Introduction
Pulmonary arterial hypertension (PAH) is
characterized by progressive increases in pulmonary vascular
resistance and pulmonary arterial pressure, which ultimately lead
to right heart failure and death (1,2).
Registry data have indicated that connective tissue disease (CTD)
is the second leading cause of PAH. Idiopathic PAH accounts for 46%
of all cases and CTD associated with PAH (CTD-PAH) accounts for
~25% (3). The prevalence of CTD-PAH
differs among different populations. For instance, several studies
from the US and Europe have investigated systemic sclerosis
(SSc)-associated PAH (SSc-PAH), which accounts for ~75% of all
CTD-PAH cases, followed by systemic lupus erythematosus (SLE)-PAH
(8-19%), mixed CTD (MCTD)-PAH (8-9%) and primary Sjögren's syndrome
(pSS)-PAH (1%) (3). However, cohort
studies from Japan (4), South Korea
(5) and China (6,7)
indicated that SLE-PAH was the most common type of CTD, followed by
SSc, pSS and MCTD. These results suggested that the clinical
characteristics and prognosis of Asian patients with CTD-PAH may
differ from those of patients with CTD-PAH in Western
countries.
PAH is one of the leading causes of morbidity and
mortality among patients with CTD-PAH (7). The 1- and 3-year survival rates among
patients with CTD-PAH are ~80 and 50%, respectively. Prognosis is
less favorable for patients with SSc-PAH compared with that for
patients with SLE-PAH in Western and Asian countries (6,7). A
retrospective review of patients with SLE from Peking Union Medical
College Hospital (PUMCH; Beijing, China) over the last 30 years
revealed that pulmonary hypertension (PH) was the third most common
cause of death, after lupus encephalopathy and renal involvement
(8). PH may occur in isolation or
in association with interstitial lung disease (ILD), the latter of
which is probably caused by pulmonary interstitial lesions, which
increase resistance in the pulmonary circulation (1). Increased resistance in the pulmonary
circulation impairs oxygen exchange, causing long-term hypoxia,
which promotes the pathogenesis of PH (1). Previous studies have indicated that
mortality is higher in patients with CTD-PH and lung involvement,
compared with that in patients with isolated PH (1,6,9). In
the modern treatment era, targeted therapies have improved the
outcomes for patients with PH, including exercise capacity, World
Health Organization-function class (WHO-FC) and hemodynamic
parameters (10,11). However, less satisfactory outcomes
were observed after treatment in Chinese patients (1-3,10,11).
Therefore, early screening for PH and expedient diagnosis are
important to improve the prognosis of patients with CTD-PAH.
Doppler echocardiography is a non-invasive
technology and has been used routinely as a tool to screen for and
detect early-stage PAH. Pulmonary arterial systolic pressure
(PASP), as measured by echocardiography, has been indicated to
correlate with hemodynamic parameters, as measured by right heart
catheterization (RHC) (1,5,12-14).
The echocardiographic method most commonly used to detect PH
estimates right ventricular systolic pressure based on maximal
tricuspid regurgitation velocity (TRV). Patients with TRV >3.4
m2 (corresponding to a PASP >50 mmHg) or with TRV
between 2.9 and 3.4 m2 (corresponding to a PASP between
35 and 49 mmHg) in the presence of other signs are considered to
have PH. Echocardiography based on the estimation of PASP has an
estimated sensitivity of 0.79-1.00 and specificity of 0.6-0.98 for
the detection of PH, as confirmed by RHC (1,3,5,9-14).
pSS was recognized as a major CTD-associated PAH in China and this
trend remained consistent in South Korea and Japan (4,5).
Though the pathology of SLE, SSc and pSS is characterized as
vasculitis in certain patterns, the difference in survival among
SSc-, SLE- and pSS-associated PAH remains unexplained. Previous
studies indicated that right cardiac insufficiency was a
significant predictor of mortality for patients with CTD-PAH
(3-5),
but there is currently no consensus regarding prognostic risk
factors such as age, Raynaud's phenomenon, pericardial effusion and
ILD. Therefore, in the present study, the clinical characteristics
of patients with three major CTDs (SLE, SSc and pSS) associated
with early diagnosed PH were investigated and the associated
controversial progressive factors were explored. Patients in a
southwestern region of China were evaluated with Doppler
echocardiography to provide a comprehensive understanding of CTD-PH
and to facilitate the early recognition, prevention and treatment
of PH in CTD.
Materials and methods
Patient selection Inclusion
criteria
A total of 4,153 hospitalized patients with one of
the three major CTDs (3,133 SLE, 497 SSc and 523 pSS) were included
in calculations of the prevalence rate. A total of 218 patients
with CTD-PH (120 patients with SLE-PH, 64 patients with SSc-PH and
34 patients with pSS-PH) encountered at the First Affiliated
Hospital of Guangxi Medical University (Nanning, China) between
October 2012 and January 2018 were retrospectively included in the
present study. SLE was diagnosed according to the American College
of Rheumatology (ACR) criteria, which were revised in 1997 and
2012(15). SSc was diagnosed
according to the American Rheumatism Association criteria
established in 1980 and confirmed by the ACR/European League
Against Rheumatism (EULAR) classification criteria developed in
2013(16). The SSc subtype was
defined as limited cutaneous disease (lcSSc) if skin thickening was
confined to distal extremities (below the elbows and knees) and
above the clavicles. SSc was defined as diffuse cutaneous disease
(dcSSc) if skin thickening involved the proximal extremities and
the torso (16). pSS was diagnosed
according to the revised criteria proposed by the American-European
Consensus Group in 2002(17). All
patients were >14 years of age.
The patients with the three major types of CTD were
assessed by Doppler echocardiography at the time of first visit to
our hospital with the presence or absence of symptoms in order to
screen for and detect early-stage CTD-PH at the initial evaluation.
PASP >35 mmHg was defined as early diagnosed CTD-PH and such
patients were enrolled in the present study. According to the PASP,
the severity of each patient's PH was classified as mild (36-50
mmHg), moderate (51-70 mmHg) or severe (≥71 mmHg) (3,9,12).
Echocardiography was performed to evaluate the right-heart
morphology in all patients. The measurements obtained included
right atrial and ventricular enlargement, as well as main pulmonary
artery widening (diameter >26 mm). Lung involvement may affect
pre-capillary arterioles in PAH or post-capillary venules in
pulmonary veno-occlusive disease (3). As no right cardiac catheterization and
pulmonary function tests were performed, it was not possible to
confirm cases of isolated PAH. Patients with SSc, pSS with ILD or
SLE who had lung involvement (such as diffuse alveolar hemorrhage
and acute lupus pneumonia) were included in the study.
Exclusion criteria
Other CTDs associated with PH, including rheumatoid
arthritis, MCTDs, Takayasu arteritis, adult-onset Still's disease
and undifferentiated CTD, were excluded from the present study, as
they were rarely encountered at our center. Patients with
overlapping syndrome were also excluded from the study.
