Introduction
Cerebral infarction is one of the leading causes of
long-term disability and death worldwide (1), accounting for more than 75% of all
stroke cases (2). Cerebral
infarction occurs when blood flow to the brain is blocked,
resulting in hypoxia and the rapid death of cerebral tissues. The
factors that induce cerebral infarction are complex, including
genetic and environmental factors (3,4).
Continuous efforts have been made to elucidate its mechanism and to
find appropriate therapeutic targets. Recently, studies have
revealed the crucial role of brain-resident immune cells in
regulating the progression of cerebral infarction, indicating that
targeting these immune cells may be an attractive therapeutic
strategy (5,6).
Microglial cells are the first line of defense of
the host against ischemic injury (5,7). After
brain injury, microglia are rapidly activated. Activated microglia
can secrete a variety of factors to regulate inflammation. Among
them, microglia releasing pro-inflammatory mediators [such as tumor
necrosis factor (TNF)-α, interleukin (IL)-1β and interferon
(IFN)-γ] are defined as the M1-type, while those releasing
neuroprotective factors (such as IL-4, IL-10 and TGF-β) are defined
as the M2-type (8,9). The polarization of activated microglia
into the M1 or M2 type depends on a variety of molecular signals in
the microenvironment. For example, IFN-γ, TNF-α, IL-2, and
lipopolysaccharide promote the polarization of microglia into the
M1 type. However, chondroitin sulfate, proteoglycan and IL-4
promote the polarization of microglia into into the M2 type
(9,10). Therefore, targeting microglial
activation is considered as a promising therapeutic strategy in the
treatment of cerebral infarction.
Long non-coding (lnc) RNAs, as noncoding transcripts
longer than 200 bp, play pivotal roles in various biological and
pathological processes, including cerebral infarction (11,12).
Multiple lncRNAs have been revealed to regulate the progression of
ischemic infarction (13). For
instance, lncRNA MEG3 modulated neuronal death following cerebral
infarction via miR21/PDCD4 signaling (14). LncRNA H19 contributed to microglial
polarization to the M1 phenotype, giving rise to post-stroke
neuroinflammation (15). However,
research on the role of lncRNAs in cerebral infarction and their
underlying mechanisms remain at an early stage (12). Recently, lncRNA X-inactive specific
transcript (XIST) has been reported to be involved in several
neurological diseases, such as spinal cord injury (16). However, the role of XIST in cerebral
ischemia is unclear.
In the present study, it was hypothesized that XIST
plays an important role in regulating the inflammatory polarization
of microglial cells in cerebral infarction and the role of the
lncRNA XIST in microglial phenotype modulation was explored using
an in vitro oxygen-glucose deprivation (OGD) model and an
in vivo middle cerebral artery occlusion (MCAO) model.
Materials and methods
Establishment of cerebral infarction
animal model
A total of 24 male C57BL/6 mice (6-8 weeks old;
weighing 20±0.3 g) were purchased from SLAC Laboratory Animal Co.,
Ltd. All experiments were carried out in accordance with the
National Institutes of Health (NIH) guidelines for the care and use
of laboratory animals and were approved by the Animal Ethics
Committee of Xingtai Medical College Second Affiliated Hospital
(Xingtai, China). Mice were fed with food and water ad
libitum and were kept at 24±2˚C and 55±2% humidity, alternating
between light and dark for 12 h. Mice were randomly divided into
two groups: Sham group and the cerebral infarction group. A total
of 12 mice were placed in each group. A cerebral infarction animal
model was established using the MCAO method according to previously
reported methods (17,18). Mice were anesthetized by
intraperitoneal injection of 5% chloral hydrate (300 mg/kg).
Briefly, common, internal and external carotid arteries were
exposed after mice were anesthetized and a nylon suture was then
inserted. Thereafter, the middle cerebral artery (MCA) was occluded
using the nylon suture after moving it forward to the origin of the
MCA. Mice in the sham operation group were only treated with neck
incision and suture after separation, without vascular ligation. At
the end of the experiment, mice were anesthetized with chloral
hydrate and then euthanized by cervical dislocation. Three mice
were randomly sacrificed in each group at 12, 36, 96 and 168 h
after MCA or Sham surgery respectively before the cerebral tissues
obtained for further investigation.
Cell culture and oxygen/glucose
deprivation (OGD) treatment
The mouse microglial cell line, BV-2, was obtained
from the Cell Bank of Chinese Academy of Sciences and cultured in
DMEM (Gibco; Thermo Fisher Scientific, Inc.) containing 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) at 37˚C
and 5% CO2. A total of two pregnant female C57BL/6 mice
(10-12 weeks old; weighing 20±0.3 g) were purchased from SLAC
Laboratory Animal Co., Ltd.. Mice were fed with food and water
ad libitum, and were kept at 24±2˚C and 55±2% humidity, in
alternating between light and dark for 12 h, whilst waiting until
the pregnant mice to give birth. Primary neurons were isolated from
the cerebral cortices of C57BL/6 mice within 24 h of birth as
previously described and cultured in neurobasal media (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 2% B-27 (Gibco;
Thermo Fisher Scientific, Inc.) at 37˚C and 5% CO2
(19).
For OGD treatment, BV-2 cells were incubated in
serum/glucose-free DMEM under the culture conditions of 94%
N2/5% CO2/1% O2 and 37˚C for 2 h
and then returned to normal culture conditions. Subsequently, 24 h
after BV-2 cells returned to normal culture conditions, the culture
supernatants of BV-2 cells were collected and used as the
conditioned media used to treat neurons for 12, 36 and 48 h at
37˚C.
Cell transfection
LncRNA XIST and inhibitor of nuclear factor κB
kinase subunit β (IKKβ) cDNAs were cloned into the pXJ40-HA-Merlin
I (Addgene plasmid no. 19699; http://n2t.net/addgene:19699; RRID:Addgene_19699) to
construct overexpression vectors. Small interfering (si)RNA-XIST,
short hairpin (sh)RNA-IKKβ, microRNA (miR)-96-5p mimics, and
miR-96-5p inhibitor were purchased from Shanghai GenePharma Co.,
Ltd. All small RNA sequences are as follows: siRNA-XIST,
5'-AUAACAGUAAGUCUGAUAGAGGACA-3'; shRNA-IKKβ,
5'-CACCGTCTTGTCGCCTAGAGCTATTCAAGAGATAGCTCTAGGCGACAAGACTTTTTTG-3';
miR-96-5p mimics, 5'-UUUGGCACUAGCACAUUUUUGCU-3'; miR-96-5p
inhibitor, 5'-AGCAAAAAUGUGCUAUGUGCCAAA-3'; siRNA negative control,
5'-UUACUCAUGUGUCAUAACACAGGUG-3'; shRNA negative control,
5'-CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG-3'; mimic
negative control, 5'-UUCUCCGAACGUGUCACGUTT-3'; inhibitor negative
control, 5'-UUGUACUACACAAAAGUACUG-3'.
BV-2 cells were seeded in 96-well plates at
5x104 cells/well. LncRNA XIST overexpression
vector/siRNA, miR-96-5p mimics/inhibitor, IKKβ overexpression
vector/shRNA and corresponding controls were transfected into BV-2
cells using Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions, respectively. Briefly, 5 µl transfection reagent and
0.5 µg plasmid or 5 pmol small RNA were mixed in 50 µl serum-free
DMEM, left to stand for 5 min and then mixed. Following incubation
at room temperature for 20 min, the mix was added to serum-starved
cells and incubated at 37˚C for 4 h. Following this, 48 h later,
cells and the culture supernatants were harvested for relative
assays.
