distributed among neurons, glial cells,
cerebral vascular smooth muscle and the endothelial cells of the
brain (65). Studies have
demonstrated that TRPV4 serves an important regulatory role in a
variety of physiological and pathological processes (66,67).
Additionally, TRPV4 is activated during cerebral ischemia when
microcirculatory disorders and energy deficiency leads to a change
in cytotoxic edema and cell membrane tension (68,69).
This suggests that TRPV4 might mediate cerebral ischemic injury. In
addition, inhibition of TRPV4 has been shown to reduce both the
expression of MMP-9 and the permeability of the BBB in IR rats
(70). A TRPV4 inhibitor also
exerted a protective effect on hippocampal carbonic anhydrase 1
(CA1) neuronal injury caused by OGD (71). Similarly, excitatory amino acids are
increased in the extracellular space following cerebral ischemia,
where oxygen free radicals and factors are also found (72,73).
In addition, abnormal expression of genes controlling apoptosis can
lead to infarction and apoptosis (74). Furthermore, TRPV4-mediated
Ca2+ influx promotes apoptosis and necrosis in OGD/R
models (74). Previous studies
demonstrated that infarction size increases following cerebral
ischemia, and TRPV4 and phosphorylated (p)-ERK levels increase
while p-Akt is downregulated, which can be blocked by TRPV4
inhibitors (75,76). Studies have also found that
regulating Ca2+, preventing inflammation and inhibiting
neurotoxicity are the predominant mechanisms by which TRPV4
protects neurons (77,78). Overall, the mechanisms of TRPV4 and
TRPV1 in cerebral ischemia are highly similar.
TRPV4 in Ca2+ influx
Ca2+ serves an important role in cerebral
ischemia
A large quantity of Ca2+ enters the cells
via the NMDAR, which causes Ca2+ overload and cell
damage (79). TRPV4 is clearly
involved in the pathological process of cerebral IR injury and,
when overexpressed, causes Ca2+ influx following
ischemic injury, thus increasing the release of presynaptic
neurotransmitters (13). TRPV4 also
improves NMDAR function to reduce neurotoxicity by increasing
glutamates and overloading Ca2+ to relieve ischemic
injury. Following hypoxia-ischemia, TRPV4 levels are significantly
increased in astrocytes of the hippocampal CA1, thus promoting the
proliferation of reactive astrocytes (13). Furthermore, TRPV4 agonists cause
intracellular Ca2+ and cation currents, which are
blocked by extracellular Ca2+ scavengers or TRPV4
antagonists (13). This suggests
that TRPV4 is involved in Ca2+ influx in
ischemic-reactive astrocytes. Preservation of microcirculation and
BBB function shortly after reperfusion is the key neuroprotective
role of TRPV4 inhibition, suggesting that TRPV4 contributed to
post-ischemic brain injury (80).
TRPV4 in inflammation and
apoptosis
TRPV4 is a non-selective, calcium-permeable cation
channel that serves a critical role in cerebral perfusion and
inflammation (81). It has been
reported that TRPV4 is activated in choroid plexus epithelia and
cerebral ischemia by cytokines and inflammatory mediators, such as
TNF-α, IL-1β and TGF-β1 (82,83). A
previous study demonstrated that the expression of TRPV4 is
significantly upregulated in MCAO rats, and that a TRPV4 inhibitor
reduces TNF-α and IL-1β levels to alleviate astrocyte OGD injury
(84). Activation of TRPV4 induces
apoptosis by downregulating the PI3K/Akt signaling pathway and
upregulating the p38 MAPK signaling pathway, which are involved in
cerebral ischemic injury (75). The
TRPV4 antagonist reduced brain infarction following reperfusion for
48 h in MCAO mice, which attenuated a decrease in the p-Akt and
Bcl-2/ Bax protein ratio and an increase in p-p38 MAPK and cleaved
caspase-3 protein levels (75).
TRPV4 in excitotoxicity
Following cerebral ischemia, the damaged BBB and
extracellular Ca2+ influx result in neurotoxicity. TRPV4
is similar to TRPV1 in regard to their role in neurotoxicity.
Previous studies have demonstrated that TRPV4 inhibits
neurotoxicity via the NMDAR 2B (13,79).
Another study indicated that TRPV4 inhibitors block the
neurotransmitter γ-aminobutyric acid of hippocampal CA1 pyramidal
neurons via the adenosine 5'-monophosphate activated protein kinase
(AMPK)-PI3K-Akt pathways to inhibit neuronal hyperexcitability
(85). Together, the existing
research indicates that blocking TRPV4 may regulate Ca2+
influxes, thus inhibiting the inflammatory response, autophagy and
apoptosis following cerebral ischemia.
6. Discussion
The mechanisms involved in brain protection
conferred by TRPV1, TRPV2 and TRPV4 are presented in Table II. Numerous studies have reported
that the activation of TRPV1 attenuates excitotoxicity injury to
inhibit Ca2+ influxes, and glutamate-induced neuronal
excitability and neuronal death (29,38,40).
Additionally, activation of TRPV1 increases endothelial nitric
oxide synthase (eNOS) phosphorylation to improve vasodilation and
induce hypothermic brain protection, ultimately reducing cerebral
infarction size and neurological scores (37-39).
While capsaicin-induced effects can be reduced or reversed in
TRPV1-knockout mice, inhibition of TRPV1 has also been revealed to
inhibit the release of inflammatory factors, thus inducing
hypothermic brain protection, reducing cerebral infarction size and
improving neuron behavior (41-46).