PH caused by other conditions described in the 2015
European Society of Cardiology and the European Respiratory Society
guidelines (1) for the Diagnosis
and Treatment of PH were excluded from the study: Definite history
of idiopathic PAH, obstructive sleep apnea, chronic obstructive
pulmonary disease, primary valvulopathy, pulmonary disease with a
mixed restrictive or obstructive pattern, portal hypertension, left
heart disease, drug/toxin exposure, HIV infection or any other
diseases known to be associated with PH (1).
Data collection
Demographic data, baseline clinical features,
laboratory results and echocardiographic parameters were collected
for analysis. The data collected at the time of PH diagnosis
included age, sex, duration of CTDs, duration from the onset of
PH-associated symptoms (exertional dyspnea, chest pain, chest
tightness and cough) to the initial diagnosis of PH, WHO-FC,
serological autoantibody profiles, N-terminal pro-brain natriuretic
peptide (NT-proBNP) levels and other biochemical parameters.
Specific clinical manifestations were recorded, including dyspnea,
cough, expectoration, edema, chest tightness, chest pain, Raynaud's
phenomenon, ischemic ulcer/pit and pericardial effusion.
High-resolution computed tomography (HRCT) scans were performed
routinely to evaluate ILD and the severity of pulmonary fibrosis
was scored individually. In brief, each lung was divided into three
zones: Upper (lung apex to aortic arch), middle (aortic arch to
inferior pulmonary veins) and lower (inferior pulmonary veins to
lung base). The extent of pulmonary abnormality in each of these
six zones was scored using a scale from 0 to 4 (0, absent; 1, 1-25;
2, 26-50; 3, 51-75; and 4, 76-100%). Pulmonary fibrosis was
characterized as pure ground-glass opacity, pulmonary fibrosis or
honeycomb cysts (18). The HRCT
images were scored according to the research of Goldin et al
(18) by two radiologists (>20
years working experience) independently, when discrepancies
occurred, a consensus was reached after discussion. For each
patient with CTD, the disease activity was scored: The SLE activity
index score (SLEDAI) for patients with SLE (12); the modified Rodan skin score (mRSS)
for SSc (19); and the EULAR
Sjögren syndrome disease activity index (ESSDAI) for pSS (20). Any history of clinical treatment for
CTD-PH, such as the use of glucocorticoids, immunosuppressants or
pulmonary vasodilators, was also recorded. The initial dose of
glucocorticoid [≤0.5 and 1-2 mg/kg/day (d), as well as 0.5-1 kg/d]
was determined based on the patient's condition and level of CTD
disease activity. The immunosuppressants used alone or in
combination among patients with CTD included cyclophosphamide pulse
[one pulse of 500 mg/m2 per month (mo) for 6-8 mo],
azathioprine (100 mg/d), cyclosporine (3-5 mg/kg/d, bid),
methotrexate (10-15 mg, weekly), leflunomide (20 mg/day),
thalidomide (50-75 mg/day), mycophenolate mofetil (75 mg, bid) and
tripterygium glycosides (50 mg, tid). The PAH-specific therapies
administered included endothelial receptor antagonists (bosentan
and ambrisentan), phosphodiesterase type 5 inhibitors (sildenafil,
tadalafil), and beraprost. The specific method used for drug
delivery and the dosage of immunosuppressants administered
throughout disease treatment were individualized according to the
patient's condition. The survival status was determined by
telephone interview or through evaluation of the medical record.
All patients were followed up at 1 year or until they met the
end-point of death up to January 2019. These patients were included
in for survival analysis. To calculate the survival rate, an
end-point of either the date of death or the last date of follow-up
at the outpatient clinic was recorded.
Statistical analysis
Statistical analysis was performed with SPSS version
24.0 (IBM Corp.). Continuous variables are expressed as the mean ±
standard deviation. Normally distributed data were compared using
Student's t-test or one-way ANOVA followed by the least-significant
difference post-hoc test. Non-normally distributed data are
suitably expressed as the median (range or 25/75th percentile) and
compared using Mann-Whitney U tests. Categorical variables are
expressed as n (%) and compared using the χ2 test.
Survival rates were calculated with the Kaplan-Meier method (log
rank test). The primary end-point was all-cause death. Rates of
survival at 1, 3 and 5 years were also determined. Univariate and
multivariate logistic regression analysis with the Cox proportional
hazards model was used to identify independent factors influencing
mortality. The results are presented as hazard ratios (HRs) with
95% confidence intervals (CIs). P<0.05 was considered to
indicate statistical significance. The figure was prepared with
GraphPad Prism 8 (GraphPad Software, Inc.).
Results
Comparison of demographic and baseline
clinical characteristics among groups
The demographic and baseline clinical
characteristics were compared among patients with SLE-PH, SSc-PH
and pSS-PH. Patients with SSc were most likely to have PH [64/497
(12.9%)], followed by those with pSS [34/523 (6.5%)] and then by
those with SLE [120/3,133 (3.8%)]. Among the 218 patients with
CTD-PH included in the present study, SLE-PH was most common
(54.8%), followed by SSc (29.2%) and pSS (16.0%). Demographic data,
baseline clinical characteristics and treatments are presented in
Table I. The mean age at the time
of PH diagnosis was 47.47±38.70 years. Patients with SLE-PH were
younger (40.54±49.82 years) than those with SSc-PH (58.02±10.93
years; P<0.05) and patients with SSc-PH were younger than those
with pSS-PH (52.09±16.08 years; P>0.05). The overall study group
included more females than males (83.9 vs. 16.1%), with a
significantly different sex ratio. There were more females in the
SLE-PH than SSc-PH the group (92.5 vs. 68.8%; P<0.05). The
overall median duration of CTDs was 12 mo (range, 1-336 mo) and the
median duration was by far the highest in patients with SSc-PH. The
median duration of symptoms prior to the diagnosis of PH was 3 mo
(range, 1-168 mo) and the median duration was highest in patients
with pSS-PH. The rate of smoking was highest in patients with
SSc-PH among all study groups. A total of 189 patients with CTD
(86.7%) had PH-related symptoms. The most common clinical symptom
of CTD-PH was dyspnea (65.6%), followed by cough (51.4%),
expectoration (39.9%), edema (34.4%), chest tightness (25.7%) and
chest pain (14.2%). Raynaud's phenomenon appeared in 38.5% of
patients with CTD-PH and was the most common symptom in patients
with SSc-PH. Among all patients with CTD-PH, 42.2% were classified
as WHO-FC III/IV (53.2% of patients with SSc-PH, 44.2% of those
with SLE-PH and 35.3% of those with pSS-PH). In addition, patients
with SSc-PH had the highest prevalence of lung involvement (81.2%),
followed by patients with pSS (58.8%) and then by patients with SLE
(30.0%). When biochemical parameters were compared among the CTD-PH
groups (data without significance were not shown), alkaline
phosphatase (ALP) levels exhibited a significant difference.