ELISA
After 24-h transfection, BV-2 cells (seeded in
96-well plates at 5x104 cells/well) were exposed to the
OGD treatment. Then TNF-α (cat. no. ab181421; Abcam) and IL-6 (cat.
no. ab222503; Abcam) levels in cell culture supernatants were
examined using ELISA kits. The absorbance was detected at 450 nm
using a microplate reader (Tecan Group, Ltd.).
Flow cytometry
Cell expression levels of inducible nitric oxide
synthase (iNOS) were detected by flow cytometry after BV-2 cells
were subjected to transfection and OGD treatment. In brief, BV-2
cells were fully digested using 0.25% trypsin at 37˚C for 5 min.
After cell counting, cells were resuspended at a density of 100 µl
4% formaldehyde per 1x106 cells and fixed at room
temperature for 15 min. Ice-cold 100% methanol was then slowly
added to a final concentration of 90% methanol and permeabilized on
ice for 10 min. The methanol was subsequently separated by
centrifugation at 500 x g for 5 min at room temperature, and
followed by incubation with 2 ng/ml PE-Cyanine7-labeled iNOS
antibody (cat. no. 25-5920-80; eBioscience; Thermo Fisher
Scientific, Inc.) in the dark at room temperature for 20 min. After
rinsing three times with PBS, these cells were were detected by a
flow cytometer (BD FACSCalibur™; BD Biosciences) and
analyzed using FlowJo v10 software (FlowJo, LLC).
Lactate dehydrogenase (LDH) assay
The neurons were seeded into 96-well plates
(5x104 cells/well) and stimulated with OGD-treated BV-2
cell conditioned media. In total, 5 ng/ml TNF-α neutralizing
antibody (cat. no. 7321; Cell Signaling Technology, Inc.) or its
IgG control (cat. no. 3900; Cell Signaling Technology, Inc.) were
added, before the cells were incubated at 37˚C and 5%
CO2 for 24 h. LDH released from apoptotic and necrotic
neurons was examined by an LDH assay kit in accordance with the
manufacturer's instructions (cat. no. C0016; Beyotime Institute of
Biotechnology). The percentage of apoptotic cells was calculated as
follows: (ODsample well-ODnegative control
well)/(ODpositive control well-ODnegative
control well) x100%.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cerebral tissues of
MCAO mice samples or BV-2 cells using TRIzol (CWbiotech, Co.,
Ltd.). Reverse transcription was performed using the
PrimeScript™ RT Master Mix (Takara Bio, Inc.). miRNA was
extracted using the miRcute Isolation Kit (Tiangen Biotech Co.,
Ltd.) and reverse transcription was conducted using the miScript II
RT Kit (Qiagen GmbH). qPCR was performed using the FastSYBR Mixture
(cat. no. CW0955; CWbiotech, Co., Ltd.). The relative expression
levels were analyzed using the 2-ΔΔCq method (20). 18S RNA and U6 were used as the
internal control. The amplification conditions for RT-PCR were as
follows: 42˚C for 40 min, followed by 85˚C for 5 min. The
amplification conditions for qPCR were as follows: 95˚C for 10 min,
followed by 40 cycles each at 95˚C for 15 sec, 60˚C for 30 sec, and
72˚C for 30 sec. The primers used were as follows: XIST forward,
5'-TAAGGACTACTTAACGGGCT-3'and reverse, 5'-TACTCAGACATTCCCTGGCA-3';
miR-96-5p forward, 5'-TTTGGCACTAGCACATTTTTGCT-3' and reverse,
5'-GTGCAGGGTCCGAGGT-3'; IKKβ forward, 5'-GACATCGCATCGGCTCTTAGA-3'
and reverse, 5'-AACGGTCACGGTGTACTTCTG-3'; U6 forward,
5'-CTCGCTTCGGCAGCACATATACT-3' and reverse,
5'-ACGCTTCACGAATTTGCGTGTC-3' and 18S forward,
5'-GTAACCCGTTGAACCCCATT-3' and reverse,
5'-CCATCCAATCGGTAGTAGCG-3'.
Western blotting
Total proteins were isolated from cell samples using
RIPA lysis buffer (Beyotime Institute of Biotechnology). Protein
concentration determination was performed using a BCA Protein Assay
Kit (Takara Bio, Inc.). A total of 20 µg of protein was
electrophoresed on 10% SDS-PAGE gels and then transferred onto PVDF
membranes; PVDF membranes were then blocked with 5% BSA (Beijing
Solarbio Science & Technology Co., Ltd.) at room temperature
for 40 min. Thereafter, the membrane was then incubated overnight
at 4˚C with anti-IKKβ (1:1,000; cat. no. ab109749; Abcam),
anti-phosphorylated (p)-p65 (Ser536) (1:1,000; cat. no. 3033; Cell
Signaling Technology, Inc.), and anti-p65 antibodies (1:1,000; cat.
no. 8242; Cell Signaling Technology, Inc.), followed by incubation
with horseradish peroxidase-conjugated anti-rabbit IgG antibody
(1:1,000; cat. no. 7074; Cell Signaling Technology, Inc.). The
protein expression levels were measured by an enhanced
chemiluminescence detection kit (Thermo Fisher Scientific, Inc.).
To quantify the protein expression, ImageJ 1.8.0 software (National
Institutes of Health) was used to analyze the gray value of the
protein bands.
Non-coding RNA target prediction and
dual-luciferase reporter assay
The putative interaction between lncRNA XIST and
miR-96-5p was searched by starBase v3.0 (http://starbase.sysu.edu.cn/) website and the target
gene of miR-96-5p was predicted in TargetScan 7.2 (http://www.targetscan.org/). The p-NF-κB-Luc plasmid
with the NF-κB response element cloned into Firefly pGL6 (Beyotime
Institute of Biotechnology) and the Renilla pRL-TK plasmid
(internal control; Promega Corporation) were used as luciferase
reporter vectors. 293T cells were purchased from the The Cell Bank
of Type Culture Collection of the Chinese Academy of Sciences and
cultured in DMEM with 10% FBS, 100 µg/ml streptomycin and
penicillin at 37˚C and 5% CO2. The vectors (50 ng for
each vector), combined with miRNA mimic/inhibitor or corresponding
negative controls (20 nM for each miRNA mimic/inhibitor or
controls), were co-transfected into 293T cells using Lipofectamine
2000 (Invitrogen; Thermo Fisher Scientific, Inc.). Then, 24 h
later, the cells were collected and the luciferase activities were
assessed using the Dual-luciferase Reporter Assay Kit (Promega
Corporation).
Statistical analysis
All quantitative data were expressed as the mean ±
SD. GraphPad Prism 7 (GraphPad Software, Inc.) was used for
statistical analysis in the present study. Unpaired Student's
t-test was employed to compare two groups. Comparison among
multiple groups was measured with one-way ANOVA followed by Tukey's
post hoc test. P<0.05 was considered to indicate a statistically
significant difference. Correlation analysis was conducted using
Spearman's rank correlation coefficient.
