Introduction
Diabetes mellitus is a common worldwide metabolic
disease, and its incidence continues to increase every year. Type 2
diabetes mellitus (T2DM) accounts for >90% of the total number
of patients with diabetes; it is a glucose and fat metabolic
disorder syndrome caused by insulin resistance and insufficient
insulin secretion (1). T2DM has
become the most prevalent chronic noncommunicable disease in the
world, the main cause of which is obesity. According to the
International Diabetes Federation, patients with T2DM in Asia
account for 60% of cases worldwide (2). A previous study (3) also indicated that the overall
prevalence of diabetes in adults is 9.1%, implying that 415 million
adults suffer from diabetes globally. The incidence of obesity in
China has rapidly increased in recent years, exceeding 42%
(4).
A number of studies have shown a significant
increase in the free fatty acid (FFA) level in the blood of obese
patients (5,6). FFA includes palmitic acid (PA) and
other fatty acids (6). The intake
of a large amount of saturated fatty acids, especially PA, can
cause disorders of glucose and lipid metabolism in the human body
(5). Therefore, excessively high
levels of FFAs in the body may be crucial in causing obesity and
T2DM (7). The recruitment,
infiltration and polarization of macrophages in adipose tissue
serve an essential role in the occurrence of obesity (8). The inflammatory response caused by
macrophages is believed to be the link between obesity and
diabetes. Inflammatory factors mediate the occurrence of insulin
resistance by influencing the insulin signaling pathway. For
example, tumor necrosis factor α (TNF-α), the first proinflammatory
cytokine found to be associated with insulin resistance, is vital
in insulin resistance (9). It can
interfere with the phosphorylation of insulin receptor substrate 1
(IRS-1) in adipocytes through JNK1 and damage the insulin signaling
pathway, thereby causing insulin resistance (10). Proinflammatory cytokines can also
downregulate the expression of IRS-1 and glucose transporter type
4, insulin-responsive (GLUT4) proteins in adipocytes and impair
insulin action (11). Inflammatory
factors can also reversely act on macrophages to expand the
inflammation response (12). This
indicates that the metabolism of macrophages is essential for
insulin resistance.
PA is widely found in nature; almost all fats and
oils contain varying amounts of PA, and it is the most abundant
saturated fatty acid in a high-fat diet (13). Moreover, it is important in fat
metabolism in macrophages. A previous study found that PA can
induce insulin resistance in liver cells through reactive oxygen
species produced by mitochondria (14). In cells, PA can activate multiple
Toll-like receptor (TLR)-dependent signaling pathways to increase
mRNA and protein expression levels of TLR and enhance the
receptor's signal transduction (13). Another study demonstrated high
expression of TLR4 in proinflammatory macrophages and in
differentiated adipocytes (15).
The expression of TLR4 in human adipocytes increases with
increasing levels of obesity and can be activated by
lipopolysaccharide to induce the production of nuclear factor-κB
(NF-κB) and cytokines (16).
Therefore, the present study aimed to explore whether PA-induced
insulin resistance was also related to the abnormal activation of
TLR4-dependent pathways and how this resistance could be
regulated.
Galectin-3 (Gal-3) is a lectin that is primarily
secreted by macrophages. Elevated levels of Gal-3 have been
reported in obese individuals and mice (17). The administration of Gal-3 to mice
can cause insulin resistance and glucose intolerance, and the
inhibition of Gal-3 can improve insulin sensitivity in obese mice.
Gal-3 treatment in vitro can directly enhance the chemotaxis
of macrophages and reduce the uptake of glucose by
insulin-stimulated muscle cells and 3T3-L1 adipocytes. Studies have
also shown that Gal-3 can directly interact with the insulin
receptor (IR) and inhibit downstream IR signaling (17,18).
These findings illustrate the vital role of Gal-3 in insulin
resistance in liver, fat and muscle cells, indicating that Gal-3
can link inflammation with decreased insulin sensitivity. The
inhibition of Gal-3 may serve as a new strategy to treat insulin
resistance. However, the specific mechanism underlying
macrophage-mediated insulin resistance is still unclear. The
findings of the present study may offer insight into the mechanism
underlying PA-mediated regulation of Gal-3 expression through TLR4,
thus providing the basis for the establishment of related
intervention methods in the future.
Materials and methods
Cell lines and reagents
Human acute monocytic leukemia cells THP-1 were
purchased from The Cell Bank of Type Culture Collection of The
Chinese Academy of Sciences (Shanghai, China). TRIzol®
RNA extraction reagent (Invitrogen), 1st Strand cDNA Synthesis kit
gDNA Purge (Novoprotein; cat. no. E042-01B), SYBR qPCR SuperMix
Plus (Novoprotein; cat. no. E096-01A) and reverse transcription
RT-PCR primers (Invitrogen) were from Thermo Fisher Scientific,
Inc. The primers for qPCR were commissioned to be synthesized by
Genewiz, Inc. Primary antibodies, including Krüppel-like factor 4
(KLF4; cat. no. R1308-1), insulin receptor (INSR; cat. no.
ET1705-22), IRS1 (cat. no. ER2001-35), Gal-3 (cat. no. ER1803-82),
NF-κB (cat. no. ER0815), phosphorylated (p)-NF-κB p65 (S529)
antibody (cat. no. ET1604-27), TLR4 (cat. no. ER1706-43) and GAPDH
(cat. no. ER1706-83), were purchased from Hangzhou Hua'an
Biotechnology Co., Ltd. The p-IRS1 (Ser1101) primary
antibody (cat. no. 2385) was from Cell Signaling Technology, Inc.