This contradiction of whether TRPV1 was activated or inhibited to
protect brain injuries may be due to the extreme complexity of the
pathogenesis of cerebral ischemia and the lack of details regarding
the role of TRPV1 in the mechanism of cerebral ischemia.
| Table IIMechanisms involved in reducing
ischemic injury via TRPV1, TRPV2 and TRPV4. |
Table II
Mechanisms involved in reducing
ischemic injury via TRPV1, TRPV2 and TRPV4.
| Mechanism | TRPV1 (channel
activation) | TRPV1 (channel
inhibition) | TRPV1-KO | TRPV2 (channel
inhibition) | TRPV4 (channel
inhibition) |
|---|
| Ca2+
influx | - | Inhibition
(29,38,40) | - | Inhibition
(61,62) | Inhibition
(79-80) |
| Inflammatory
reaction | - | Inhibition
(41-44) | NR | NR | Inhibition
(81-84) |
| Neurotoxicity | - | Inhibition
(29,38) | - | NR | Inhibition
(13,79,85) |
| Neuron growth | NR | NR | NR | Activation
(61,63) | NR |
| Hypothermic brain
protection | Activation
(45-55) | - | - | NR | NR |
| Vasodilation | - | Activation
(34-39) | - | NR | NR |
These contrasting outcomes may be the result of the
following: i) Variable TRPV1 agonists and inhibitors. In the
collected literature, TRPV1 agonists included capsaicin (29), evodiamine (30) and dihydrocapsaicin (DHC) (51,54).
The TRPV1 inhibitors included capsazepine (33) and AMG-9810(42). Those drugs may have slightly
different effects via different pathways. ii) Variable dosages may
impact the results. For example, the dosages of capsaicin used were
1 or 3 nmol (29) or 1 mg/kg
(42), and Huang et al
(29) found that 1 or 3 nmol
capsaicin reduced cerebral infarction size and improved motor
function. Hakimizadeh et al (42) used capsaicin in measures of 0.1, 0.5
or 1 mg/kg, and Cao et al (51) used 1.25 mg/kg DHC with no obvious
neuroprotective effect. Furthermore, the study by Cao et al
indicated that a TRPV1 agonist (25 mg/kg rinvanil) induced
hypothermia with a neuroprotective effect on ischemic brain injury.
Meanwhile, a high dosage of rinvanil (50 mg/kg) demonstrated no
significant effect on brain injury (33,54).
The dosage of capsazepine (TRPV1 inhibitor) was 20 nmol (33) and that of AMG-9810 was 0.5 mg/kg
(42). In addition, the varied
selection of animal models, including Sprague-Dawley rats, Wistar
rats and C57B/L6 mice among others, may have impacted the results.
Therefore, due to the differing results, a more rigorous
experimental design is required, along with an increased number of
samples, to clarify the mechanisms by which TRPV1 channels confer
their effects in order to provide a solid theoretical foundation
for the development of therapeutic strategies targeting cerebral
ischemia.
The existing literature has established that TRPV1
and TRPV4 demonstrate similar effects on Ca2+ influx,
inflammation and neurotoxicity. The mode of action for TRPV2 in
regard to relieving ischemic injury is unique, as it includes four
main signaling pathways, Ca2+/JNK, PI3K/Akt, MAPK and
Ca2+. Table III
compares the results of each pathway. TRPV1 and TRPV4 serve similar
roles in blocking Ca2+ influx, inhibiting the
inflammatory response and reducing neurotoxicity. An ion channel
blocker (such as capsazepine) effectively reduces Ca2+
influx, the inflammatory response and neurotoxicity induced by
cerebral ischemia injury, the mechanism of which is related to the
MAPK-JNK pathways. In contrast to the pharmacological effects of
TRPV4, TRPV1 is also likely a sensor that participates in the
regulation of body temperature, which could serve to decrease IR
injury, but the pathway by which it functions is unique. When TRPV1
is activated, body temperature and metabolism are reduced. The
existing literature has established that TRPV1 activation reduces
IR injury (54,55); however, there have been reports
indicating conflicting results, as inhibition of TRPV1 resulted in
a decrease in body temperature, thus reducing IR injury (42). As such, it is clear that TRPV1
regulates body temperature and this is a fundamental difference in
relation to TRPV4.
| Table IIIRelief of ischemic injury by TRPV1,
TRPV2 and TRPV4. |
Table III
Relief of ischemic injury by TRPV1,
TRPV2 and TRPV4.
| Channel | Process of
relieving ischemic injury |
|---|
| TRPV1 | Therapeutic
hypothermia, Ca2+, NMDAR, JNK pathway |
| TRPV2 | MAPK-JNK signaling
pathway, JNK-dependent pathway |
| TRPV4 | Ca2+,
NMDAR, Akt signaling pathway, PI3K/Akt pathway, p38MAPK pathway,
AMPK-PKC pathway |
TRPVs are important ion channels associated with