Results
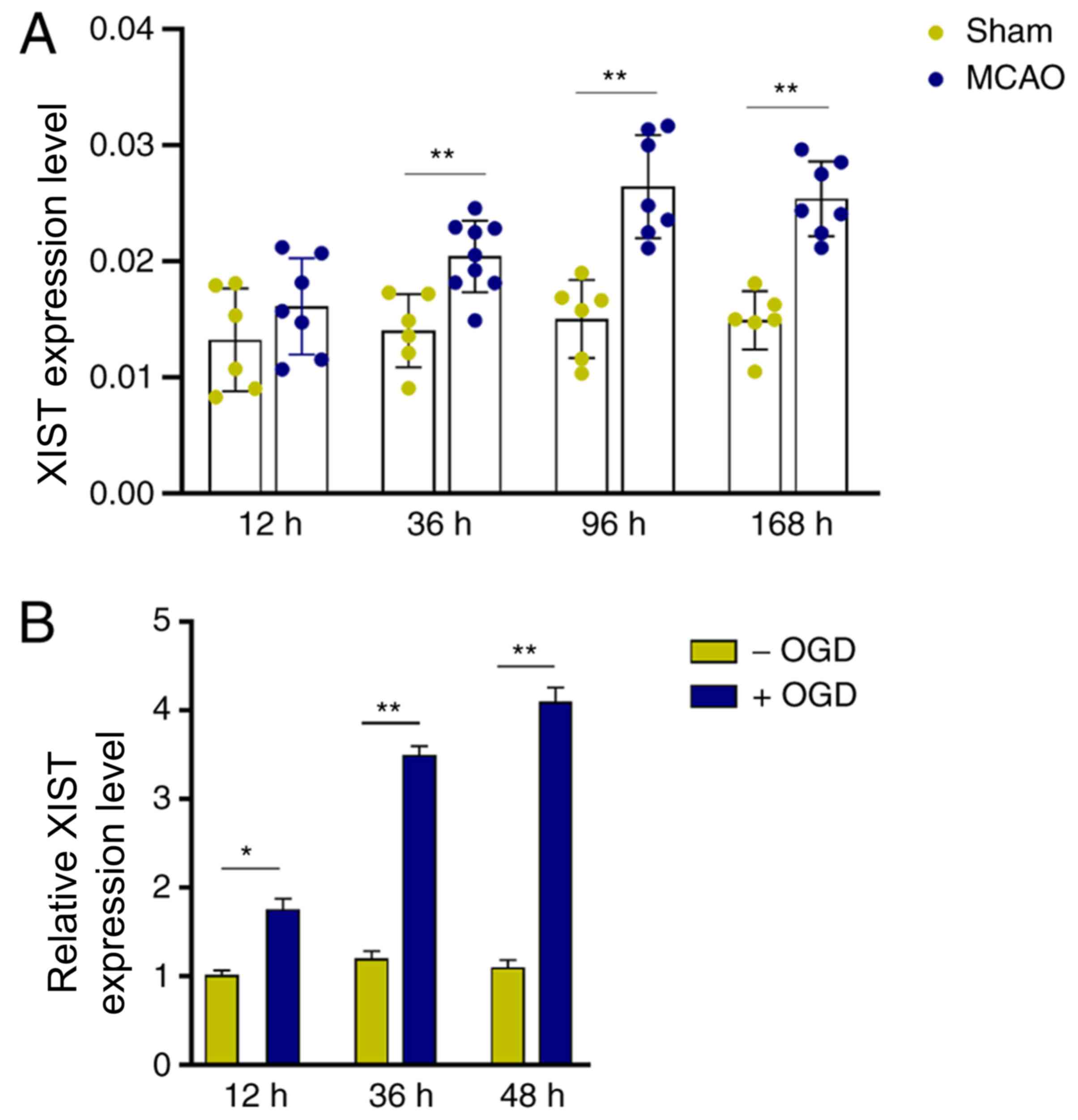
LncRNA XIST is upregulated in MCAO
mice and OGD-activated microglia
To profile the expression of XIST in cerebral
infarction, an in vivo mouse MCAO ischemic model was
established. Compared with the sham group, XIST expression was
significantly increased in mouse cerebral tissues 36 h after MCAO
treatment, and this upregulation could also be observed 168 h after
MCAO treatment (Fig. 1A).
Furthermore, it was observed that the in vitro OGD treatment
significantly increased XIST expression in BV-2 cells in a
time-dependent manner (Fig. 1B).
These results suggested that lncRNA XIST may be involved in the
pathological process of cerebral infarction by modulating
microglial function.
LncRNA XIST promotes pro-inflammatory
mediator production and mediates TNF-α-mediated neuronal
apoptosis
To investigate the role of lncRNA XIST in
microglia-mediated neuroinflammation, TNF-α and IL-6 were examined
after altering XIST expression (Fig.
2A-C). Fig. 2A demonstrated the
transfection efficiency of si-XIST and XIST overexpression XIST in
microglia. XIST overexpression enhanced the production of TNF-α
(1.8-fold increase) and IL-6 (1.68-fold increase) in OGD-challenged
microglia (Fig. 2B and C). However, XIST inhibition downregulated
the levels of TNF-α (45% decrease) and IL-6 (14% decrease)
(Fig. 2B and C). Similarly, OGD-induced iNOS expression
was augmented (1.45-fold increase) by XIST overexpression, but was
attenuated (34% decrease) after XIST inhibition (Fig. 2D and E).
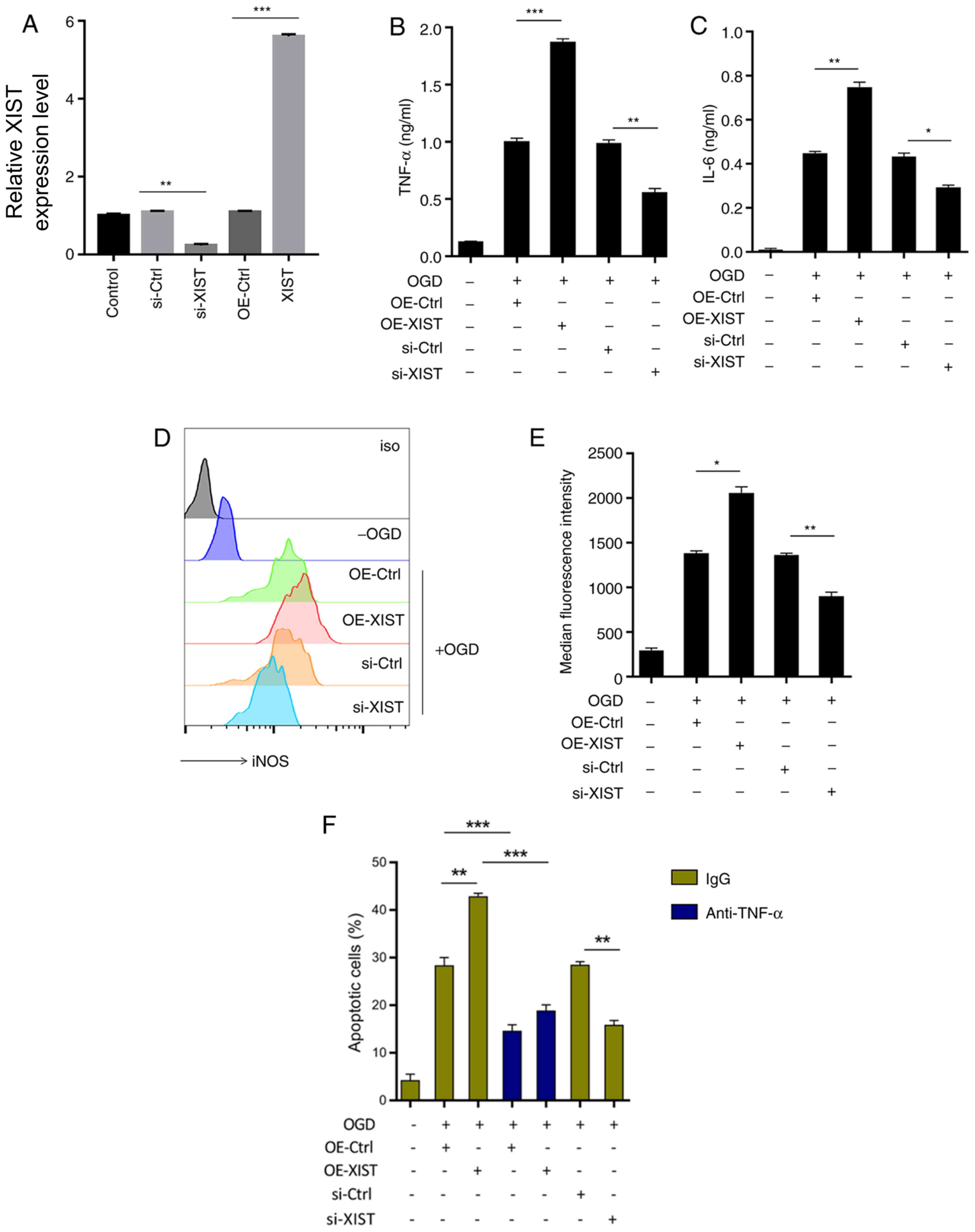 | Figure 2XIST promotes the release of
proinflammatory cytokines and controls TNF-α-mediated neuronal
apoptosis. After transfection with the XIST overexpression vector,
siRNA or corresponding controls, BV-2 cells were exposed to OGD for
24 h. (A) Confirmation of knockdown and overexpression of XIST by
reverse transcription-quantitative PCR. (B) TNF-α and (C) IL-6