The following secondary antibodies were used: Horseradish
peroxidase-conjugated goat anti-rabbit IgG (cat. no. HA1001;
Hangzhou HuaAn Biotechnology Co., Ltd.) and FITC-conjugated goat
anti-rabbit antibody, (cat. no. HA1004; Hangzhou HuaAn
Biotechnology Co., Ltd.). DAPI was purchased from Beyotime
Institute of Biotechnology, and the (3R,4R,5S,6R)6-(hydroxymethyl)-
3-[(7-nitrobenzo(c) (1,2,5)
oxadiazol-4-yl) amino] tetrahydro-2H pyran-2,4,5-triol (2-NBDG)
glucose uptake/transport fluorescent probe (cat. no. MX4511-1MG)
was from Shanghai Mao Kang Biological Technology, Co., Ltd. The
TLR4 protein inhibitor (TAK 242; cat. no. 243984-11-4) was also
obtained from Merck KGaA. All protocols and the use of human
peripheral blood were approved by the Affiliated Hospital of
Medical School of Ningbo University (Ningbo, China; approval no.
KY20190102). A total of 10 men (40-50 years old; including 5
healthy individuals and 5 patients with type 2 diabetes) and 10
women (40-50 years old; including 5 healthy individuals and 5
patients with type 2 diabetes) were admitted to the Affiliated
Hospital of Ningbo University from January 2018 to December 2018 to
detect the expression level of Gal-3 protein in the peripheral
blood of patients. All subjects provided oral informed consent.
Induction of THP-1 cells by phorbol
12-myristate 13-acetate (PMA) to differentiate into
macrophages
THP-1 cells were grown in a 6-well plate
(5x106 cells/well) and grown in an incubator at 37˚C and
5% CO2 in RPMI-1640 (cat. no. ZQ-230) medium containing
10% FBS (cat. no. ZQ500; Zhejiang Ruyao Biotechnology Co., Ltd.).
Cells between passage four to eight were selected for the
experiments based on the cell growth state and cell morphology. A
total of 1x106 cells/well were inoculated in a 6-well
plate and grown in an incubator at 37˚C and 5% CO2 in
RPMI-1640 medium containing 10% FBS. The differentiated macrophages
were identified after treatment with 200 ng/ml
12-O-Tetradecanoyl-phorbol 13-Acetate (PMA; cat. no. P6741; Beijing
Solarbio Science & Technology Co., Ltd.) for 3 days.
Identification of macrophages
A total of 2x106 cells/well were
inoculated in a 24-well plate and grown in an incubator at 37˚C and
5% CO2 in RPMI-1640 medium containing 10% FBS for 2 h.
Cultured macrophages were washed with PBS and fixed with methanol
at 4˚C for 20 min. After fixation, all subsequent steps were
carried out at room temperature. Samples were washed 1X PBS and
stained with hematoxylin for 3 min and eosin for 5 sec. After
dehydration, xylene was added and neutral gum was used for sealing.
Cultured THP-1 cells that did not receive PMA induction were
centrifuged at 800 x g room at temperature for 5 min, resuspend 100
µl in 1X PBS and then dropped onto a slide. After air drying, the
cells were fixed with methanol at 4˚C for 20 min, followed by HE
staining using the same method as described above. Prior to HE
staining, both groups of cells were photographed under a phase
contrast microscope to observe their morphology (magnification,
x100 and x400). In addition, THP-1 cells were cultured in a 24-well
plate with the inoculation density of 2x106 cells per
well, and used for microsphere phagocytosis test. A drop of
polystyrene microspheres (cat. no. P107780; Shanghai Aladdin
Biochemical Technology Co., Ltd.) was added to 2.5 ml PBS, mixed
thoroughly and then 5 µl diluted beads were added to the
PMA-treated THP-1 cells. The cells were incubated at 37˚C for 50
min and then washed twice with 1x PBS. The phagocytosed
microspheres were counted under an inverted microscope
(magnification, x100). Phagocytosis rate (%)=(the number of
phagocytic pellets/the number of cells in one field).
Induction of insulin resistance and
inflammatory factors in macrophages
Macrophages in good growth condition and in the
logarithmic growth phase were treated with 0, 200, 400 or 600 µM PA
(cat. no. P0500; Merck KGaA) for 24 h. Subsequently, the cells were
tested for glucose uptake ability according to the glucose uptake
assay protocol described below. The cell supernatant was collected
via centrifugation at 800 x g for 20 min at 4˚C and the
inflammatory factors, including interleukin (IL)-1β (cat. no.
SEKH-0002), IL-6 (cat. no. SEKH-0013) and TNF-α (cat. no.
SEKS-0003), were detected by ELISA (all from Beijing Solarbio
Science & Technology Co., Ltd.) according to the manufacturer's
protocol.
Glucose uptake assay
For the glucose uptake assay, 1x105
macrophages were used following treatment with PA (0, 200, 400 or
600 µM), inoculated (1x105 cells/well) in a 6-well
plate, cultured at 37˚C for 48 h, washed three times with PBS and
incubated with 100 µM 2-NBDG at 37˚C for 20 min. The cells were
washed three times with precooled PBS, digested with 0.25% trypsin,
collected via centrifugation at 800 x g for 5 min at 4˚C and
examined using an Attune NxT flow cytometer (Thermo Fisher
Scientific, Inc.) with FlowJo 10 software (FlowJo LLC).