Although ALP levels were higher in the pSS-PH group than those in
the SLE-PH and SSc-PH groups (P<0.05), the mean values remained
within normal limits. Levels of NT-proBNP were elevated to various
degrees in all three groups, with the greatest increase observed in
the SLE-PH group. The level of IgG was higher in the pSS-PH group
than that in the other two groups (P<0.05), likely due to the
hyperglobulinemia associated with pSS. The antinuclear antibody
(Ab) and anti-extractable nuclear antigen Ab staining patterns were
consistent with the characteristics of CTD. Pulmonary arterial
pressure was mildly to moderately elevated in 80.3% of patients
with CTD-PH. PASP was higher in the pSS group than in the SLE or
SSc groups (P<0.05). Pericardial effusion was more common in
patients with SLE-PH due to the nature of the disease. However, no
differences among the groups were observed in the degree of PH
divided by the PASP or parameters of right-heart morphology (right
atrial and ventricular enlargement, main pulmonary artery
widening). The rate of left ventricular compliance decrease was
significantly different among the groups (especially in the SSc-PH
group; P<0.05) (Table II).
 | Table IComparison of demographic and baseline
clinical characteristics among patients with different types of
CTD-PH. |
Table I
Comparison of demographic and baseline
clinical characteristics among patients with different types of
CTD-PH.
| Item | Total (n=218) | SLE-PH (n=120) | SSc-PH (n=64) | pSS-PH (n=34) | P-value |
|---|
| Prevalence of
PHa | 218/4,153 (5.2) | 120/3,133 (3.8) | 64/497 (12.9) | 34/523 (6.5) | |
| Age (years) | 47.47±38.70 | 40.54±49.82 |
58.02±10.93b | 52.09±16.08 | 0.001 |
| Female | 183 (83.9) | 111 (92.5) | 44 (68.8) | 28 (82.4) | <0.001 |
| Duration of CTDs
(mo) | 12 (1-336) | 9.5 (1-336) | 21.5
(1-240)b | 12 (1-108) | 0.007 |
| Duration from
symptom onset to PH diagnosis (mo) | 3 (1-168) | 2 (1-84) | 7 (1-108) | 12 (1-168) | <0.001 |
| Smoking | 20 (9.2) | 3 (1.7) | 15
(23.4)b | 2
(5.9)c | <0.001 |
| BMI
(kg/m2) | 20.63±2.97 | 20.65±3.04 | 20.50±2.93 | 20.79±2.84 | 0.977 |
| Hypertension | 45 (20.6) | 21 (17.5) | 18 (28.1) | 6 (17.6) | 0.213 |
| Dyspnea | 143 (65.6) | 71 (49.7) | 46 (71.9) | 26 (65.6) | 0.078 |
| Cough | 112 (51.4) | 63 (52.5) | 34 (53.1) | 15 (44.1) | 0.652 |
| Expectoration | 87 (39.9) | 44 (36.7) | 31 (48.4) | 12 (36.7) | 0.250 |
| Raynaud's
phenomenon | 84 (38.5) | 25 (20.8) | 52
(81.2)b | 7
(20.6)c | <0.001 |
| Edema | 75 (34.4) | 56 (46.7) | 16
(25.0)b | 3
(8.8)b | <0.001 |
| Chest
tightness | 56 (25.7) | 35 (29.2) | 13 (20.3) | 8 (23.5) | 0.404 |
| Chest pain | 31 (14.2) | 20 (16.7) | 5 (7.8) | 6 (17.6) | 0.215 |
| Syncope | 6 (2.8) | 5 (42.0) | 0 (0.0) | 1 (2.9) | 0.259 |
| WHO-FC | | | | | 0.297 |
|
I | 79 (36.2) | 48 (40.0) | 23 (35.9) | 8 (23.5) | |
|
II | 47 (21.6) | 19 (15.8) | 14 (21.9) | 14 (41.2) | |
|
III | 53 (24.3) | 26 (21.7) | 16 (25.0) | 11 (32.4) | |
|
IV | 39 (17.9) | 27 (22.5) | 1 (28.2) | 1 (2.9) | |
| Lung
involvement | 108 (49.5) | 36 (30.0) | 52
(81.2)b | 20
(58.8)b,c | <0.001 |
| ALP (U/l) | 73.82±58.21 | 66.94±39.93 | 75.62±37.38 |
95.81±116.13b,c | 0.046 |
| NT-proBNP
(pg/ml) | 2,761.5 (966.3,
10,816.0) | 4,411.5 (2,303.5,
14,065.0) | 1,333.0 (181.2,
10,342. 0)b | 811.0 (472.8,
2,682.0)a | 0.004 |
| IgG (g/l) | 15.18 (11.64,
21.06) | 15.02 (11.33,
21.61) | 14.75 (11.55,
17.61) | 18.13 (13.83,
28.69)b,c | 0.065 |
| Auto-Abs | | | | | |
|
ANA | 211 (96.8) | 117 (97.5) | 61 (91.3) | 33 (97.1) | 0.722 |
|
Anti-centromere
Ab | 13 (6.0) | 1 (0.8) | 9
(14.1)b | 3
(8.8)b,c | 0.001 |
|
Anti-nRNP/Sm
Ab | 81 (37.2) | 65 (54.2) | 5
(7.8)b | 11
(32.4)b,c | <0.001 |
|
Anti-phospholipid
Ab | 5 (2.3) | 4 (3.3) | 0 (0) | 1 (3.3) | 0.335 |
|
Glucocorticoids | 211 (96.8) | 118 (98.3) | 61 (95.3) | 32 (94.1) | 0.343 |
|
Immunosuppressants | 201 (92.2) | 112 (93.3) | 59 (92.2) | 30 (88.2) | 0.646 |
| Vasodilator
therapy | | | | | |
|
Use of
medications | 94 (43.1) | 48 (40.0) | 32(50) | 14 (41.2) | 0.414 |
|
Combination
therapy | 19 (20.4) | 11 (23.4) | 3 (9.4) | 5 (35.7) | 0.097 |
| Table IIComparison of baseline
echocardiographic characteristics among groups of patients with
connective tissue disease-PH. |
Table II
Comparison of baseline
echocardiographic characteristics among groups of patients with
connective tissue disease-PH.
| Item | Total (n=218) | SLE-PH (n=120) | SSc-PH (n=64) | pSS-PH (n=34) | P-value |
|---|
| PASP (mmHg) | 56.28±21.54 | 56.54±21.29 |
50.16±14.39a |
66.91±28.82a,b | 0.001 |
| Degree of PH | | | | | 0.146 |
|
Mild | 114 (52.3) | 62 (51.7) | 39 (60.9) | 13 (38.2) | |
|
Moderate | 61 (28.0) | 31 (25.8) | 18 (28.1) | 12 (35.3) | |
|
Severe | 43 (19.7) | 27 (22.5) | 7 (10.9) | 9 (26.5) | |
| Pericardial
effusion | 134 (61.5) | 93 (77.5) | 29
(45.3)a | 12
(35.3)a | <0.001 |
| Right atrium
enlargement | 58 (27.1) | 36 (30.0) | 12 (18.8) | 11 (32.4) | 0.197 |
| Right ventricle
enlargement | 52 (23.9) | 30 (25.0) | 11 (17.2) | 11 (32.4) | 0.223 |
| Main pulmonary
artery widening | 73 (33.5) | 47 (39.2) | 22 (34.4) | 13 (38.2) | 0.813 |
| Decreased left
ventricular compliance | 103 (47.2) | 46 (55.5) | 44
(68.8)a | 14
(38.2)b | <0.001 |
| EF (%) | 67.7±10.47 | 66.65±9.81 | 69.56±11.74 | 66.40±5.31 | 0.251 |
Comparison of demographic and baseline
clinical characteristics between survivors and non-survivors
Symptoms of dyspnea, expectoration and chest
tightness were more common among non-survivors than survivors. The
duration of symptoms prior to diagnosis of PH was longer in
non-survivors (median, 8 mo; range, 1-168 mo) than in survivors
(median, 2 mo; range, 1-144 mo). The proportion of patients with
WHO-FC III/IV in the non-survivor group (58.9%) was significantly
higher than the proportion in the survivor group (35.1%; P=0.003).