expression levels in the culture supernatants were assayed by
ELISA. iNOS expression in the BV-2 cells was assessed by (D) flow
cytometry and expressed as (E) median fluorescence intensity. (F)
Apoptosis of mouse primary cerebral neurons was examined using a
lactate dehydrogenase assay after incubation with the culture
supernatants containing TNF-α neutralization antibody or IgG
control. *P<0.05, **P<0.01 and
***P<0.001. XIST, X-inactive specific transcript;
TNF, transforming growth factor; IL, interleukin; iNOS, inducible
nitric oxide synthase; si-, small interfering; Ctrl, control; OE,
overexpression. |
Compared with neurons grown in the supernatants of
OGD-BV-2 cells, culture supernatant from XIST-overexpressed
OGD-BV-2 cells exacerbated neuronal apoptosis (1.5-fold increase),
while TNF-α neutralization rescued this effect (50% decrease)
(Fig. 2F). Compared with the
OGD-BV-2 cell supernatant culture, the lncRNA XIST-silenced
OGD-BV-2 cell supernatant significantly inhibited neuronal
apoptosis (45% decrease; Fig. 2F).
These results indicated that lncRNA XIST contributed to
TNF-α-mediated neuronal apoptosis by promoting microglial
proinflammatory activation.
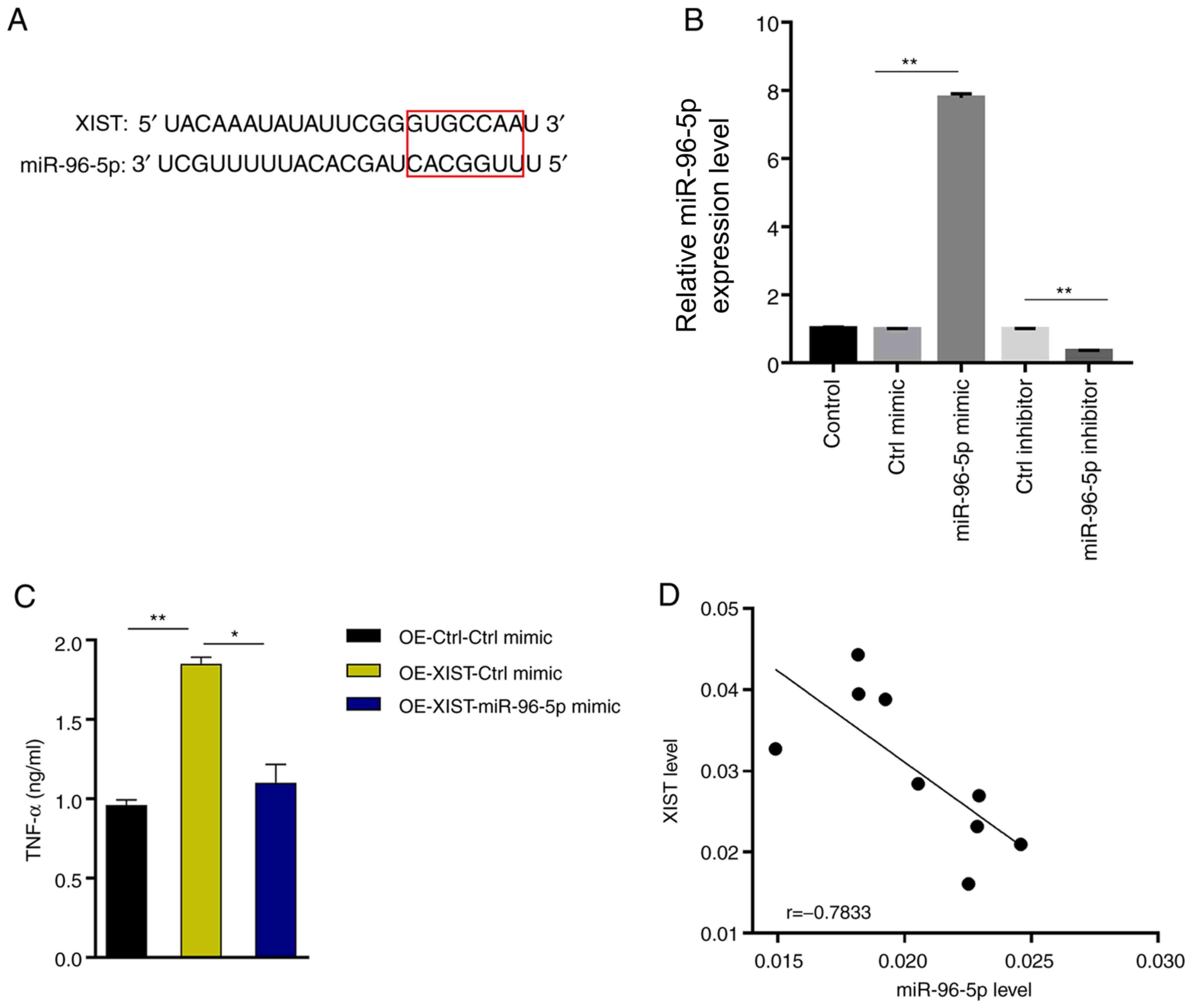
LncRNA XIST enhances TNF-α production
by directly suppressing miR-96-5p
To further elucidate the underlying molecular
mechanism of lncRNA XIST function, the starBase website was
searched and a putative interaction between lncRNA XIST and
miR-96-5p (Fig. 3A) was found.
miR-96-5p was successfully knocked down or overexpressed in BV-2
cells (Fig. 3B). miR-96-5p
overexpression antagonized the ability of XIST to enhance TNF-α
production (37% decrease, OE-XIST + miR-96-5p mimic vs. OE-XIST +
Ctrl mimic; Fig. 3C). Further
analysis of the expression profiles revealed a negative correlation
between lncRNA XIST and miR-96-5p in MCAO-treated mouse brain
tissues (Fig. 3D), indicating that
XIST acts as a sponge for miR-96-5p to antagonize its function.