Cell viability test
Macrophages were collected in the logarithmic growth
phase and cell viability was detected using the MTT method. A total
of 100 µl macrophages were added to each well, and the density of
the cells was adjusted to 5x103 macrophages/well (the
edge holes were filled with sterile PBS). The cells were incubated
at 37˚C and 5% CO2 until the macrophage monolayer
covered the bottom of the well (96-well flat-bottom plate). After
the adherent growth of macrophages, cells were treated with 100 µl
drug-containing complete medium (PA, 0, 200, 400 and 600 µM) in
each well. After incubation at 37˚C for 48 h, 20 µl 5% MTT was
added to each well for 4 h. The culture medium in the well was
removed, 150 µl DMSO was added. The absorbance was measured at an
optical density of 490 nm using a microplate reader (MultiskanGO;
Thermo Fisher Scientific, Inc.).
Western blot analysis
The macrophages (1x106) were removed from
the CO2 incubator after 24 h treatment with PA and
washed twice with precooled PBS. The remaining PBS was aspirated,
the cells were lysed with RIPA lysis buffer (cat. no. R0010;
Beijing Solarbio Science & Technology Co., Ltd.) on ice and
then the macrophage lysate was pipetted to destroy the DNA. A total
of 3 µl lysate was used for BCA quantification. Further, 4X protein
loading buffer was added to the remaining samples, incubated at
95˚C for 10 min to denature and stored at -20˚C until further
analysis. Proteins (40 µg) were separated by 10% SDS-PAGE,
transferred to a PVDF membrane at a constant current of 200 mA for
2 h and blocked with 5% skimmed milk for 1 h at room temperature.
The membranes were incubated with the primary antibody (rabbit anti
p-IRS1/IRS1/INSR/KLF4/Gal-3; all, 1:1,000) overnight at 4˚C. The
internal reference antibody GAPDH was added and incubated for 1 h
at room temperature (cut according to marker size). The membrane
was washed three times with 1X TBST (0.15% Tween-20), then
incubated with the secondary antibody at room temperature for 2 h,
followed by three washes with TBST. Proteins were visualized using
ECL solution (cat. no. PE0010; Beijing Solarbio Science &
Technology Co., Ltd.). The ChemiDoc-It Imaging System (UVP, LLC)
was used for observation and ImageJ 8.0 (National Institutes of
Health) was used for quantification. When assessing the protein
levels of the downstream TLR4 pathway, four groups were
constructed: The control group; the PA group, which received
induction with 400 µm PA; the TAK-242 group, in which macrophages
were treated with 1 µM TAK-242; and the TAK-242 + PA group, where
macrophages were treated with 400 µM PA and 1 µM TAK-242. After
administration, macrophages were cultured for 24 h under 5%
CO2 and 37˚C. Western blot analysis was performed at the
end of treatment using the same procedures as described above.
RT-qPCR
After 24 h of treatment with PA, the treated
macrophages were collected (1x106) and total RNA was
extracted by the TRIzol® method. An
ultramicro-spectrophotometer (NanoDrop One/OneC; Thermo Fisher
Scientific, Inc.) was used to detect nucleic acid concentration,
and the integrity of the extracted mRNA was verified by agarose gel
electrophoresis. After incubating samples at 25˚C for 5 min, the
1st Strand cDNA Synthesis kit (cat. no. E047-01A; Novoprotein) was
applied and used according to the manufacturer's protocol,
incubating samples at 42˚C for 60 min for RT. The reaction was
terminated by heating at 70˚C for 5 min, and the mRNA was reverse
transcribed into cDNA. qPCR was conducted using SYBR qPCR SuperMix
Plus for analysis, along with the 7500 Fast Real-Time PCR System
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The reaction
mixture contained 20 ng cDNA, 1.5 mmol of each primer and 2.5 mM
MgCl2, and the thermocycling conditions were as follows:
Initial denaturation at 95˚C for 2 min; followed by 40 cycles of
denaturation at 95˚C for 10 sec, annealing at 60˚C for 10 sec and
extension at 72˚C for 30 sec. The relative expression of Gal-3 was
calculated using the 2-ΔΔCq method (19). Primer 3 (Premier Biosoft
International.) was used to design primer pairs; the sequences were
as follows: Gal-3, forward 5'-CGGAGCCAGCCAACGAG-3' and reverse,
5'-AACGCATCATGGAGCGAAAA-3'; GAPDH forward,
5'-GCACCGTCAAGGCTGAGAAC-3' and reverse,
5'-TGGTGAAGACGCCAGTGGA-3'.
Immunofluorescence
THP-1 cells (1x106) were inoculated on a
6-well plate with a cover glass slide placed on the plate. After
inducing macrophages with PMA, the cells were then slip-fed and
cultured at 37˚C and 5% CO2 for 24 h until a 70% fusion
degree was reached. The next day, 400 µM PA and 37 nM GB1107
(Shanghai ZrBiorise Biotechnology Co., Ltd.; cat. no. 1978336-61-6)
were added, and set up the experimental group with the above two
reagents, the standard medium was used for the negative control.
The groups were divided into four: The control group; the PA group,
where macrophages were treated with 400 µM PA; the GB1107 group,
where macrophages were treated with 37 nM GB1107; and the PA+GB1107
group, where macrophages were treated with 400 µM PA and 37 nM
GB1107. Cells were incubated for 8 h at 37˚C with 5%
CO2. The cells were then fixed with 4% paraformaldehyde
for 20 min at room temperature and incubated with rabbit antibody-3
antibodies (1:100) at room temperature for 2 h. Cells were then
incubated with the FITC-labeled secondary antibody (goat anti
rabbit; 1:500). The nuclei were stained and sealed with
anti-fluorescence quenching solution (including DAPI; cat. no.