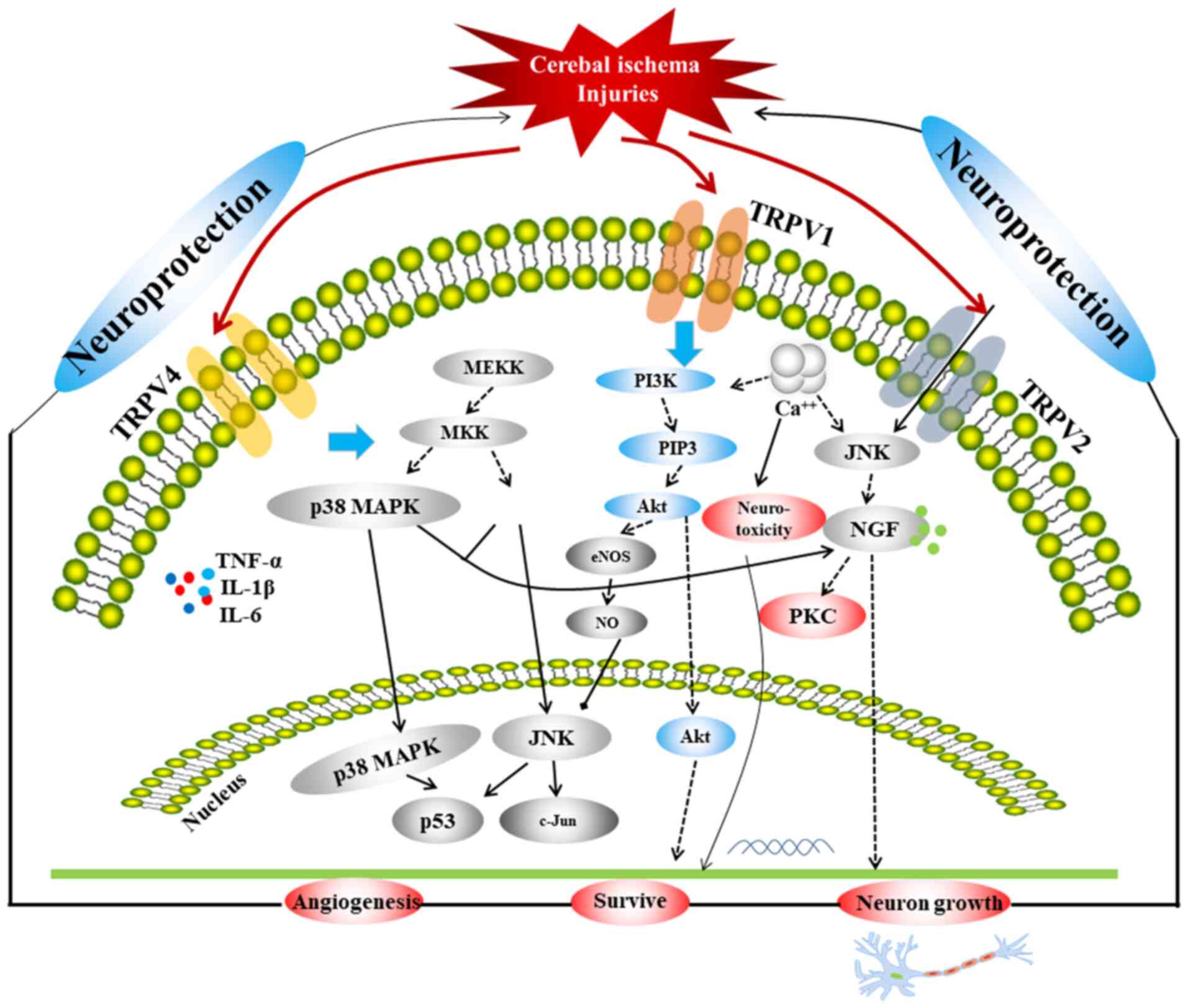
ischemic stroke. A comprehensive schematic detailing the
correlation among the three TRPVs in relation to ischemia or
hypoxic injury is provided in Fig.
1, which includes three main signaling pathways, p38 MAPK, JNK
and PI3K/Akt. The MAPK pathway mediates the proliferation,
differentiation and apoptosis of neuronal cells under different
pathophysiological conditions and is related to neurological
diseases (86,87). It has been revealed that the MAPK
pathway is a key pathway involved in the protection of the BBB
(88-90).
Furthermore, JNK and p38 are key proteins in the MAPK pathway. It
has been confirmed that the JNK/p38 MAPK pathway involves almost
all physiological and pathological processes of ischemic and IR
injury, and it is one of the most critical pathways involved in BBB
injuries (89). The activity of p38
in neurons is significantly enhanced in the ischemic region of rats
exhibiting IR injury, and the expression of apoptosis genes, such
as Bcl and Bax, is increased, thereby inducing the apoptosis of
neuronal cells (91). A previous
study determined that under a variety of stimuli, the p38 MAPK
pathway is involved in mediating the production of inducible NO
synthase (92). Following cerebral
IR, a large quantity of NO is produced, which regulates MMP-9
through guanylate cyclase (93),
promoting the development of ischemic brain injuries. In addition,
a previous study demonstrated that the JNK pathway, which is a MAPK
signaling pathway, induces apoptosis following IR injury via the
mitochondrial and death receptor pathways (94); as such, inhibiting the JNK pathway
could protect neurons from injury. The PI3K/Akt signaling pathway
is widely distributed in various cells and is a signal transduction
pathway involved in cell survival, proliferation and
differentiation regulation (95-97).
Activation of the PI3K/Akt signaling pathway inhibits apoptosis via
multiple pathways, reduces IR damage and exerts neuroprotective
effects (98,99). A previous study found that in the
development of ischemic brain injury, the PI3K/Akt cell survival
signaling pathway predominates during the early stage, whereas the
JNK apoptotic signaling pathway predominates during the later stage
in MCAO rats. The expression of p-Akt (Ser473) in the penumbra was
significantly downregulated 0.5 h after ischemia, significantly
upregulated at 1.5-5 h, downregulated at 9-24 h and returned to
baseline at 48 h, after which the JNK apoptosis signaling pathway
began to dominate (100).
Furthermore, p-Akt expression in neurons is increased 1-3 h after
IR injury, which indicates that the PI3K/Akt signaling pathway is
involved in the early-stage stress response (101). These three pathways are linked by
the TRPVs, and are closely associated with autophagy and apoptosis;
therefore, they serve an important regulatory role in cerebral
ischemic injury.
Ca2+ also serves a pivotal role in
cerebral ischemia, as intracellular Ca2+ overloaded via
NMDAR triggers a series of harmful events and is the final common
pathway leading to nerve cell death (102). Therefore, inhibition of
Ca2+ overload could also be an important approach for
reducing the extent of cerebral ischemic injury. A series of
cascades occur due to increased Ca2+ influx and
glutamate levels following cerebral ischemia, which are important
factors that serve to aggravate ischemic or IR injury. Following
cerebral ischemia, increased levels of glutamate activate the
glutamate receptors and Ca2+ influx occurs, which could
induce neurotoxicity via NMDAR. TRPV1 and TRPV4 inhibit
Ca2+ influx and reduce neurotoxicity, which might be
related to the inhibition of NMDAR expression and the regulation of
the AMPK pathway in the relief of ischemic injury.
TRPV1 and TRPV4 inhibit inflammatory responses by
reducing inflammatory factors and by linking the JNK, Akt and ERK
pathways to promote apoptosis and autophagy (43,75,76).
Therapeutic hypothermia induced by TRPV1 reduces the consumption of
sugar and oxygen, and the metabolism of brain cells to exert
additional neuroprotective effects (45-57).
TRPV1 also enhances vasodilation to alleviate ischemic injury via
the AMPK-eNOS pathway (37-39).
The mechanism of TRPV2 in attenuating ischemic injury is different
from that of TRPV1 or TRPV4 in that it promotes the release of NGF,
thereby promoting neuronal growth via the MAPK-JNK/ERK pathways
(61,63).