MiR-96-5p downregulates NF-κB
signaling and reduces TNF-α production by inhibiting IKKβ
expression
Next, the target gene of miR-96-5p was predicted in
TargetScan and it was revealed that miR-96-5p could directly bind
to the IKKβ (Fig. 4A), whose
activation is a vital event upstream of NF-κB signaling (21,22).
To validate this, miR-96-5p mimic or inhibitor was transfected into
microglia, and IKKβ expression was examined using RT-qPCR. The
results revealed that IKKβ expression was significantly inhibited
by miR-96-5p overexpression (41% decrease; Fig. 4B). In contrast, IKKβ expression was
significantly enhanced upon miR-96-5p inhibition (2.3-fold
increase) (Fig. 4B). At the protein
level, miR-96-5p overexpression downregulated the expression of
IKKβ (47% decrease), and thus decreased the level of p-p65 (the
active subunit of NF-κB) (75% decrease), which was upregulated by
OGD treatment (Fig. 4C). In
addition, compared with the OGD+/Ctrl mimic group (p-p65/total
p65=0.44), miR-96-5p overexpression significantly reduced the ratio
of p-p65/total p65 (p-p65/total p65=0.16) (Fig. 4C). In order to further verify the
miR-96-5p/IKKβ interaction, miR-96-5p mimic/inhibitor and NF-κB
luciferase reporter were co-transfected into 293 cells. Luciferase
activity was decreased upon miR-96-5p overexpression (63%
decrease), but increased upon miR-96-5p inhibition (1.8-fold
increase) (Fig. 4D). These results
indicated that miR-96-5p downregulated NF-κB signaling by
suppressing IKKβ expression.
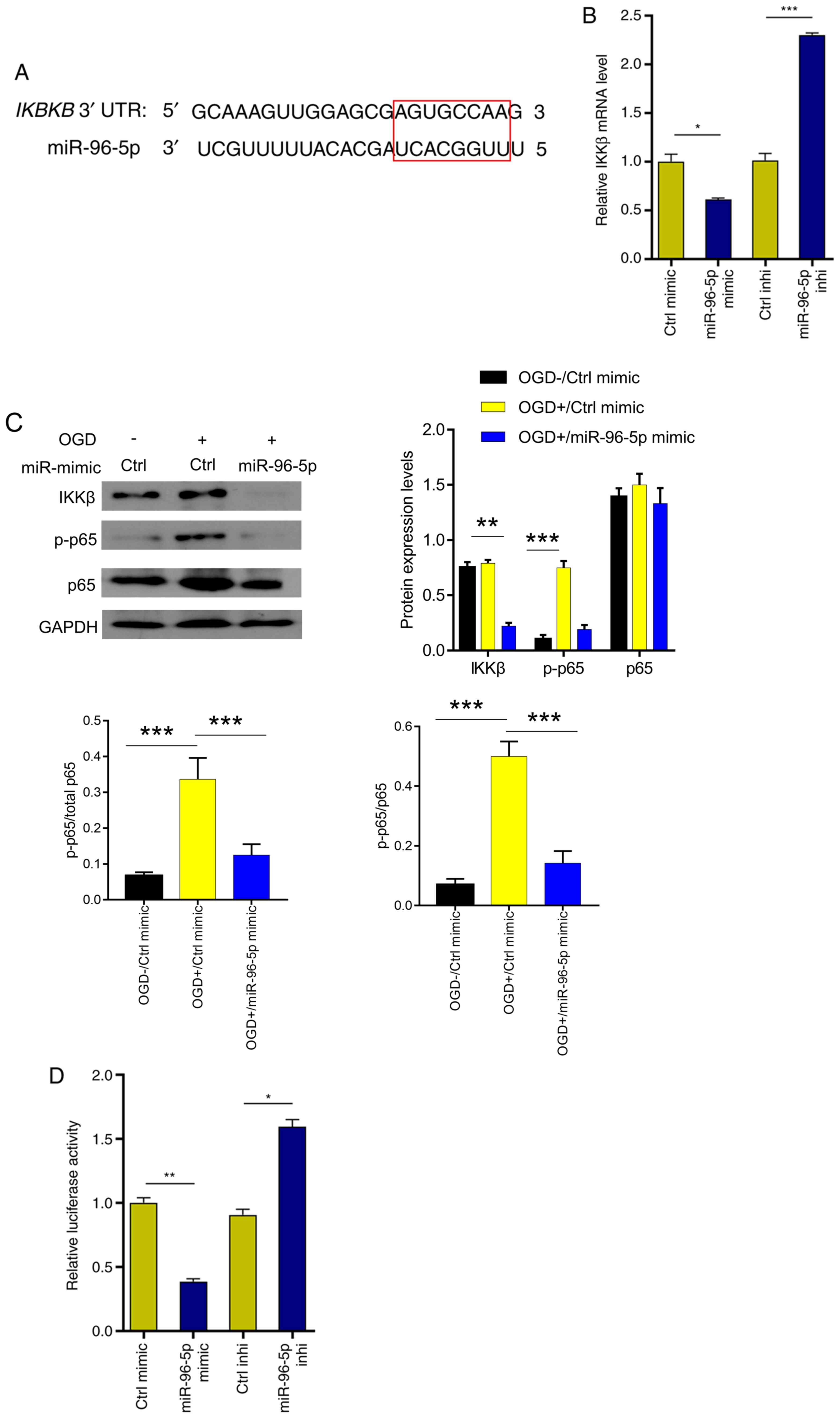 | Figure 4miR-96-5p inactivates NF-κB signaling
by targeting and inhibiting IKKβ. (A) IKKβ was predicted to be a