P0131; Beyotime Institute of Biotechnology) was added. A Leica
DM500 fluorescence microscope was used to observe and obtain images
(Leica Microsystems GmbH). Image-Pro Plus 6.0 (Media Cybernetics,
Inc.) software was used for the quantification of results.
Bioinformatics analysis
The names of all proteins were put into the string
website to predict the relationship between proteins (https://string-db.org/).
Statistical analysis
All experiments were repeated three times
separately, and the data are expressed as the mean ± standard
deviation, differences between two groups were measured by unpaired
Student's t-test using SPSS 22.0 software (IBM Corp.). One-way
ANOVA was used to analyze differences between multiple groups,
followed by Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
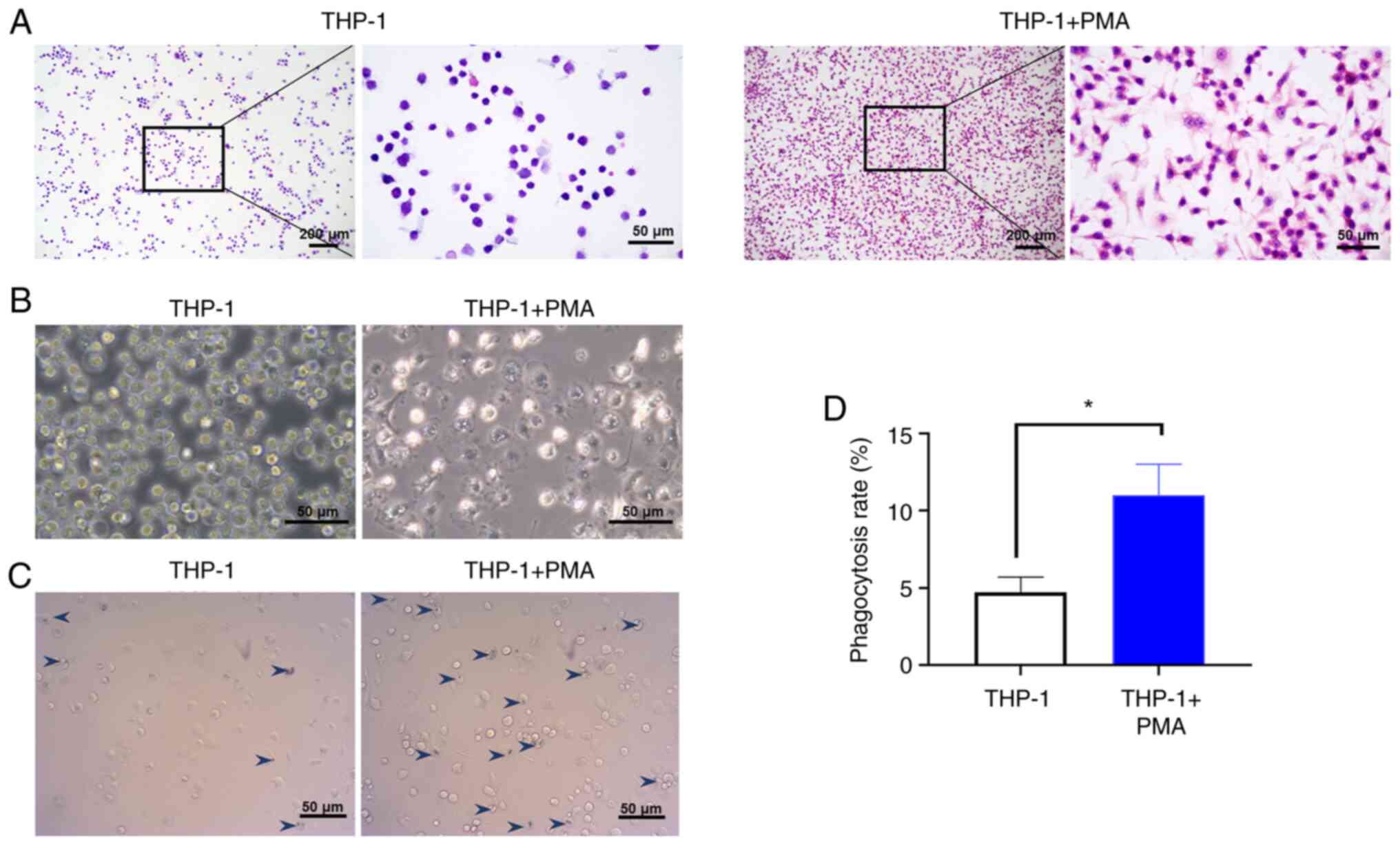
Induction of macrophage
differentiation
The adherent macrophages induced by PMA were
observed directly under a light microscope and phase contrast
microscope after HE staining (Fig.
1A and B). The adherent cells
exhibited clear boundaries and blunt round protrusions. The nuclei
were large and irregularly shaped at one end of the cell. THP-1
cells without PMA induction demonstrated a round karyotype and less
cytoplasm. In addition, the macrophages phagocytosed the
microspheres, indicating that the PMA-induced THP-1 cells exhibited
phagocytic ability (Fig. 1C), which
was statistically significant compared with the number of
phagocytosed microspheres in the uninduced THP-1 cells (P<0.05;
Fig. 1D). The results indiacted
that PMA could induce macrophage differentiation successfully.