Compared with survivors, non-survivors had higher levels of
NT-proBNP (Though with was not significant) and ALP, likely due to
right ventricular dysfunction being more common among non-survivors
than among survivors (6,7). Lung involvement was also more common
among non-survivors compared with survivors (44.4 vs. 58.9%,
P=0.075; Table III). In the
subgroup analysis of SSc-PH and pSS-PH, ILD HRCT scores were higher
among non-survivors than among survivors (SSc, 20.33±3.92 vs.
12.72±8.95; pSS, 31.43±14.95 vs. 13.5±10.24; P<0.05; data not
shown). Compared with CTD-PH survivors, non-survivors had higher
PASP (P=0.001) and a higher proportion of cases with moderately to
severely elevated pulmonary arterial pressure (P=0.001).
Non-survivors also had a higher prevalence of right atrial
enlargement and right ventricular enlargement (P<0.05; Table IV).
| Table IIIComparison of demographic and
baseline clinical characteristics in CTD-PH survivors and
non-survivors. |
Table III
Comparison of demographic and
baseline clinical characteristics in CTD-PH survivors and
non-survivors.
| Item | Total (n=173) | Survivors
(n=117) | Non-survivors
(n=56) | P-value |
|---|
| Age (years) | 47.47±38.70 | 48.04±50.07 | 50.46±18.61 | 0.727 |
| Female sex | 146 (84.4) | 100 (85.5) | 46 (82.1) | 0.573 |
| Duration of CTDs
(mo) | 12 (1-336) | 12 (1-336) | 14 (1-132) | 0.215 |
| Duration from
symptom onset to PH diagnosis (mo) | 3 (1-168) | 2 (1-144) | 8 (1-168) | 0.024 |
| Smoking | 16 (9.2) | 7 (6.0) | 9 (16.1) | 0.032 |
| BMI
(kg/m2) | 20.63±2.97 | 20.75±2.80 | 20.45±3.10 | 0.523 |
| Hypertension | 39 (22.5) | 21 (17.9) | 18 (32.1) | 0.037 |
| Dyspnea | 112 (64.7) | 68 (58.1) | 44 (78.6) | 0.008 |
| Cough | 87 (50.3) | 54 (46.2) | 33 (58.9) | 0.116 |
| Expectoration | 67 (38.7) | 38 (32.5) | 29 (51.8) | 0.015 |
| Raynaud's
phenomenon | 62 (35.8) | 38 (32.5) | 24 (42.9) | 0.183 |
| Edema | 54 (31.2) | 34 (29.1) | 20 (35.7) | 0.377 |
| Chest
tightness | 44 (25.4) | 24 (20.5) | 20 (35.7) | 0.032 |
| Chest pain | 25 (14.5) | 18 (15.4) | 7 (12.5) | 0.614 |
| Syncope | 5 (2.9) | 2 (1.7) | 3 (5.4) | 0.330 |
| WHO-FC | | | | 0.013 |
|
I | 65 (37.6) | 49 (41.9) | 16 (28.6) | |
|
II | 34 (19.7) | 27 (23.1) | 7 (12.5) | |
|
III | 44 (25.4) | 27 (23.1) | 17 (30.4) | |
|
IV | 30 (17.3) | 14 (12.0) | 16 (28.6) | |
| Lung
involvement | 85 (49.1) | 52 (44.4) | 33 (58.9) | 0.075 |
| ALP (U/l) | 73.82±58.21 | 63.98±38.45 | 86.32±91.73 | 0.026 |
| NT-proBNP
(pg/ml) | 2,761.5 (966.3,
10,800.0) | 2,586.0 (811.0,
5,731.0) | 3,277.0 (1,062.0,
15,000) | 0.305 |
| IgG (g/l) | 15.18
(11.64,21.06) | 3.37 (1.62,
3.37) | 15.01 (10.67,
20.05) | 0.419 |
| Auto-Ab | | | | |
|
ANA | 166 (96.0) | 111 (94.9) | 55 (98.2) | 0.297 |
|
Anti-centromere
Ab | 9 (5.2) | 6 (5.1) | 3 (5.4) | 0.950 |
|
Anti-nRNP/Sm
Ab | 67 (38.4) | 48 (41.0) | 19 (33.9) | 0.370 |
|
Anti-phospholipid
Ab | 5 (2.9) | 4 (3.4) | 1 (1.8) | 1.000 |
|
Glucocorticoids | 167 (96.5) | 115 (98.3) | 52 (92.9) | 0.087 |
|
Immunosuppressants | 160 (92.5) | 111 (94.9) | 49 (87.5) | 0.121 |
| Vasodilator
therapy | | | | |
|
Use of
medications | 72 (41.6) | 49 (41.9) | 23 (41.1) | 0.920 |
|
Combination
therapy | 14 (8.1) | 9 (7.8) | 5 (8.9) | 0.793 |
| Table IVComparison of baseline
echocardiographic characteristics between connective tissue
disease-PH survivors and non-survivors. |
Table IV
Comparison of baseline
echocardiographic characteristics between connective tissue
disease-PH survivors and non-survivors.
| Item | Total (n=173) | Survivors
(n=117) | Non-survivors
(n=56) | P-value |
|---|
| PASP (mmHg) | 56.28±21.54 | 51.85±18.61 | 63.46±23.02 | 0.001 |
| Degree of PH | | | | 0.009 |
|
Mild | 95 (55.2) | 74 (63.2) | 21 (37.5) | |
|
Moderate | 46 (26.7) | 26 (22.2) | 20 (35.7) | |
|
Severe | 31 (18.0) | 17 (14.6) | 15 (26.8) | |
| Pericardial
effusion | 103 (59.5) | 72 (61.5) | 31 (55.4) | 0.438 |
| Right atrium
enlargement | 49 (28.3) | 27 (23.1) | 22 (39.3) | 0.027 |
| Right ventricle
enlargement | 44 (25.4) | 21 (17.9) | 23 (41.1) | 0.001 |
| Main pulmonary
artery widening | 66 (38.2) | 42 (35.9) | 24 (42.9) | 0.378 |
| Decreased left
ventricular compliance | 81 (46.8) | 49 (41.9) | 32 (57.1) | 0.060 |
| EF (%) | 67.70±10.48 | 66.49±10.33 | 67.93±12.55 | 0.511 |
Analysis of the impact of CTD disease activity on
prognosis for patients with PH indicated that the SLEDAI scores
were suggestive of high activity in both survivors and
non-survivors, with no significant difference between groups
(15.32±6.66 vs. 16.40±1.48; P=0.528; data not shown). Among the 64
patients with SSc-PH included in the study, 22 (34.3%) had lcSSc
and 42 (65.6%) had dcSSc. Among the 47 patients with SSc-PH who
were followed up, there was no significant difference in mortality
between the lcSSc and dcSSc cases [43.8% (7/16) vs. 45.2% (14/31),
respectively; P=0.927], nor was there any significant difference
between survivors and non-survivors in terms of mRSS scores for the
degree of skin fibrosis (26.88±10.45 vs. 24.65±11.18, respectively;
P=0.493) (above data not shown). Compared with non-survivors of
pSS-PH, survivors of pSS-PH had higher levels of immunoglobulin G
(IgG) [24.93 (15.16,32.43) vs. 13.33 (11.69,15.11) g/l]; P=0.020]
and ESSDAI [(22.88±10.90 vs. 13.38±2.92; P=0.025] (data not
shown).