target of miR-96-5p in starBase (the red rectangle indicates the
binding sequence). (B) IKKβ mRNA levels were examined after
transfection with miR-96-5p mimic, inhibitor or control. (C)
Immunoblots of IKKβ, p65, and p-p65 proteins in OGD-induced BV-2
cells after transfection with miR-96-5p mimic or control. (D) The
luciferase activities were determined at 24 h after cotransfection
of p-NF-κB-Luc plasmid, Renilla pRL-TK plasmid (internal
control), and miR-96-5p mimic/inhibitor or corresponding negative
controls into 293 cells. *P<0.05,
**P<0.01 and ***P<0.001. miR-96-5p,
microRNA-96-5p; IKKβ, inhibitor of nuclear factor κB kinase subunit
β; p-, phosphorylated; Ctrl, control; OGD, oxygen/glucose
deprivation. |
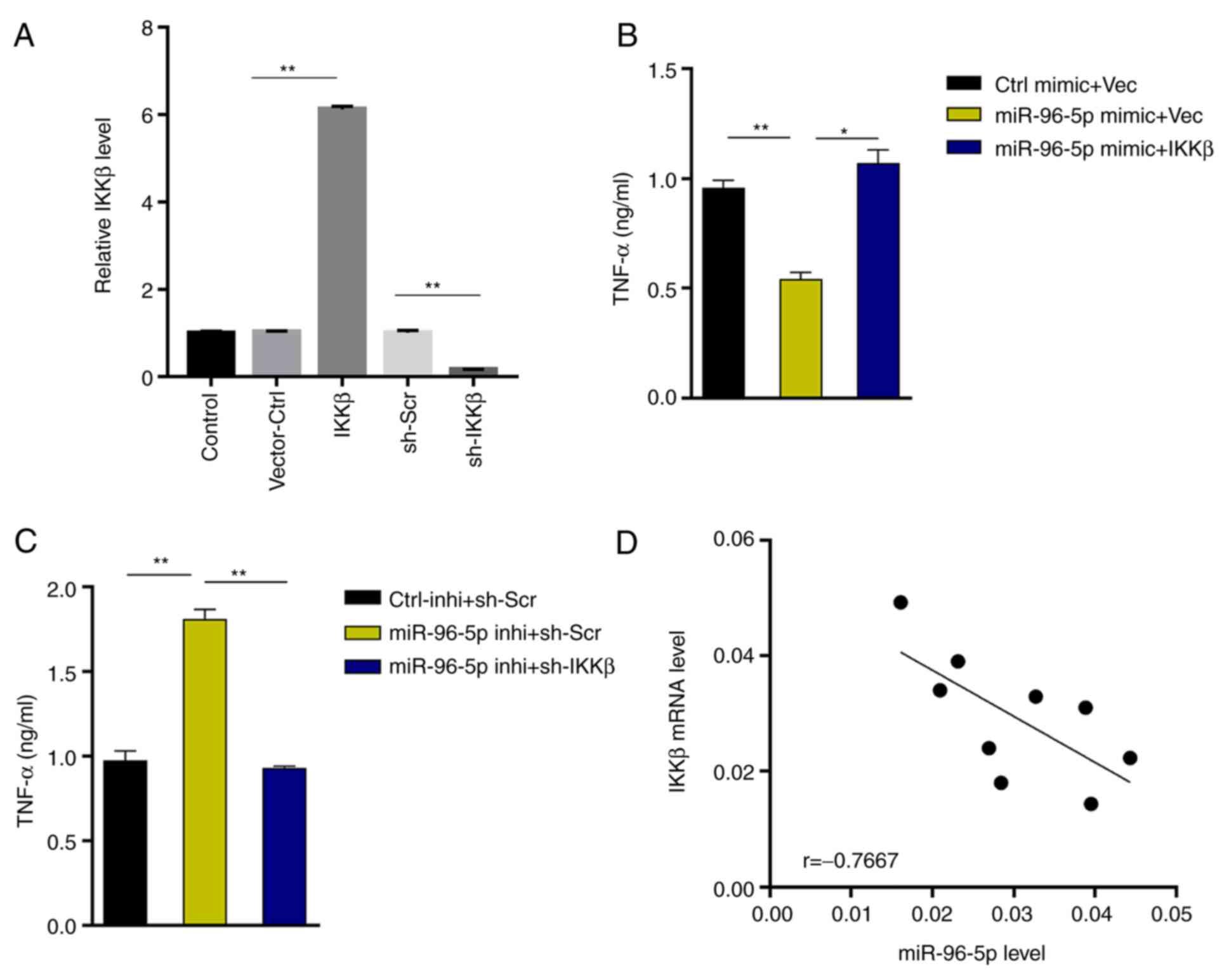
To further substantiate the results, a rescue assay
was performed. IKKβ was successfully knocked down or overexpressed
in BV-2 cells (Fig. 5A). The
results revealed that IKKβ overexpression reversed the decreased
TNF-α production in miR-96-5p mimic-transfected microglia (2.1-fold
increase, miR-96-5p mimic + IKKβ vs. miR-96-5p mimic + Vec), while
IKKβ silencing inhibited the upregulation of TNF-α production in
miR-96-5p inhibitor-transfected microglia (48% decrease, miR-96-5p
inhibitor + sh-IKKβ vs. miR-96-5p inhibitor + sh-Scr) (Fig. 5B and C). Furthermore, IKKβ expression was
inversely associated with miR-96-5p expression in MCAO-treated
mouse brain tissues (Fig. 5D),
supporting the IKKβ-targeting role of miR-96-5p.
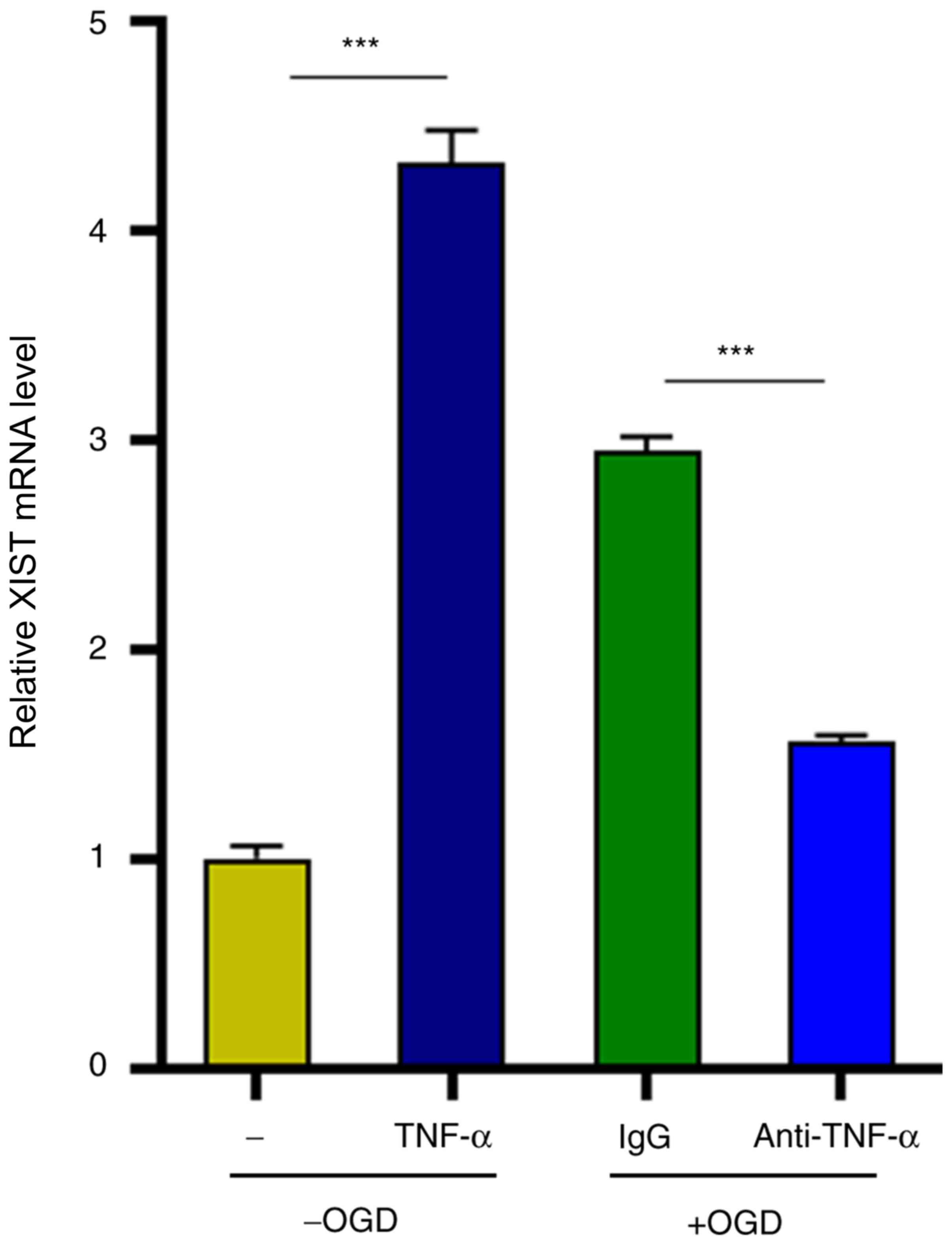
TNF-α in turn positively regulates
lncRNA XIST expression
Finally, the possible factors that contributed to
the augmented XIST expression in microglia were investigated.