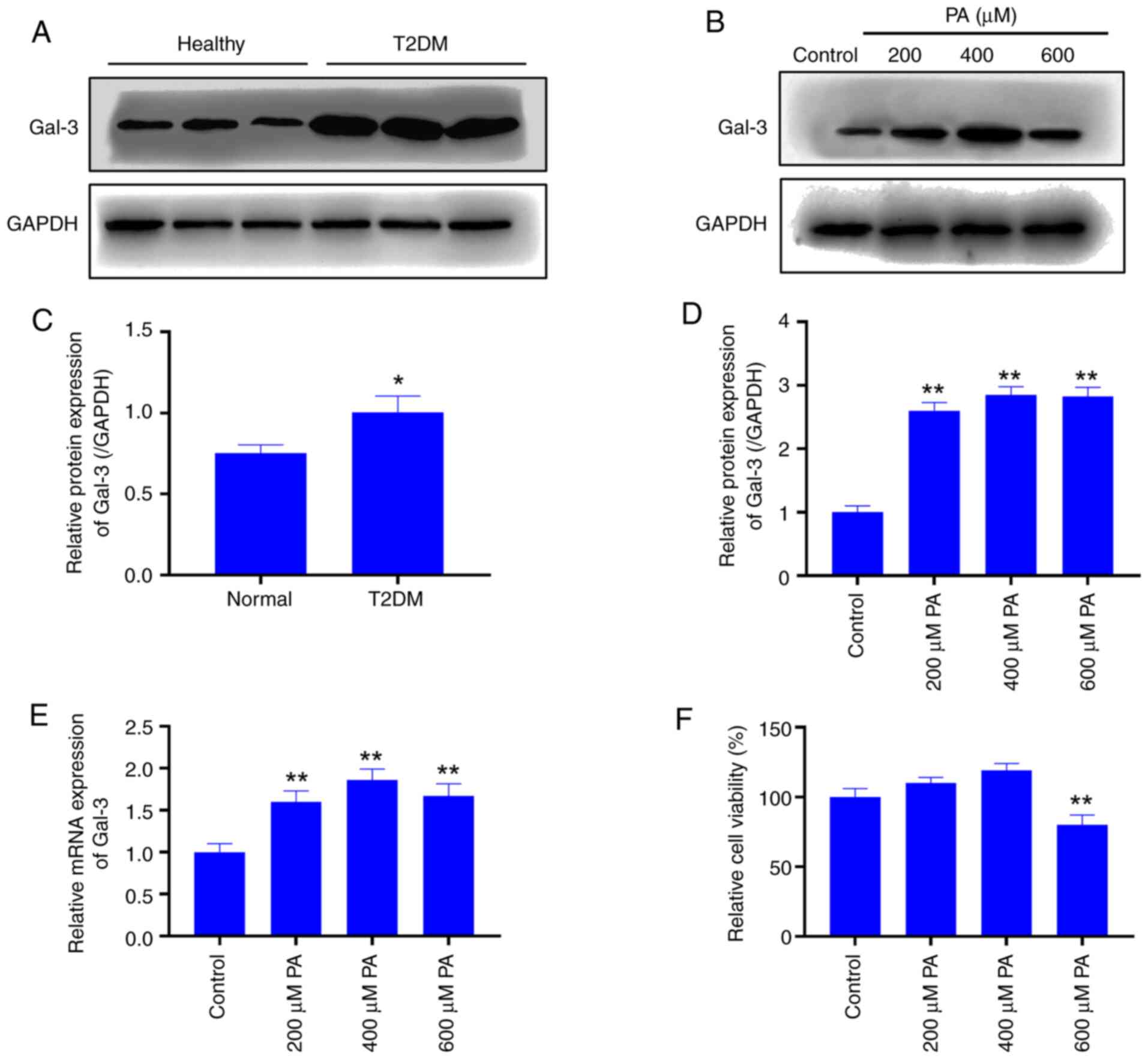
PA induces macrophages to produce
insulin resistance and promote the secretion of inflammatory
factors
Macrophages were treated with 0, 200, 400 or 600 µM
PA, and 2-NBDG was used to detect the sugar uptake ability of
macrophages. The results showed that the glucose uptake was
significantly lower in the PA-treated groups compared with uptake
in the control group (P<0.01; Fig.
2A). In addition, the protein expression levels of
p-INSR, INSR, and IRS1 were detected by western blotting.
The results demonstrated that the expression levels of INSR were
downregulated following PA treatment, and p-IRS1 was
significantly decreased (P<0.05) (Fig. 2B and C), indicating that PA induced insulin
resistance in macrophages. At the same time, the relative protein
expression level of KLF4 was significantly decreased in PA-treated
cells compared with the control group (P<0.01; Fig. 2D and E). After treatment with different PA
concentrations, the levels of inflammatory cytokines IL-6, IL-1β
and TNF-α in the cells increased significantly compared with
untreated control cells (P<0.01; Fig. 2F-H, respectively).