Determination of prognosis and
predictors of mortality in patients with CTD-PH
Among the 218 patients included in the present
study, 96.5% received glucocorticoids and 92.5% received
immunosuppressive agents at the time of their diagnosis with CTD. A
total of 72 patients (41.6%) in 173 follow-up patients received
specific pulmonary vasodilator therapy for PH and 14 patients
(8.1%) received combination therapy. No significant difference in
the frequency with which these treatments were administered was
observed between non-survivors and survivors (Table III). Short-term usage of targeted
treatments was common and patients with CTD-PH were typically
unable to continue targeted treatment for >6 mo.
The mean duration of follow-up after PH diagnosis
was 26.17±20.66 months. A total of 173 patients were followed up,
including 98 patients with SLE-PH, 47 with SSc-PH and 28 with
pSS-PH. The rates of mortality during the follow-up period were
26.3, 48.8 and 41.7%, respectively. The overall survival rates at
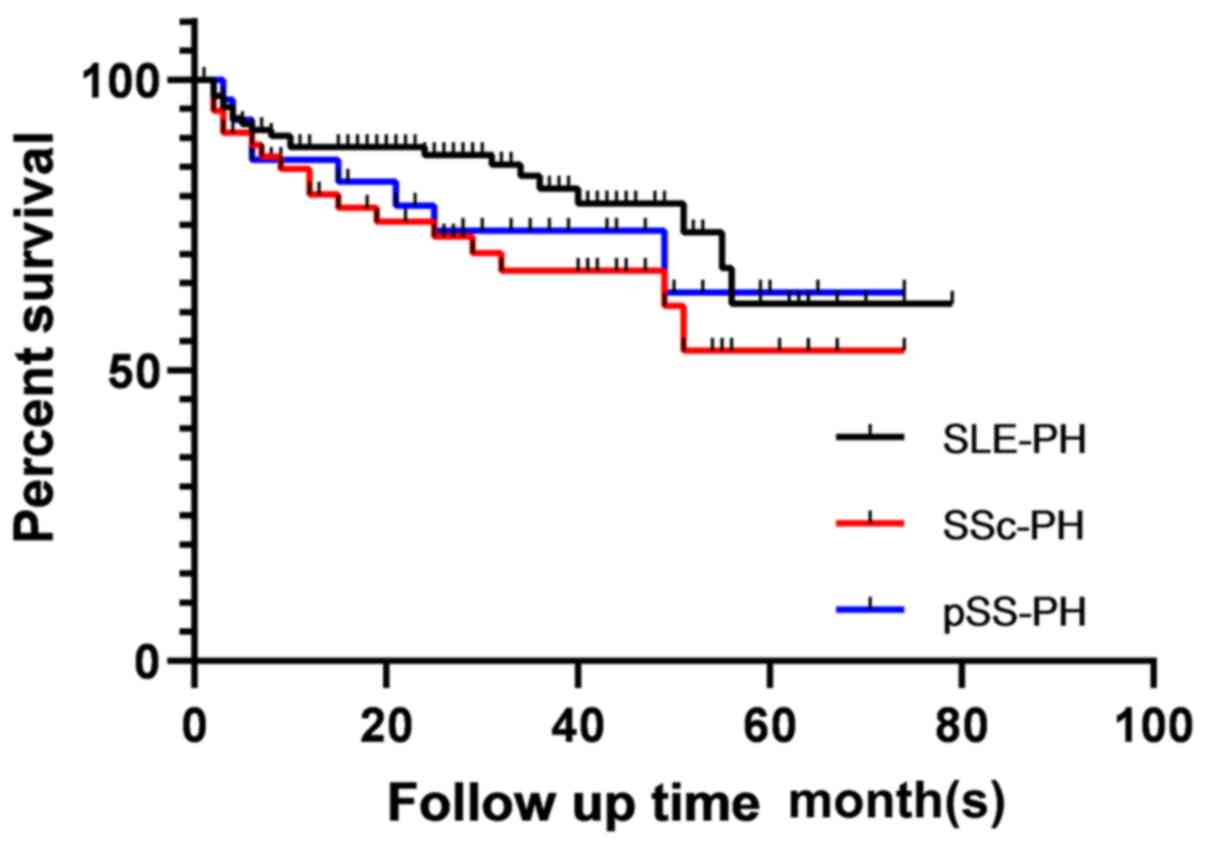
1, 3 and 5 year were 81.4, 72.4 and 56.9%, respectively. The rate
of survival tended to be lowest among patients with SSc (75.3,
63.0, 50.1%), followed by patients with pSS (73.7, 59.7 and 59.7%)
and then by patients with SLE (84.7, 77.9 and 58.9%); however,
there was no significant difference among these three subgroups of
patients with CTD-PH (P=0.177; Fig.
1).
Univariate Cox regression analysis of risk factors
for mortality among patients with CTD-PH identified age ≥50 years,
PH severity, enlargement of the right ventricle, WHO-FC III/IV,
dyspnea and expectoration as influencing variables. However, in the
multivariate analysis, only age ≥50 years was an independent risk
factor for mortality (Table V).
| Table VUnivariate and multivariate analysis
of predictors of mortality in patients with CTD-PH. |
Table V
Univariate and multivariate analysis
of predictors of mortality in patients with CTD-PH.
| | Univariate
analysis | Multivariate
analysis |
|---|
| Factor | HR (95% CI) | P-value | HR (95% CI) | P-value |
|---|
| Female sex | 0.79
(0.40-1.56) | 0.496 | | |
| Age ≥50 years | 1.721
(1.018-2.911) | 0.043 | 1.766
(1.107-3.065) | 0.043 |
| Duration of CTDs ≥5
years | 0.630
(0.308-1.286) | 0.204 | | |
| Duration of symptom
onset to PH diagnosis | 1.003
(0.994-1.013) | 0.501 | | |
| Type of CTDs | | | | |
|
SLE, SSc and
pSS | 1.251
(0.899-1.74) | 0.184 | | |
|
SLE vs.
non-SLE | 1.594
(0.914-2.700) | 0.083 | | |
|
SSc vs.
non-SSc | 1.568
(0.913-2.695) | 0.103 | | |
| Degree of PH (high,
moderate and mild) | 1.588
(1.160-2.172) | 0.004 | 1.333
(0.086-2.065) | 0.199 |
| Raynaud's
phenomenon | 0.764
(0.450-1.298) | 0.320 | | |
| Dyspnea | 2.037
(1.075-3.860) | 0.029 | 1.131
(0.506-2.577) | 0.764 |
| Expectoration | 1.704
(1.008-2.881) | 0.047 | 1.372
(0.797-2.363) | 0.254 |
| Pericardial
effusion | 0.799
(0.471-1.354) | 0.799 | | |
| Right atrium
enlargement | 1.683
(0.984-2.88) | 0.057 | | |
| Right ventricle
enlargement | 2.164
(1.269-3.689) | 0.005 | 1.433
(0.684-2.999) | 0.340 |
| Main pulmonary
artery widening | 1.2
(0.692-2.081) | 0.515 | | |
| Lung
involvement | 1.456
(0.854-2.484) | 0.168 | | |
| SSc and
pSS-ILD | 1.167
(0.502-2.711) | 0.720 | | |
| WHO-FC III/IV vs.