Notably, it was determined that TNF-α stimulation significantly
upregulated XIST expression in microglia in the absence of OGD
treatment (4.2-fold increase) (Fig.
6). Concordant with this, the TNF-α neutralizing antibody
counteracted OGD-induced enhancement of XIST expression (47%
decrease) (Fig. 6). Collectively,
these results indicated that lncRNA XIST supported the expression
of TNF-α, which in turn positively regulated XIST expression in
microglia and thus formed a feedback to promote the proinflammatory
activation of microglia.
Discussion
In the present study, it was revealed that lncRNA
XIST was significantly upregulated in MCAO-treated mice and
OGD-treated microglia. In addition, it was also demonstrated that
XIST overexpression enhanced the expression and release of
pro-inflammatory mediators in microglia. LncRNA XIST has been
reported to participate in multiple diseases, including cancer and
neurological diseases (16,23-26).
In spinal cord injury, XIST promoted neuronal apoptosis by
regulating AKT phosphorylation (16). In the cellular model of Alzheimer's
disease, lncRNA XIST was involved in oxidative stress and apoptosis
of hippocampal neurons by functioning as a sponge for
miR-132(27). In line with this,
data from GEO database revealed that XIST expression in a patient
was augmented within 6 months after ischemic stroke (28). However, the regulatory mechanism of
XIST on the inflammatory polarization of microglia in cerebral
infarction remains unclear.
Inflammation plays an important role in the
pathophysiological process of cerebral infarction and has a dual
role of protection and harm to the brain tissue. LncRNA XIST was
confirmed to promote neuroinflammation by promoting the expression
of TNF-α and IL-6(28).
Downregulation of XIST repressed inflammatory cytokine expression
(approximately 60% decrease for COX-2, 50% decrease for TNF-α and
IL-6) in microglias (26). XIST
silencing was revealed to counteract LPS-induced TNF-α and IL-6
production (approximately 60% decrease for TNF-α and IL-6) in
microglial cells (29). The present
research revealed that XIST knockdown inhibited pro-inflammatory
mediator production (45% decrease for TNF-α, 14% decrease for IL-6,
and 34% decrease for iNOS) and TNF-α-mediated neuronal apoptosis
(45% decrease). These results were consistent with previous
research results (26,28,29).
To investigate the regulatory mechanism of lncRNA XIST, the miRNAs
that bind to it were explored and it was revealed that XIST could
act as a sponge for miR-96-5p, counteracting its inhibitory effect
on TNF-α production. The present research revealed that miR-96-5p
overexpression repressed TNF-α production (37% decrease). A
previous study revealed that downregulation of miR-96 markedly
increased the level of TNF-α in BV2 cells (approximately 2.0-fold
increase) (30). miR-96-5p has an
inhibitory effect on autophagy and apoptosis of breast tumor cells
(31). Kinoshita et al
revealed that the diurnal rhythm of miR-96-5p played a protective
role in dopaminergic neurons in Parkinson's disease (32). Collectively, these studies and our
findings indicate the crucial role of miR-96-5p in neurological
diseases and neuronal apoptosis.
IKKβ/IkB/NF-κB signaling plays a protective or toxic
role in neuroinflammation depending on the differential external
stimuli, cell types, and activation of NF-κB dimers (33,34).
Microglial activation of the NF-κB p50/p65 subunit downstream of
IKKβ was correlated with the production of pro-inflammatory
mediators, including IFN-γ, TNF-α, and IL-6, resulting in secondary
injury after the initial onset of cerebral infarction (21,33). A
previous study showed that targeting XIST induced apoptosis of
human osteosarcoma cells by activating NF-κB (35). Another previous study revealed that
miR-96 overexpression directly inhibits IKKβ expression, but the
upstream regulation of miR-96 has not been explored (30). The present results revealed that
IKKβ was directly targeted and silenced by miR-96-5p. It was also
revealed that p-p65 (the active subunit of NF-κB) accompanied by
pro-inflammatory cytokines was downregulated (75% decrease)
accordingly when miR-96-5p was overexpressed, demonstrating the
modulation of lncRNA XIST/miR-96-5p on IKKβ/IkB/NF-κB signaling in
microglia. IKKβ silencing inhibited TNF-α production (48%
decrease), which was consistent with a previous study (IKKβ
silencing resulted in a decrease in the TNF-α level of
approximately 35%) (30). Notably,
it was revealed that TNF-α, whose production could be induced by
the upstream lncRNA XIST/miR-96-5p/IKKβ/NF-κB axis, which in turn
augmented XIST expression (4.2-fold increase), indicating that
there could be a positive feedback loop between XIST and TNF-α
production through miR-96-5p/IKKβ/NF-κB signaling. In the future,
the detailed modulation mechanism of the positive feedback loop
between XIST and TNF-α in cerebral infarction remains to be
elucidated.
In conclusion, it was revealed that the lncRNA XIST
promoted inflammatory polarization of microglia in cerebral
ischemia by regulating the miR-96-5p/IKKβ/NF-κB axis, leading to
enhanced production of proinflammatory mediators and aggravated
neuronal apoptosis. The lncRNA XIST may be a potential target in
lncRNA-based ischemia therapy. The present findings provide novel
insights into the functional involvement of microglia in cerebral
infarction and its modulation mechanism.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by the Youth Grant from the
Science Foundation of Hebei Province (grant no. H2018206232).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
MZ and JKY designed the original research. MZ
conducted the statistical analysis and drafted the manuscript. MZ,
JKY and JM participated in the data collection and interpretation.
All authors read and approved the final manuscript. MZ and JKY can
authenticate the raw data in this study.
Ethics approval and consent to
participate
The experiments were carried out in accordance with
the National Institutes of Health (NIH) guidelines for the care and
use of laboratory animals approved by the Animal Ethics Committee
of Xingtai Medical College Second Affiliated Hospital (Xingtai
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lozano R, Naghavi M, Foreman K, Lim S,
Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, et
al: Global and regional mortality from 235 causes of death for 20
age groups in 1990 and 2010: A systematic analysis for the global
burden of disease study 2010. Lancet. 380:2095–2128.
2012.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Wang C, Li N, Wang H, Yin H and Zhao Y:
Study on essential drug use status and its influencing factors
among cerebral infarction inpatients in county level hospitals of
Anhui Province, China. PLoS One. 13(e0193513)2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Holliday EG, Traylor M, Malik R, Bevan S,
Falcone G, Hopewell JC, Cheng YC, Cotlarciuc I, Bis JC, Boerwinkle
E, et al: Genetic overlap between diagnostic subtypes of ischemic
stroke. Stroke. 46:615–619. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Puthanveetil P, Chen S, Feng B, Gautam A
and Chakrabarti S: Long non-coding RNA MALAT1 regulates
hyperglycaemia induced inflammatory process in the endothelial
cells. J Cell Mol Med. 19:1418–1425. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Benakis C, Garcia-Bonilla L, Iadecola C
and Anrather J: The role of microglia and myeloid immune cells in
acute cerebral ischemia. Front Cell Neurosci. 8(461)2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Seifert HA and Pennypacker KR: Molecular
and cellular immune responses to ischemic brain injury. Transl
Stroke Res. 5:543–553. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Faustino JV, Wang X, Johnson CE, Klibanov
A, Derugin N, Wendland MF and Vexler ZS: Microglial cells
contribute to endogenous brain defenses after acute neonatal focal
stroke. J Neurosci. 31:12992–13001. 2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hu X, Li P, Guo Y, Wang H, Leak RK, Chen
S, Gao Y and Chen J: Microglia/macrophage polarization dynamics
reveal novel mechanism of injury expansion after focal cerebral
ischemia. Stroke. 43:3063–3070. 2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Orihuela R, McPherson CA and Harry GJ:
Microglial M1/M2 polarization and metabolic states. Br J Pharmacol.