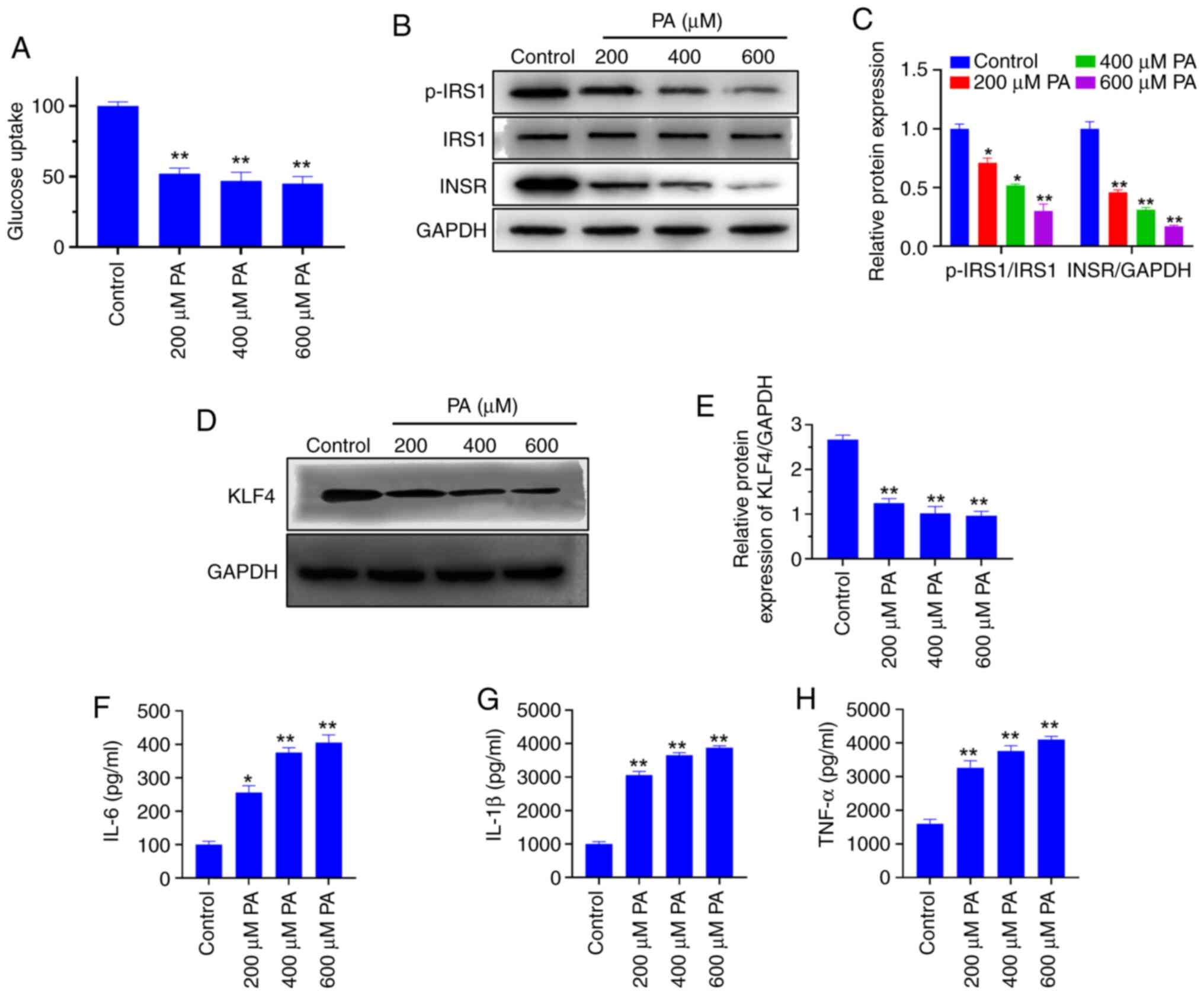 | Figure 2PA-induced insulin resistance
downregulates the expression of KLF4 in macrophages. (A)
2-deoxy-2-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-D-glucose
uptake detection. (B) Representative western blots and (C)
semi-quantification of the protein expression levels of insulin
resistance-related proteins. (D) Representative western blots and
(E) semi-quantification of KLF4 protein expression levels. ELISA
detection of the levels of (F) IL-6, (G) IL-1β and (H) TNF-α
following treatment with different concentrations of PA.
*P<0.05 and **P<0.01 vs. control. IL,
interleukin; INSR, insulin receptor; IRS1, insulin receptor
substrate 1; KLF4, Krüppel-like factor 4; p, phosphorylated; PA,
palmitic acid; TNF-α, tumor necrosis factor α. |
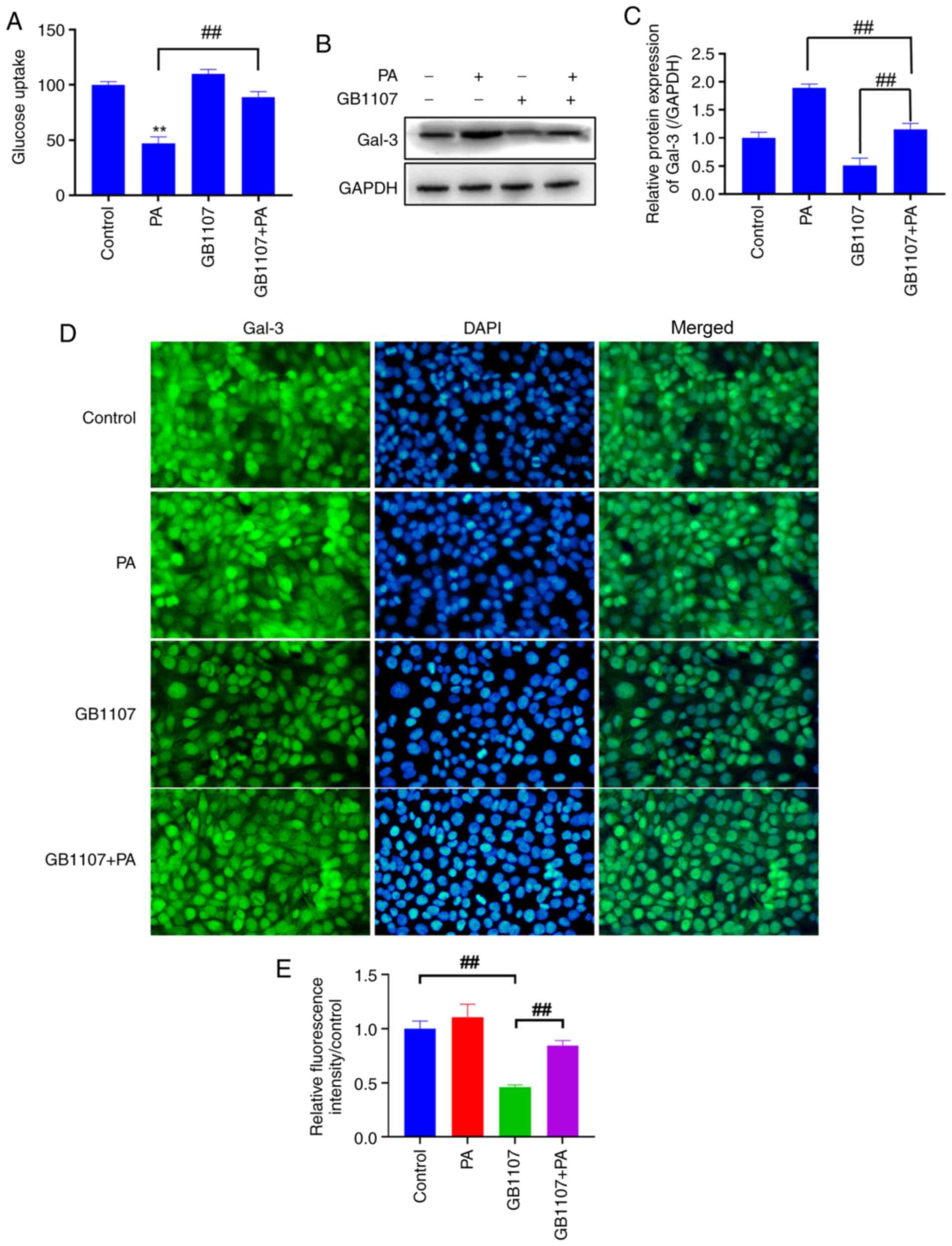
PA mediates macrophage insulin
resistance through Gal-3
Western blotting analysis of the peripheral blood
showed that Gal-3 was significantly upregulated in T2DM patients
compared with healthy individuals (P<0.05; Fig. 3A and C). Moreover, compared with the control
group, different concentrations of PA significantly increased the
expression of Gal-3 in treated macrophages (P<0.01; Fig. 3B and D). Therefore, it was speculated that Gal-3
may be involved in the regulation of insulin resistance by
macrophages. Fig. 3E suggested that
100, 400 or 600 µM PA significantly increased Gal-3 expression when
compared with the control group. The MTT viability assay was
performed on macrophages treated with different concentrations of
PA. When the PA treatment concentration was 400 µM, cell viability
was not significantly affected compared with that in the control
group (P>0.05; Fig. 3F).
However, at 600 µM, cell viability decreased significantly compared
with that in the control group (P<0.01). Taking into account the
effects of different concentrations of PA on the protein and gene
expression levels and cell viability of GAL-3 in macrophages, 400
µM PA was selected as the appropriate treatment concentration for
further experiments.
Following PA treatment, macrophages were treated