I/II | 2.213
(1.298-3.773) | 0.004 | 1.414
(0.715-2.798) | 0.320 |
| Use of vasodilator
therapy | 0.943
(0.553-1.608) | 0.829 | | |
| Combination
vasodilator therapy | 1.153
(0.460-2.891) | 0.762 | | |
Discussion
In the present study, the clinical manifestations
and survival rates associated with three major CTDs accompanied by
PH were investigated. The study included patients from southwestern
China who had been diagnosed with the condition based on the
results of echocardiography, with the aim to screen and detect
early-stage PH. The prevalence of PH was highest among patients
with SSc, while the SLE group accounted for the largest population
among patients with CTD-PH. Patients with CTD-PH who were screened
early with Doppler echocardiography, particularly those with lung
involvement, had less favorable prognoses. Therefore, early
recognition by echocardiography and early treatment (particularly
with vasodilators) should be attempted to counteract the poor
prognosis exhibited by patients with CTD-PH.
The distribution of types of CTD associated with PAH
varies across different regions. For instance, SSc-PAH predominates
in Western cohorts (3). In Asian
cohorts, SLE-PAH is the most common type of CTD associated with PAH
and SSc-PAH is least common. One study of 129 Chinese patients with
CTD-PAH reported that the type of CTD most commonly associated with
PAH was SLE (49%), followed by pSS (16%), MCTD (9%) and SSc (6%)
(6). A study performed at PUMCH
suggested that patients with PAH were more likely to have SLE
(58.4%) than to have SSc (26.3%) or pSS (15.3%) (7). Similar results were reported in cohort
studies based on data obtained from national registries in Japan
(4) and South Korea (5). Consistent with the aforementioned
results of Asian studies, the present study determined that the
most common type of CTD-PH was SLE-PH (120/218, 54.8%), while PH
was most common among patients with SSc (64/497, 12.9%).
In terms of survival, the present results indicated
that disparities in disease distribution did not affect the risk of
death. The 1-, 3- and 5-year survival rates among patients with
CTD-PH were 81.4, 72.4 and 56.9%, respectively, in accordance with
the results previously reported by Hao et al and Zhao et
al (6,7). However, the survival rates reported in
the present study are higher than those reported in the REVEAL
study (21) and in the study by
Condliffe et al (9). These
differences in survival rates may reflect variations in the
prevalence of CTDs.
Prognosis appeared to be most favorable among
patients with SLE-PH and poorer among patients with SSc-PH as
compared with patients with PH with other types of CTDs. Several
reasons may account for this difference. In the present study, mean
patient age and CTD disease duration were lower among patients with
SLE-PH as compared with those in patients with SSc-PH. These
patterns may reflect differences in the pathogenesis of
inflammation and immune system dysfunction or differences in the
response to immunosuppression therapies (7,22). The
pathological features of SLE are inflammatory lesions in small
vessels throughout the circulatory system. Pulmonary hypertension
in patients with SLE may be caused by acute inflammatory cell
infiltration, vasospasm or microthrombus formation. Therefore,
pulmonary arterial pressure may decrease or return to normal after
active immunosuppression and anticoagulation in patients with
SLE-PH. Patients with SSc-PH exhibit vascular lesion reactions,
including chronic inflammatory cell infiltration, vascular
endothelial damage, vascular remodeling and angiostenosis, all of
which result in irreversible pathologic changes in pulmonary
vessels. These patients may also respond poorly to
immunosuppressive treatment (22).
Finally, lung involvement has varying effects on prognosis in
patients with CTD-PH (6,7). Lung involvement that occurs in SLE is
more likely to appear as acute interstitial pneumonia, diffuse
alveolar hemorrhage with lupus pneumonia or acute pulmonary
embolism caused by antiphospholipid syndrome. These conditions may
be relieved or reversed by immunosuppressive treatment (22). However, chronic pulmonary fibrosis
is the chronic lung lesion type that most commonly leads to pSS-PH
and SSc-PH. This condition is also less responsive to
immunosuppressive therapy (6,7,22).
The present study indicated that pulmonary arterial
pressure was typically mildly to moderately elevated in patients
with CTD-PH. In addition, PASP was higher in the pSS group than in
the SLE and SSc groups. Higher levels of IgG and ESSDAI were
observed in pSS-PH survivors; CTD disease activity (SLEDAI in SLE,
mRSS in SSc and ESSDAI in pSS) decreased after the administration
of immunosuppressive therapy. It is noteworthy that PASP was higher
in the pSS group than in the SLE and SSc groups, which was similar
to the result in the study by Zhao et al (7), in which patients with pSS-PAH had
worse hemodynamic profiles on diagnostic RHC than the SLE-PAH and
SSc-PAH groups, with significantly higher mean right atrial
pressure, mean pulmonary artery pressure and pulmonary vascular
disease values and a lower cardiac index. The underlying mechanism
is likely related to immune pathogenesis. An immunofluorescence
study by Zhao et al (7)
demonstrated the deposition of immune system components in the
muscular-type arteries of patients with pSS-PAH, indicating the
presence of an immune complex-mediated injury. This former study
postulated the hypothesis that endothelial damage, immune complex
accumulation, necrotic vasculitis and imbalances in the levels of
endothelium-derived vasoactive molecules contribute to the
pathogenesis of pSS-PAH. The pathogenesis of pSS-PAH is similar to
that of the acute inflammatory lesions present in patients with
SLE-PAH, which may respond well to treatment (7). As with the results of previous studies
(6,7), patients with SLE were younger than
those with pSS and SSc in the current study, and age was an
important contributor to mortality, as older patients had a higher
mortality rate. Delays in diagnosis may also account for
differences in survival rates between patients with SLE-PH and
pSS-PH. Additional studies will be necessary to better understand
existing barriers to the early diagnosis of PH in patients with pSS
(7).
Regarding predictors of mortality, previous studies
suggested that poor heart function indices, poor hemodynamics,
lower exercise capacity, lower mixed venous oxygen saturation,
WHO-FC III/IV and elevated NT-proBNP were all significant risk
factors among patients with CTD-PAH (3-5).
The present results indicated that age ≥50 years, severity of PH,
enlargement of the right ventricle, WHO-FC III/IV, dyspnea and
expectoration were associated with poor survival. Age ≥50 years was
the only independent risk factor for mortality. There is currently
no consensus regarding prognostic risk factors such as age,
Raynaud's phenomenon, pericardial effusion and ILD.