In the present review, the reason for the different
effects of TRPV1 and TRPV2, (which are non-selective ion channels)
were analyzed. Previous studies demonstrated that Ca2+
serves an important role in ischemia and IR injuries (28,31).
Therefore, the majority of studies conducted related experiments,
which revealed that TRPV1 and TRPV2 are highly permeable to
Ca2+ (29,38,61).
However, their roles in injuries are vastly different, which the
present study hypothesizes is due to their different expression
sites. TRPV1 is expressed in numerous cell types, such as neural,
vascular endothelial and glial cells, whereas TRPV2 is primarily
expressed in neural cells. Thus, studies on TRPV1, including blood
vessels, neurotoxicity and inflammation are extensive. There were
few studies on TRPV2 and ischemia or ischemic reperfusion. The
existing data predominantly refer to NGF and TRPV2. NGF is a key
factor in neuron growth, which might be the entry point for
studying them. In future studies, further investigation into other
factors or pathways and how they are associated with TRPV2 could be
investigated, which would enhance research data and provide a basis
for the treatment of cerebral ischemia. Among the TRPVs, TRPV1 is
expressed in cerebrovascular endothelial cells, neurons and
astrocytes (103), and the TRPV1
channel has been revealed to be a potential target influencing the
efficacy of treating strokes (16).
7. Conclusions
In addition to their crucial function in regulating
Ca2+ influx, TRPVs also exert a neuroprotective effect
via other signaling pathways, such as the p38 MAPK, JNK and
PI3K-Akt signaling pathways. The physiological effects accompanying
the TRPV channels and their associated signal transduction pathways
still require further investigation. Furthermore, additional in
vivo and in vitro experiments are required to rigorously
evaluate the role of TRPVs in neurological injury following
cerebral ischemia, as well as to provide targets for the
development of novel drugs and to inform strategies for the
clinical treatment of cerebral ischemia, as the pathogenesis of
cerebral ischemic injury is complex. In the future, the related
pathways of angiogenesis and TRPVs require further investigation to
determine whether the mechanism regulating TRPVs can help increase
axon signal transduction in neurons and enhance brain
protection.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the National Natural
Science Foundation of China (grant nos. 81873023 and 81473371), the
Innovation Team in Chengdu University of Traditional Chinese
Medicine (grant no. CXTD2018004) and the Open Research Fund of the
Key Laboratory of Southwestern Characteristic Chinese Medicine
Resources, Chengdu University of Traditional Chinese Medicine
(grant no. 2020XSGG025).
Availability of data and materials
Not applicable.
Authors' contributions
QX and RM collected the data, wrote the manuscript
and confirm the authenticity of all the raw data. JW and HC
collected literature and oversaw article writing. XG and HL
accessed the literature and edited the manuscript. All authors read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Vos T, Allen C, Arora M, Barber RM, Bhutta
Z, Brown A, Carter AR, Charlson FJ, Chen A, Coggeshall M, et al:
Global, regional, and national incidence, prevalence, and years
lived with disability for 310 diseases and injuries, 1990-2015: A
systematic analysis for the global burden of disease study 2015.
Lancet. 388:1545–1602. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Zhou M, Wang H, Zhu J, Chen W, Wang L, Liu
S, Li Y, Wang L, Liu Y, Yin P, et al: Cause-specific mortality for
240 causes in China during 1990-2013: A systematic subnational
analysis for the global burden of disease study 2013. Lancet.
387:251–272, 10015. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Huang ZY, Zhang XX, Sun WL, Chen C, Li DF,
Fang J, Fu MH, Liu QS, Yan TH and Li SJ: Research progress of
inflammation reaction related to endoplasmic reticulum stress in
ischemic endoplasmic reticulum stress. Chin Pharmacol Bull.
31:23–26. 2015.(In Chinese).
|
|
4
|
Dirnagl U, Iadecola C and Moskowitz MA:
Pathobiology of ischaemic stroke: An integrated view. Trends
Neurosci. 22:391–397. 1999.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kovac S, Kostova Dinkova AT, Herrmann AM,
Melzer N, Meuth SG and Gorji A: Metabolic and homeostatic changes
in seizures and acquired epilepsy-mitochondria, calcium dynamics
and reactive oxygen species. Int J Mol Sci. 18(1935)2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Fahrner JA, Liu R, Perry MS, Klein J and
Chan DC: A novel de novo dominant negative mutation in DNM1L
impairs mitochondrial fission and presents as childhood epileptic
encephalopathy. Am J Med Genet A. 170:2002–2011. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Chan PH: Reactive oxygen radicals in
signaling and damage in the ischemic brain. J Cereb Blood Flow
Metab. 21:2–14. 2001.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Chan PH: Role of oxidants in ischemic
brain damage. Stroke. 27:1124–1129. 1996.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Venkatachalam K and Montell C: TRP
channels. Annu Rev Biochem. 76:387–417. 2007.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Rakers C, Schmid M and Petzold GC: TRPV4
channels contribute to calcium transients in astrocytes and neurons
during peri-infarct depolarizations in a stroke model. Glia.
65:1550–1561. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Han J, Xu HH, Chen XL, Hu HR, Hu KM, Chen
ZW and He GW: Total flavone of rhododendron improves cerebral
ischemia injury by activating vascular TRPV4 to induce
endothelium-derived hyperpolarizing factor-mediated responses. Evid
Based Complement Alternat Med. 2018(8919867)2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Seki T, Goto K, Kiyohara K, Kansui Y,
Murakami N, Haga Y, Ohtsubo T, Matsumura K and Kitazono T:
Downregulation of endothelial transient receptor potential
vanilloid type 4 channel and small-conductance of Ca2+-activated K+
channels underpins impaired endothelium-dependent hyperpolarization
in hypertension. Hypertension. 69:143–153. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Li L, Qu W, Zhou L, Lu Z, Jie P and Chen L
and Chen L: Activation of transient receptor potential vanilloid 4
increases NMDA-activated current in hippocampal pyramidal neurons.