with the Gal-3 inhibitor GB1107 (37 nM), and the results showed
that GB1107 could significantly slow down the weakening effect of
PA on the glucose uptake capacity of macrophages (P<0.01;
Fig. 4A), thereby alleviating the
PA-induced insulin resistance of macrophages. In addition, after
adding GB1107, the relative protein expression level of Gal-3 in
PA-treated macrophages was significantly reduced, but the protein
of Gal-3 was not affected (20)
(P<0.01; Fig. 4B and C). Western blotting analysis and
immunofluorescence demonstrated that compared with the control,
Gal-3 protein was significantly down regulated in the GB1107 group
(Fig. 4D), whereas Gal-3 protein
expression in the PA + GB1107 group returned to the same level as
in the control group. These data indicated that PA may mediate the
occurrence of macrophage insulin resistance by affecting the
expression of Gal-3.
PA affects downstream KLF4, p-NF-κB
and Gal-3 protein expression and mediates insulin resistance
through TLR4 in macrophages
The expression levels of TLR4 protein and the
downstream proteins and p-NF-κB, as well as Gal-3 were detected by
western blot analysis (Fig. 5A).
The results showed that compared with the control group, the
expression of TLR4 in the PA group was significantly upregulated
(P<0.01), and the expression of the protein KLF4 decreased
(Fig. 5B). In addition, compared
with the control group, the p-NF-κB significantly increased
(P<0.01) and the expression of Gal-3 was significantly
upregulated (P<0.01) in the PA group The macrophages were
cultured for 24 h at 5% CO2 and 37˚C after
co-administration of PA (400 µM) and the TLR4 protein inhibitor
(TAK-242; 1 µM), the expression levels of KLF4 and Gal-3 protein
returned to control levels, and the p-NF-κB protein level was
significantly inhibited (P<0.01). Bioinformatics analysis showed
an interactive relationship between KLF4, Gal-3 and TLR4 proteins
(Fig. 5C), The results suggested
that PA may affect TLR4, p-NF-κB and Gal-3 by inhibiting the
expression of KLF4, thus mediating the development of insulin
resistance in macrophages.