Age is well recognized as a significant predictor of
survival in patients with CTD-PH. The UK's national registry of all
incident cases of CTD-PAH indicated that patients <60 years of
age had better survival rates than patients aged ≥70 years
(9). A study based on data from the
French SSc-PAH registry that was published in 2012 suggested that
the risk of death of patients increased by 1.05-fold for each
additional year of age at the time of PAH diagnosis (23). Analysis of the patient data in South
Korea's nationwide registry of patients with CTD-PH (as detected by
echocardiography) suggested that age >60 years was a risk factor
for mortality (5). In the present
study, multivariate analysis revealed that age ≥50 years was the
only independent risk factor for mortality. These discrepancies
among studies may reflect differences in sample size, CTD
distribution and diversity in populations. Hence, further
large-cohort studies that distinguish between CTD-PAH patient
subgroups are required to verify the effect of age on survival of
patients with CTD-PAH.
Raynaud's phenomenon, fingertip ulcer/pit and
gangrene are examples of direct evidence of vascular disease.
Raynaud's phenomenon is considered to be associated with CTD-PAH
and is an independent risk factor for the onset of PAH.
Mechanistically, Raynaud's phenomenon is associated with systolic
and diastolic dysfunction of the acral arteriole. When it affects
the pulmonary artery, the condition is called ‘pulmonary Raynaud's
phenomenon’ (24). The persistent
vasospasm of pulmonary arterioles may result in pulmonary vascular
remodeling and increased pulmonary vascular resistance (24). In the present study, Raynaud's
phenomenon appeared to be more common in the group of CTD-PH
non-survivors compared with survivors. However, this difference was
not statistically significant, which was in accordance with the
report by Zhao et al (7).
Therefore, whether Raynaud's phenomenon is a prognostic risk factor
for CTD-PH remains controversial.
Pleural effusion is considered to be a risk factor
for PAH among patients with CTD. However, whether pleural effusion
predicts survival remains controversial. A study by Hao et
al (6) and one study based on
data from the South Korean nationwide registry (5) suggested that patients with pleural
effusion had poorer survival outcomes than patients without pleural
effusion, highlighting that the condition may be a predictor of
mortality among patients with CTD-PAH. However, one study of
patients with SSc-PAH from France (23) and the study by Zhao et al
(7) indicated no significant
association between the prevalence of pleural effusion and survival
of patients with CTD-PAH. The present study also failed to identify
a significant association between pleural effusion and survival
among patients with CTD-PAH. Additional studies will be required to
further clarify these discrepant findings.
Lung involvement is common in patients with CTD. SLE
tends to be complicated by ILD (usually non-specific interstitial
pneumonia), diffuse hemorrhagic alveoli and acute lupus pneumonia,
all of which respond well to immunosuppressive therapy. ILD
(typically in the form of interstitial pneumonia) is more
frequently encountered in patients with SSc or pSS, among whom
immunosuppressive therapy is less effective. Mild PH is common
among patients with severe interstitial lung disease. Hao et
al (6) defined ILD-PH as
moderate ILD based on the results of HRCT (1/3-2/3 of the lung
field involved) in combination with a total lung capacity of 60%
predicted via pulmonary function testing, or severe ILD based on
the results of HRCT (>2/3 of the lung field involved). The
results of Hao et al (6)
indicated that the prognosis of patients with CTD with ILD-PH was
significantly less favorable than that of patients with isolated
CTD-PAH in Chinese cohorts. Studies from the UK (9), US (21) and France (23) obtained similar results after
comparing the survival of patients with ILD-PH or
respiratory-associated PH with the survival of patients with
isolated SSc-PAH. In the present study, lung involvement with
CTD-PH appeared to be more common among non-survivors than among
survivors, but the difference was not statistically significant.
Further subgroup analysis of the combination of SSc-PAH and pSS-PAH
suggested that the HRCT scores observed in non-survivors were
higher than those in survivors. Hence, it may be concluded that
severe ILD is associated with a poor prognosis.
Of note, the present study had certain
limitations. First, PH was defined on the basis of PASP
>35 mmHg, as measured by Doppler echocardiography but not by
RHC. However, pulmonary arterial pressure (as measured by
echocardiography) was indicated to be consistently associated with
the hemodynamic index measured by RHC (3,5,9,12-14).
Echocardiography is a non-invasive diagnostic tool that provides a
reasonably reliable and comprehensive assessment of the right heart
and pulmonary circulation and may be used routinely for screening
and diagnosing early PH. Furthermore, due to the lack of pulmonary
function tests, it was not possible to compare ILD-PH and isolated
PH. In addition, although 43.1% of patients included in the present
study used pulmonary vasodilators and 20.4% received combination
therapy, drug withdrawal was common. Beraprost was adopted as a
specific pulmonary treatment in the present study; however,
beraprost is less effective than other pulmonary vasodilators or
combination therapy. No significant difference in the frequency
with which these treatments were administered was observed between
non-survivors and survivors. This result may reflect selection
bias, as patients at higher risk of mortality were selected because
they had higher rates of PASP and right ventricle dysfunction. It
is also possible that short-term specific PAH treatment improved
symptoms in patients with CTD only transiently without altering the
overall prognosis. The poorer prognosis may reflect the fact that
the patients in the present study who received pulmonary
vasodilators were typically older patients with CTD-PH who had
systemic disease, particularly myocardial involvement and
comorbidities. However, even if a patient's condition worsened,
there was no increase in the dose or administration of combined
treatment due to the cost that would accrue to the patient and
their family. Further investigation will be necessary to determine
whether more aggressive management is able to improve the overall
prognosis in such cases (23).
In conclusion, among these three major types of
CTD-PH, SSc-PH had the highest prevalence. The overall prognosis
for patients with CTD-PH remains poor. Patients with SLE-PH have
the most favorable prognosis. Among the 3 types of CTD-PH, patients
with SSc-PH have the worst outcomes. Age ≥50 years is the only
independent risk factor for mortality.
Acknowledgements
The authors would like to thank our radiology
colleagues, Dr Kai Li and Dr Xuechun Guan (Department of Radiology,
the First Affiliated Hospital of Guangxi Medical University), whose
experience in interstitial lung diseases helped examine the HRCT
results and assign HRCT scores.
Funding
Funding: This study was supported by grants from the National
Natural Science Foundation (grant nos. 81860292 and 81760295) and
Guangxi Medical and Health Technology Development and Promotion
Project (grant no. S2018084).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JP collected and analyzed the data, and wrote the
manuscript. JW, FQ and FD collected the data, including demographic
data, baseline clinical features, laboratory results and
echocardiographic parameters. LL and CZ contributed to the
conception, design and organization of the current study, and
reviewed and revised the manuscript. LL and CZ confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript. All authors are fully responsible
for all aspects of the study and the final manuscript. All authors
agree to be accountable for all aspects of the work in ensuring
that questions related to the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
The study protocol was approved by the Ethics
Committee of the First Affiliated Hospital of Guangxi Medical
University (approval no. 2018-KY-National Natural Science
Foundation-012). Written informed consent was obtained from all
patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Galiè N, Humbert M, Vachiery JL, Gibbs S,
Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A,
Beghetti M, et al: 2015 ESC/ERS guidelines for the diagnosis and
treatment of pulmonary hypertension: The joint task force for the
diagnosis and treatment of pulmonary hypertension of the European
society of cardiology (ESC) and the European respiratory society
(ERS): Endorsed by: Association for European paediatric and
congenital cardiology (AEPC), international society for heart and
lung transplantation (ISHLT). Eur Heart J. 37:67–119.