Front Cell Neurosci. 7(17)2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Lau A and Tymianski M: Glutamate
receptors, neurotoxicity and neurodegeneration. Pflugers Arch.
460:525–542. 2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wong CO, Chen K, Lin YQ, Chao Y, Duraine
L, Lu Z, Yoon WH, Sullivan JM, Broadhead GT, Sumner CJ, et al: A
TRPV channel in Drosophila motor neurons regulates presynaptic
resting Ca2+ levels, synapse growth, and synaptic transmission.
Neuron. 84:764–777. 2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Satheesh NJ, Uehara Y, Fedotova J, Pohanka
M, Büsselberg D and Kruzliak P: TRPV currents and their role in the
nociception and neuroplasticity. Neuropeptides. 57:1–8.
2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Hoenderop JG, Nilius B and Bindels RJ:
Molecular mechanism of active Ca2+ reabsorption in the distal
nephron. Annu Rev Physiol. 64:529–549. 2002.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Barley NF, Howard A, Callaghan DO, Legon S
and Walters JRF: Epithelial calcium transporter expression in human
duodenum. Am J Physiol Gastrointest Liver Physiol. 2:G285–G290.
2001.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Everaerts W, Gees M, Alpizar YA, Farre R,
Leten C, Apetrei A, Dewachter I, van Leuven F, Vennekens R, De
Ridder D, et al: The capsaicin receptor TRPV1 is a crucial mediator
of the noxious effects of mustard oil. Curr Biol. 21:316–321.
2011.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Khairatkar-Joshi N and Szallasi A: TRPV1
antagonists: The challenges for therapeutic targeting. Trends Mol
Med. 15:14–22. 2009.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Cui M, Honore P, Zhong C, Gauvin D, Mikusa
J, Hernandez G, Chandran P, Gomtsyan A, Brown B, Bayburt EK, et al:
TRPV1 receptors in the CNS play a key role in broad-spectrum
analgesia of TRPV1 antagonists. J Neurosci. 26:9385–9393.
2006.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ward NJ, Ho KW, Lambert WS, Weitlauf C and
Calkins DJ: Absence of transient receptor potential vanilloid-1
accelerates stress-induced axonopathy in the optic projection. J
Neurosci. 34:3161–3170. 2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wen YD, Sheng R, Zhang LS, Han R, Zhang X,
Zhang XD, Han F, Fukunaga K and Qin ZH: Neuronal injury in rat
model of permanent focal cerebral ischemia is associated with
activation of autophagic and lysosomal pathways. Autophagy.
4:762–769. 2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Khan MM, Ishrat T, Ahmad A, Hoda MN, Khan
MB, Khuwaja G, Srivastava P, Raza SS, Islam F and Ahmad S: Sesamin
attenuates behavioral, biochemical and histological alterations
induced by reversible middle cerebral artery occlusion in the rats.
Chem Biol Interact. 183:255–263. 2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Li L, Tan J, Miao Y, Lei P and Zhang Q:
ROS and autophagy: Interactions and molecular regulatory
mechanisms. Cell Mol Neurobiol. 35:615–621. 2015.PubMed/NCBI View Article : Google Scholar : Yang Y, Gao K, Hu
Z, Li W, Davies H, Ling S, Rudd JA and Fang M: Autophagy
upregulation and apoptosis downregulation in DAHP and triptolide
treated cerebral ischemia. Mediators Inflamm 2015: 120198,
2015.
|
|
26
|
Charriaut-Marlangue C: Apoptosis: A target
for neuroprotection. Therapie. 59:185–190. 2004.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Dai Z, Xiao J, Liu S, Cui L, Hu G and
Jiang D: Rutaecarpine inhibits hypoxia/reoxygenation-induced
apoptosis in rat hippocampal neurons. Neuropharmacology.
55:1307–1312. 2008.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Golech SA, McCarron RM, Chen Y, Bembry J,
Lenz F, Mechoulam R, Shohami E and Spatz M: Human brain
endothelium: Coexpression and function of vanilloid and
endocannabinoid receptors. Brain Res Mol Brain Res. 132:87–92.
2004.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Huang M, Cheng G, Tan H, Qin R, Zou Y,
Wang Y and Zhang Y: Capsaicin protects cortical neurons against
ischemia/reperfusion injury via down-regulating NMDA receptors. Exp
Neurol. 295:66–76. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Liu A, Wang SH, Hou SY, Lin CJ, Chiu WT,
Hsiao SH, Chen TH and Shih CM: Evodiamine induces transient
receptor potential vanilloid-1-mediated protective autophagy in
U87-MG astrocytes. Evid Based Complement Alternat Med.
2013(354840)2013.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Wang Z, Sun L, Yu H, Zhang Y, Gong W, Jin
H, Zhang L and Liang H: Binding mode pediction of evodiamine within
vanilloid receptor TRPV1. Int J Mol Sci. 13:8958–8969.
2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Hale AN, Ledbetter DJ, Gawriluk TR and
Rucker EB III: Autophagy: Regulation and role in development.
Autophagy. 7:951–972. 2013.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Miyanohara J, Shirakawa H, Sanpei K,
Nakagawa T and Kaneko S: A pathophysiological role of TRPV1 in
ischemic injury after transient focal cerebral ischemia in mice.
Biochem Biophys Res Commun. 467:478–483. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Yang D, Luo Z, Ma S, Wong WT, Ma L, Zhong
J, He H, Zhao Z, Cao T, Yan Z, et al: Activation of TRPV1 by
dietary capsaicin improves endothelium-dependent vasorelaxation and
prevents hypertension. Cell Metab. 12:130–141. 2010.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Breyne J and Vanheel B: Methanandamide
hyperpolarizes gastric arteries by stimulation of TRPV1 receptors
on perivascular CGRP containing nerves. J Cardiovasc Pharmacol.
47:303–309. 2006.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Xu X, Wang P, Zhao Z, Cao T, He H, Luo Z,
Zhong J, Gao F, Zhu Z, Li L, et al: Activation of transient
receptor potential vanilloid 1 by dietary capsaicin delays the
onset of stroke in stroke-prone spontaneously hypertensive rats.