173:649–665. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Tang Y and Le W: Differential roles of M1
and M2 microglia in neurodegenerative diseases. Mol Neurobiol.
53:1181–1194. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Bao MH, Szeto V, Yang BB, Zhu SZ, Sun HS
and Feng ZP: Long non-coding RNAs in ischemic stroke. Cell Death
Dis. 9(281)2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Ren W and Yang X: Pathophysiology of long
non-coding RNAs in ischemic stroke. Front Mol Neurosci.
11(96)2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Dykstra-Aiello C, Jickling GC, Ander BP,
Shroff N, Zhan X, Liu D, Hull H, Orantia M, Stamova BS and Sharp
FR: Altered expression of long noncoding RNAs in blood after
ischemic stroke and proximity to putative stroke risk loci. Stroke.
47:2896–2903. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Yan H, Rao J, Yuan J, Gao L, Huang W, Zhao
L and Ren J: Long non-coding RNA MEG3 functions as a competing
endogenous RNA to regulate ischemic neuronal death by targeting
miR-21/PDCD4 signaling pathway. Cell Death Dis.
8(3211)2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wang J, Zhao H, Fan Z, Li G, Ma Q, Tao Z,
Wang R, Feng J and Luo Y: Long noncoding RNA H19 promotes
neuroinflammation in ischemic stroke by driving histone deacetylase
1-dependent M1 microglial polarization. Stroke. 48:2211–2221.
2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Gu S, Xie R, Liu X, Shou J, Gu W and Che
X: Long coding RNA XIST contributes to neuronal apoptosis through
the downregulation of AKT phosphorylation and is negatively
regulated by miR-494 in rat spinal cord injury. Int J Mol Sci.
18(732)2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Xu LX, Lv Y, Li YH, Ding X, Wang Y, Han X,
Liu MH, Sun B and Feng X: Melatonin alleviates brain and peripheral
tissue edema in a neonatal rat model of hypoxic-ischemic brain
damage: The involvement of edema related proteins. BMC Pediatr.
17(90)2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Blanco S, Hernández R, Franchelli G,
Ramos-Álvarez MM and Peinado MÁ: Melatonin influences NO/NOS
pathway and reduces oxidative and nitrosative stress in a model of
hypoxic-ischemic brain damage. Nitric Oxide. 62:32–43.
2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Ni J, Wang X, Chen S, Liu H, Wang Y, Xu X,
Cheng J, Jia J and Zhen X: MicroRNA let-7c-5p protects against
cerebral ischemia injury via mechanisms involving the inhibition of
microglia activation. Brain Behav Immun. 49:75–85. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ridder DA and Schwaninger M: NF-kappaB
signaling in cerebral ischemia. Neuroscience. 158:995–1006.
2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Herrmann O, Baumann B, de Lorenzi R,
Muhammad S, Zhang W, Kleesiek J, Malfertheiner M, Köhrmann M,
Potrovita I, Maegele I, et al: IKK mediates ischemia-induced
neuronal death. Nat Med. 11:1322–1319. 2005.PubMed/NCBI View
Article : Google Scholar
|
|
23
|
Zhang YL, Li XB, Hou YX, Fang NZ, You JC
and Zhou QH: The lncRNA XIST exhibits oncogenic properties via
regulation of miR-449a and Bcl-2 in human non-small cell lung
cancer. Acta Pharmacol Sin. 38:371–381. 2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Yao Y, Ma J, Xue Y, Wang P, Li Z, Liu J,
Chen L, Xi Z, Teng H, Wang Z, et al: Knockdown of long non-coding
RNA XIST exerts tumor-suppressive functions in human glioblastoma
stem cells by up-regulating miR-152. Cancer Lett. 359:75–86.
2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Jin H, Du XJ, Zhao Y and Xia DL:
XIST/miR-544 axis induces neuropathic pain by activating STAT3 in a
rat model. J Cell Physiol. 233:5847–5855. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yan XT, Lu JM, Wang Y, Cheng XL, He XH,
Zheng WZ, Chen H and Wang YL: XIST accelerates neuropathic pain
progression through regulation of miR-150 and ZEB1 in CCI rat
models. J Cell Physiol. 233:6098–6106. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wang X, Wang C, Geng C and Zhao K: LncRNA
XIST knockdown attenuates Aβ25-35-induced
toxicity, oxidative stress, and apoptosis in primary cultured rat
hippocampal neurons by targeting miR-132. Int J Clin Exp Pathol.
11:3915–3924. 2018.PubMed/NCBI
|
|
28
|
Li WX, Qi F, Liu JQ, Li GH, Dai SX, Zhang
T, Cheng F, Liu D and Zheng SG: Different impairment of immune and
inflammation functions in short and long-term after ischemic
stroke. Am J Transl Res. 9:736–745. 2017.PubMed/NCBI
|
|
29
|
Zhao Q, Lu F, Su Q, Liu Z, Xia X, Yan Z,
Zhou F and Qin R: Knockdown of long noncoding RNA XIST mitigates
the apoptosis and inflammatory injury of microglia cells after
spinal cord injury through miR-27a/Smurf1 axis. Neurosci Lett.
715(134649)2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Huang Y, Zhu N, Chen T, Chen W, Kong J,
Zheng W and Ruan J: Triptolide suppressed the microglia activation
to improve spinal cord injury through miR-96/IKKβ/NF-κB pathway.
Spine (Phila Pa 1976). 44:E707–E714. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Shi Y, Zhao Y, Shao N, Ye R, Lin Y, Zhang
N, Li W, Zhang Y and Wang S: Overexpression of microRNA-96-5p
inhibits autophagy and apoptosis and enhances the proliferation,
migration and invasiveness of human breast cancer cells. Oncol
Lett. 13:4402–4412. 2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Kinoshita C, Aoyama K, Matsumura N,
Kikuchi-Utsumi K, Watabe M and Nakaki T: Rhythmic oscillations of
the microRNA miR-96-5p play a neuroprotective role by indirectly
regulating glutathione levels. Nat Commun. 5(3823)2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Schneider A, Martin-Villalba A, Weih F,
Vogel J, Wirth T and Schwaninger M: NF-kappaB is activated and
promotes cell death in focal cerebral ischemia. Nat Med. 5:554–559.
1999.PubMed/NCBI View
Article : Google Scholar
|
|
34
|
Shih RH, Wang CY and Yang CM: NF-kappaB
signaling pathways in neurological inflammation: A mini review.
Front Mol Neurosci. 8(77)2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Gao W, Gao J, Chen L, Ren Y and Ma J:
Targeting XIST induced apoptosis of human osteosarcoma cells by
activation of NF-kB/PUMA signal. Bioengineered. 10:261–270.
2019.PubMed/NCBI View Article : Google Scholar
|