2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Simonneau G, Montani D, Celermajer DS,
Denton CP, Gatzoulis MA, Krowka M, Williams PG and Souza R:
Haemodynamic definitions and updated clinical classification of
pulmonary hypertension. Eur Respir J. 53(1801913)2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Thakkar V and Lau EM: Connective tissue
disease-related pulmonary arterial hypertension. Best Pract Res
Clin Rheumatol. 30:22–38. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Shirai Y, Yasuoka H, Okano Y, Takeuchi T,
Satoh T and Kuwana M: Clinical characteristics and survival of
Japanese patients with connective tissue disease and pulmonary
arterial hypertension: A single-centre cohort. Rheumatology
(Oxford). 51:1846–1854. 2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kang KY, Jeon CH, Choi SJ, Yoon BY, Choi
CB, Lee CH, Suh CH, Lee CW, Cho CS, Nam EJ, et al: Survival and
prognostic factors in patients with connective tissue
disease-associated pulmonary hypertension diagnosed by
echocardiography: Results from a Korean nationwide registry. Int J
Rheum Dis. 20:1227–1236. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Hao YJ, Jiang X, Zhou W, Wang Y, Gao L,
Wang Y, Li GT, Hong T, Huo Y, Jing ZC and Zhang ZL: Connective
tissue disease-associated pulmonary arterial hypertension in
Chinese patients. Eur Respir J. 44:963–972. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zhao J, Wang Q, Liu Y, Tian Z, Guo X, Wang
H, Lai J, Huang C, Yang X, Li M and Zeng X: Clinical
characteristics and survival of pulmonary arterial hypertension
associated with three major connective tissue diseases: A cohort
study in China. Int J Cardiol. 236:432–437. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Fei Y, Shi X, Gan F, Li X, Zhang W, Li M,
Hou Y, Zhang X, Zhao Y, Zeng X and Zhang F: Death causes and
pathogens analysis of systemic lupus erythematosus during the past
26 years. Clin Rheumatol. 33:57–63. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Condliffe R, Kiely DG, Peacock AJ, Corris
PA, Gibbs JS, Vrapi F, Das C, Elliot CA, Johnson M, DeSoyza J, et
al: Connective tissue disease-associated pulmonary arterial
hypertension in the modern treatment era. Am J Respir Crit Care
Med. 179:151–157. 2009.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Fischer A, Denton CP, Matucci-Cerinic M,
Gillies H, Blair C, Tislow J and Nathan SD: Ambrisentan response in
connective tissue disease-associated pulmonary arterial
hypertension (CTD-PAH)-A subgroup analysis of the ARIES-E clinical
trial. Respir Med. 117:254–263. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Coghlan JG, Galiè N, Barberà JA, Frost AE,
Ghofrani HA, Hoeper MM, Kuwana M, McLaughlin VV, Peacock AJ,
Simonneau G, et al: Initial combination therapy with ambrisentan
and tadalafil in connective tissue disease-associated pulmonary
arterial hypertension (CTD-PAH): Subgroup analysis from the
AMBITION trial. Ann Rheum Dis. 76:1219–1227. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Weatherald J, Montani D, Jevnikar M, Jaïs
X, Savale L and Humbert M: Screening for pulmonary arterial
hypertension in systemic sclerosis. Eur Respir Rev.
28(190023)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Shahane A: Pulmonary hypertension in
rheumatic diseases: Epidemiology and pathogenesis. Rheumatol Int.
33:1655–1667. 2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Barst RJ, McGoon M, Torbicki A, Sitbon O,
Krowka MJ, Olschewski H and Gaine S: Diagnosis and differential
assessment of pulmonary arterial hypertension. J Am Coll Cardiol.
43 (Suppl 12):40S–47S. 2004.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Fortuna G and Brennan MT: Systemic lupus
erythematosus: Epidemiology, pathophysiology, manifestations, and
management. Dent Clin North Am. 57:631–655. 2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
van den Hoogen F, Khanna D, Fransen J,
Johnson SR, Baron M, Tyndall A, Matucci-Cerinic M, Naden RP,
Medsger TA Jr, Carreira PE, et al: 2013 classification criteria for
systemic sclerosis: An American College of rheumatology/European
league against rheumatism collaborative initiative. Arthritis
Rheum. 65:2737–2747. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Vitali C, Bombardieri S, Jonsson R,
Moutsopoulos HM, Alexander EL, Carsons SE, Daniels TE, Fox PC, Fox
RI, Kassan SS, et al: Classification criteria for Sjögren's
syndrome: A revised version of the European criteria proposed by
the American-European consensus group. Ann Rheum Dis. 61:554–558.
2002.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Goldin JG, Lynch DA, Strollo DC, Suh RD,
Schraufnagel DE, Clements PJ, Elashoff RM, Furst DE, Vasunilashorn
S, McNitt-Gray MF, et al: High-resolution CT scan findings in
patients with symptomatic scleroderma-related interstitial lung
disease. Chest. 134:358–367. 2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Tang J, Lei L, Pan J, Zhao C and Wen J:
Higher levels of serum interleukin-35 are associated with the
severity of pulmonary fibrosis and Th2 responses in patients with
systemic sclerosis. Rheumatol Int. 38:1511–1519. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Seror R, Theander E, Brun JG, Ramos-Casals
M, Valim V, Dörner T, Bootsma H, Tzioufas A, Solans-Laqué R, Mandl
T, et al: Validation of EULAR primary Sjögren's syndrome disease
activity (ESSDAI) and patient indexes (ESSPRI). Ann Rheum Dis.
74:859–866. 2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Chung L, Liu J, Parsons L, Hassoun PM,
McGoon M, Badesch DB, Miller DP, Nicolls MR and Zamanian RT:
Characterization of connective tissue disease-associated pulmonary
arterial hypertension from REVEAL: Identifying systemic sclerosis
as a unique phenotype. Chest. 138:1383–1394. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zanatta E, Polito P, Famoso G, Larosa M,
De Zorzi E, Scarpieri E, Cozzi F and Doria A: Pulmonary arterial
hypertension in connective tissue disorders: Pathophysiology and
treatment. Exp Biol Med (Maywood). 244:120–131. 2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Launay D, Sitbon O, Hachulla E, Mouthon L,
Gressin V, Rottat L, Clerson P, Cordier JF, Simonneau G and Humbert
M: Survival in systemic sclerosis-associated pulmonary arterial
hypertension in the modern management era. Ann Rheum Dis.
72:1940–1946. 2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Callejas-Rubio JL, López-Pérez L,
Moreno-Escobar E and Ortego-Centeno N: Raynaud's phenomenon and
pulmonary arterial hypertension. Lupus. 17(355)2008.PubMed/NCBI View Article : Google Scholar
|