Stroke. 42:3245–3251. 2011.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Ching LC, Chen CY, Su KH, Hou HH, Shyue
SK, Kou YR and Lee TS: Implication of AMP-activated protein kinase
in transient receptor potential vanilloid type 1-mediated
activation of endothelial nitric oxide synthase. Mol Med.
18:805–815. 2012.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Hurtado-Zavala JI, Ramachandran B, Ahmed
S, Halder R, Bolleyer C, Awasthi A, Stahlberg MA, Wagener RJ,
Anderson K, Drenan RM, et al: TRPV1 regulates excitatory
innervation of OLM neurons in the hippocampus. Nat Commun.
8(15878)2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Zhang MJ, Yin YW, Li BH, Liu Y, Liao SQ,
Gao CY, Li JC and Zhang LL: The role of TRPV1 in improving VSMC
function and attenuating hypertension. Prog Biophys Mol Biol.
117:212–216. 2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Szydlowska K and Tymianski M: Calcium,
ischemia and excitotoxicity. Cell Calcium. 47:122–129.
2010.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Chen L, Liu C, Liu L and Cao X: Changes in
osmolality modulate voltage-gated sodium channels in trigeminal
ganglion neurons. Neurosci Res. 64:199–207. 2009.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Hakimizadeh E, Shamsizadeh A, Roohbakhsh
A, Arababadi MK, Hajizadeh MR, Shariati M, Rahmani MR and
Allahtavakoli M: Inhibition of transient receptor potential
vanilloid-1 confers neuroprotection, reduces tumor necrosis
factor-alpha, and increases IL-10 in a rat stroke model. Fund Clin
Pharmacol. 31:420–428. 2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Long M, Wang Z, Zheng D, Chen J, Tao W,
Wang L, Yin N and Chen Z: Electroacupuncture pretreatment elicits
neuroprotection against cerebral ischemia-reperfusion injury in
rats associated with transient receptor potential vanilloid
1-mediated anti-oxidant stress and anti-inflammation. Inflammation.
42:1777–1787. 2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Yang XL, Wang X, Shao L, Jiang GT, Min JW,
Mei XY, He XH, Liu WH, Huang WX and Peng BW: TRPV1 mediates
astrocyte activation and interleukin-1β release induced by hypoxic
ischemia (HI). J Neuroinflammation. 16(114)2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Gavva NR, Bannon AW, Surapaneni S, Hovland
DN Jr, Lehto SG, Gore A, Juan T, Deng H, Han B, Klionsky L, et al:
The vanilloid receptor TRPV1 is tonically activated in vivo and
involved in body temperature regulation. J Neurosci. 27:3366–3374.
2007.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Steiner AA, Turek VF, Almeida MC,
Burmeister JJ, Oliveira DL, Roberts JL, Bannon AW, Norman MH, Louis
JC, Treanor JJ, et al: Nonthermal activation of transient receptor
potential vanilloid-1 channels in abdominal viscera tonically
inhibits autonomic cold-defense effectors. J Neurosci.
27:7459–7468. 2007.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Gavva NR: Body-temperature maintenance as
the predominant function of the vanilloid receptor TRPV1. Trends
Pharmacol Sci. 29:550–557. 2008.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Li J and Zhang S: The effect of mild
hypothermia on mice cerebral ischemia-reperfusion nerve cell
apoptosis. Chin J Neurosurg. 26:904–907. 2010.(In Chinese).
|
|
49
|
Ye X, Yu S, Li C and Guo L:
Neuroprotective role of mild hypothermia on cerebral
ischemia-reperfusion injury in rats. Clin Med China. 22:124–126.
2006.(In Chinese).
|
|
50
|
Xue BS, Feng PH, Wei ND, Li ZF, Li YY, Lv
XL and Hou WJ: The effect and mechanism of hypothermia on repair of
cerebral ischemia-reperfusion injury rats. Prog Anat Sci.
22:654–657. 2016.(In Chinese).
|
|
51
|
Cao Z, Balasubramanian A, Pedersen SE,
Romero J, Pautler RG and Marrelli SP: TRPV1-mediated
pharmacological hypothermia promotes improved functional recovery
following ischemic stroke. Sci Rep. 7(17685)2017.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Lay C and Badjatia N: Therapeutic
hypothermia after cardiac arrest. Curr Atheroscler Rep. 12:336–342.
2010.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Ai L, Qiao Q, Chen N, Yang T, Tang X and
Yue J: Neuroprotective effect of therapeutic hypothermia induced by
dihydrocapsaicin on cerebral ischemia reperfusion injury in mice. J
Xinxiang Med Univ. 34:1058–1062. 2017.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
54
|
Muzzi M, Felici R, Cavone L, Gerace E,
Minassi A, Appendino G, Moroni F and Chiarugi A: Ischemic
neuroprotection by TRPV1 receptor-induced hypothermia. J Cereb
Blood Flow Metab. 32:978–982. 2012.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Cao Z, Balasubramanian A and Marrelli SP:
Pharmacologically induced hypothermia via TRPV1 channel agonism
provides neuroprotection following ischemic stroke when initiated
90 min after reperfusion. Am J Physiol Regul Integr Comp Physiol.
306:R149–R156. 2014.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Shibasaki K: Physiological significance of
TRPV2 as a mechanosensor, thermosensor and lipid sensor. J Physiol
Sci. 66:359–365. 2016.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Shibasaki K, Ishizaki Y and Mandadi S:
Astrocytes express functional TRPV2 ion channels. Biochem Biophys
Res Commun. 441:327–332. 2013.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Deng Q: Efficacy evaluation of Erigeron
breviscapus on neurological function recovery after minimally
invasive procedures for removal of intracranial hematoma. Chin J
Pract Nerv Dis. 12:82–84. 2009.
|
|
59
|
Kojima I and Nagasawa M: TRPV2. Handb Exp
Pharmacol. 222:247–272. 2014.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Park HJ, Kwon H, Lee S, Jung JW, Ryu JH,
Jang DS, Lee YC and Kim DH: Echinocystic acid facilitates neurite
outgrowth in neuroblastoma Neuro2a cells and enhances spatial
memory in aged mice. Biol Pharm Bull. 40:1724–1729. 2017.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Cohen MR, Johnson WM, Pilat JM, Kiselar J,
DeFrancesco-Lisowitz A, Zigmond RE and Moiseenkova-Bell VY: Nerve
growth factor regulates transient receptor potential vanilloid 2
via extracellular signal-regulated kinase signaling to enhance
neurite outgrowth in developing neurons. Mol Cell Biol.
35:4238–4252. 2015.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Zhang H, Xiao J, Hu Z, Xie M, Wang W and
He D: Blocking transient receptor potential vanilloid 2 channel in
astrocytes enhances astrocyte-mediated neuroprotection after
oxygen-glucose deprivation and reoxygenation. Eur J Neurosci.
44:2493–2503. 2016.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Xiao J, Yang F, Zhang H, Wang W and He D:
TRPV2 activation enhances the expression of nerve growth factor in
primary cultured astrocytes under oxygen-glucose
deprivation/reoxygenation. Chin J Cell Biol. 36:773–779. 2014.
|
|
64
|
Luo H, Rossi E, Saubamea B, Chasseigneaux
S, Cochois V, Choublier N, Smirnova M, Glacial F, Perrière N,
Bourdoulous S, et al: Cannabidiol increases proliferation,
migration, tubulogenesis, and integrity of human brain endothelial
cells through TRPV2 activation. Mol Pharm. 16:1312–1326.
2019.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Garcia-Elias A, Mrkonjic S, Jung C,
Pardo-Pastor C, Vicente R and Valverde MA: The TRPV4 channel. Handb
Exp Pharmacol. 222:293–319. 2014.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Nilius B and Voets T: The puzzle of TRPV4
channelopathies. EMBO Rep. 14:152–163. 2013.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Liedtke W, Choe Y, Martí-Renom MA, Bell
AM, Denis CS, Sali A, Hudspeth AJ, Friedman JM and Heller S:
Vanilloid receptor-related osmotically activated channel (VR-OAC),
a candidate vertebrate osmoreceptor. Cell. 103:525–535.
2000.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Li L, Liu C and Chen L and Chen L:
Hypotonicity modulates tetrodotoxin-sensitive sodium current in
trigeminal ganglion neurons. Mol Pain. 7(27)2011.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Chen NN, Wang JP, Jiang C, Liu C, Li X,
Zhao Y and Hao Y: Research about the influence of progesterone on
expression of COX-2 and the water content of injured brain in
cerebral ischemia in rats. J Apoplexy Nerv Dis. 6:671–673.
2009.
|
|
70
|
Lu WC, Ma YJ, Xie H, Dig XH, Su QX, Xing
HH, Meng YH, Fan J and Tian JH: Effect of TRPV4 channel on focal
cerebral ischemic reperfusion injury in rats. Prog Anat Sci.
23:353–355. 2017.(In Chinese).
|
|
71
|
Lipski J, Park TI, Li D, Lee SC, Trevarton
AJ, Chung KK, Freestone PS and Bai JZ: Involvement of TRP-like
channels in the acute ischemic response of hippocampal CA1 neurons
in brain slices. Brain Res. 1077:187–199. 2006.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Zacharia BE, Hickman ZL, Grobelny BT,
DeRosa PA, Ducruet AF and Connolly ES: Complement inhibition as a
proposed neuroprotective strategy following cardiac arrest.
Mediators Inflamm. 2009(124384)2009.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Bernard SA, Gray TW, Buist MD, Jones BM,
Silvester W, Gutteridge G and Smith K: Treatment of comatose
survivors of out-of-hospital cardiac arrest with induced
hypothermia. N Engl J Med. 346:557–563. 2002.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Wu Q, Qian C, Zhao N, Dong Q, Li J, Wang
BB, Chen L, Yu L, Han B, Du YM and Liao YH: Activation of transient
receptor potential vanilloid 4 involves in hypoxia/reoxygenation
injury in cardiomyocytes. Cell Death Dis. 8(e2828)2017.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Jie P, Hong Z, Tian Y, Li Y, Lin L, Zhou
L, Du Y and Chen L and Chen L: Activation of transient receptor
potential vanilloid 4 induces apoptosis in hippocampus through
downregulating PI3K/Akt and upregulating p38 MAPK signaling
pathways. Cell Death Dis. 6(e1775)2015.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Jie P, Lu Z, Hong Z, Li L, Zhou L, Li Y,
Zhou R, Zhou Y, Du Y and Chen L and Chen L: Activation of transient
receptor potential vanilloid 4 is involved in neuronal injury in
middle cerebral artery occlusion in mice. Mol Neurobiol. 53:8–17.
2016.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Butenko O, Dzamba D, Benesova J, Honsa P,
Benfenati V, Rusnakova V, Ferroni S and Anderova M: The increased
activity of TRPV4 channel in the astrocytes of the adult rat
hippocampus after cerebral hypoxia/ischemia. PLoS One.
7(e39959)2012.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Dunn KM, Hill-Eubanks DC, Liedtke WB and
Nelson MT: TRPV4 channels stimulate Ca2+-induced Ca2+ release in
astrocytic endfeet and amplify neurovascular coupling responses.
Proc Natl Acad Sci USA. 110:6157–6162. 2013.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Zhang YF, Fan XJ, Li X, Peng LL, Wang GH,
Ke KF and Jiang ZL: Ginsenoside Rg1 protects neurons from
hypoxic-ischemic injury possibly by inhibiting Ca2+ influx through
NMDA receptors and L-type voltage-dependent Ca2+ channels. Eur J
Pharmacol. 586:90–99. 2008.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Tanaka K, Matsumoto S, Yamada T, Yamasaki
R, Suzuki M, Kido MA and Kira JI: Reduced post-ischemic brain
injury in transient receptor potential vanilloid 4 knockout mice.
Front Neurosci. 14(453)2020.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Diaz-Otero JM, Yen TC, Ahmad A,
Laimon-Thomson E, Abolibdeh B, Kelly K, Lewis MT, Wiseman RW,
Jackson WF and Dorrance AM: Transient receptor potential vanilloid
4 channels are important regulators of parenchymal arteriole
dilation and cognitive function. Microcirculation.
26(e12535)2019.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Simpson S, Preston D, Schwerk C, Schroten
H and Blazer-Yost B: Cytokine and inflammatory mediator effects on
TRPV4 function in choroid plexus epithelial cells. Am J Physiol
Cell Physiol. 317:C881–C893. 2019.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Emsley HC and Tyrrell PJ: Inflammation and
infection in clinical stroke. J Cereb Blood Flow Metab.
22:1399–1419. 2002.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Zhu D, Yin L and Liu Q: The role of TRPV4
in OGD damage of cultured in vitro astrocytes. Chin J Clin Res.
26:885–888. 2013.
|
|
85
|
Hong Z, Tian Y, Qi M, Li Y, Du Y and Chen
L, Liu W and Chen L: Transient receptor potential vanilloid 4
inhibits γ-aminobutyric acid-activated current in hippocampal
pyramidal neurons. Front Mol Neurosci. 9(77)2016.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Ashraf MI, Ebner M, Wallner C, Haller M,
Khalid S, Schwelberger H, Koziel K, Enthammer M, Hermann M,
Sickinger S, et al: A p38MAPK/MK2 signaling pathway leading to
redox stress, cell death and ischemia/reperfusion injury. Cell
Commun Signal. 12(6)2014.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Morente V, Pérez-Sen R, Ortega F,
Huerta-Cepas J, Delicado EG and Miras-Portugal MT: Neuroprotection
elicited by P2Y13 receptors against genotoxic stress by inducing
DUSP2 expression and MAPK signaling recovery. Biochim Biophys Acta.
1843:1886–1898. 2014.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Maddahi A, Chen Q and Edvinsson L:
Enhanced cerebrovascular expression of matrix metalloproteinase-9
and tissue inhibitor of metalloproteinase-1 via the MEK/ERK pathway
during cerebral ischemia in the rat. Bmc Neurosci.
10(56)2009.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Kovalska M, Kovalska L, Pavlikova M,
Janickova M, Mikuskova K, Adamkov M, Kaplan P, Tatarkova Z and
Lehotsky J: Intracellular signaling MAPK pathway after cerebral
ischemia-reperfusion injury. Neurochem Res. 37:1568–1577.
2012.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Rumbaut RE, McKay MK and Huxley VH:
Capillary hydraulic conductivity is decreased by nitric oxide
synthase inhibition. Am J Physiol. 268:H1856–H1861. 1995.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Piao CS, Kim JB, Han PL and Lee JK:
Administration of the p38 MAPK inhibitor SB203580 affords brain
protection with a wide therapeutic window against focal ischemic
insult. J Neurosci Res. 73:537–544. 2003.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Kang J, Zhang Y, Cao X, Fan J, Li G, Wang
Q, Diao Y, Zhao Z, Luo L and Yin Z: Lycorine inhibits
lipopolysaccharide-induced iNOS and COX-2 up-regulation in RAW264.7
cells through suppressing P38 and STATs activation and increases
the survival rate of mice after LPS challenge. Int Immunopharmacol.
12:249–256. 2012.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Ridnour LA, Windhausen AN, Isenberg JS,
Yeung N, Thomas DD, Vitek MP, Roberts DD and Wink DA: Nitric oxide
regulates matrix metalloproteinase-9 activity by
guanylyl-cyclase-dependent and -independent pathways. Proc Natl
Acad Sci USA. 104:16898–16903. 2007.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Okuno S, Saito A, Hayashi T and Chan PH:
The c-Jun N-terminal protein kinase signaling pathway mediates Bax
activation and subsequent neuronal apoptosis through interaction
with Bim after transient focal cerebral ischemia. J Neurosci.
24:7879–7887. 2004.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Gao X, Zhang H, Takahashi T, Hsieh J, Liao
J, Steinberg GK and Zhao H: The Akt signaling pathway contributes
to postconditioning's protection against stroke; the protection is
associated with the MAPK and PKC pathways. J Neurochem.
105:943–955. 2008.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Sun T, Li YJ, Tian QQ, Wu Q, Feng D, Xue
Z, Guo YY, Yang L, Zhang K, Zhao MG and Wu YM: Activation of liver
X receptor β-enhancing neurogenesis ameliorates cognitive
impairment induced by chronic cerebral hypoperfusion. Exp Neurol.
304:21–29. 2018.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Weiss HR, Chi OZ, Kiss GK, Liu X, Damito S
and Jacinto E: Akt activation improves microregional oxygen
supply/consumption balance after cerebral ischemia-reperfusion.
Brain Res. 1683:48–54. 2018.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Gao F, Gao E, Yue TL, Ohlstein EH, Lopez
BL, Christopher TA and Ma XL: Nitric oxide mediates the
antiapoptotic effect of insulin in myocardial ischemia-reperfusion:
The roles of PI3-kinase, Akt, and endothelial nitric oxide synthase
phosphorylation. Circulation. 105:1497–1502. 2002.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Rocha-Ferreira E, Rudge B, Hughes MP,
Rahim AA, Hristova M and Robertson NJ: Immediate remote ischemic
postconditioning reduces brain nitrotyrosine formation in a piglet
asphyxia model. Oxid Med Cell Longev. 2016(5763743)2016.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Kamada H, Nito C, Endo H and Chan PH: Bad
as a converging signaling molecule between survival PI3-K/Akt and
death JNK in neurons after transient focal cerebral ischemia in
rats. J Cereb Blood Flow Metab. 27:521–533. 2007.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Li F, Omori N, Jin G, Wang SJ, Sato K,
Nagano I, Shoji M and Abe K: Cooperative expression of survival
p-ERK and p-Akt signals in rat brain neurons after transient MCAO.
Brain Res. 962:21–26. 2003.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Berliocchi L, Bano D and Nicotera P: Ca2+
signals and death programmes in neurons. Philos Trans R Soc Lond B
Biol Sci. 360:2255–2258. 2005.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Zhang E and Liao P: Brain transient
receptor potential channels and stroke. J Neurosci Res.
93:1165–1183. 2015.PubMed/NCBI View Article : Google Scholar
|