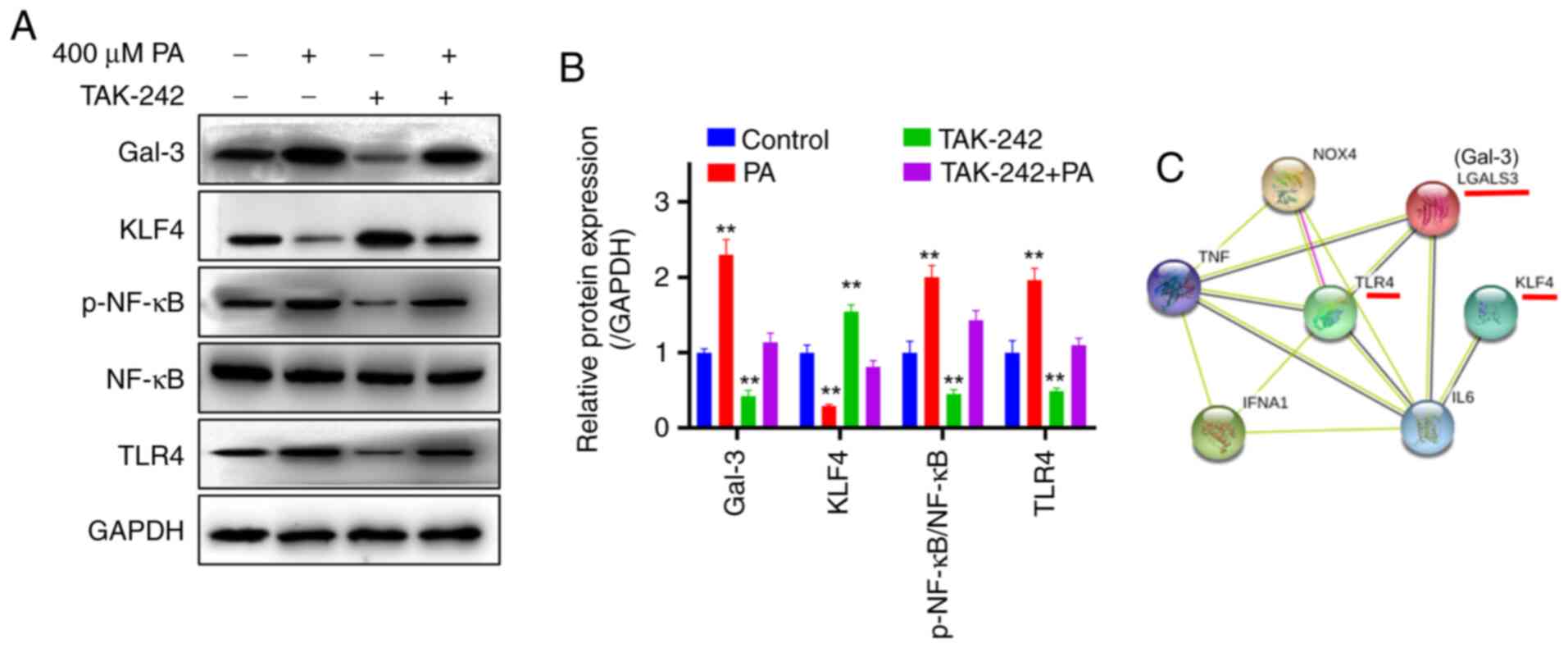 | Figure 5PA promotes the activation of the
TLR4/NF-κB pathway by downregulating KLF4 and promoting Gal-3
expression. Macrophages were treated with PA, TAK-242 and both. (A)
Representative western blots and (B) semi-quantification of protein
expression levels. (C) Main protein interaction map.
**P<0.01 vs. control. Gal-3, galectin-3; KLF4,
Krüppel-like factor 4; LGALS3, galectin-3; NF-κB, nuclear
factor-κB; p, phosphorylated; PA, palmitic acid; TLR4, toll-like
receptor 4; IFNA1, Interferon α 1; NOX4, NADPH οxidase 4. |
Discussion
During the development of T2DM, macrophages and
other immune cells accumulate in adipose tissue. Obesity affects
the biological characteristics of adipose tissue (21). However, the molecular mechanism and
the key mediators of macrophage-induced inflammation and insulin
resistance in T2DM are still poorly understood. A previous study
(21) indicated that the cytokines
secreted by THP-1 (myeloid leukemia cell line) monocytes can
communicate with a variety of cell types, such as human
microvascular endothelial cells and tumor-related macrophages.
Furthermore, the analysis of clinical data revealed excessive
macrophage production and infiltration in patients with T2DM
(22,23). Therefore, the present study used
THP-1-derived macrophages as a cell model to test the effects of
macrophage-like THP-1 cells on insulin resistance.
In the current study, THP-1 cells were induced to
differentiate into macrophages. The phagocytic microsphere
experiment showed that the phagocytic ability was significantly
improved in PMA-induced THP-1 cells compared with untreated cells.
The experimental results demonstrated that THP-cells changed from
suspension cells to adherent cells after being induced by PMA, and
had various shapes, clear boundaries and blunt round protrusions.
This morphology was similar to that shown by Sasidhar et al
(24). The phagocytosis experiment
of THP-1 cells induced by PMA showed that the phagocytosis ability
of THP-1 cells was significantly increased after induction, which
is also an important characteristic of macrophages (25). Therefore, the success of macrophage
induction was assessed from morphology and function. Furthermore,
the glucose uptake rate of PA-treated macrophages decreased
significantly compared with the control group. Insulin resistance
was detected at the protein level, and the results were consistent
with expectations. For example, INSR proteins and the ratio of
p-IRS1/IRS1 (Ser1101) also showed a decrease after PA induction. A
previous study also demonstrated that a decrease in p-IRS1
expression results in insulin resistance (26). Combined with the above glucose
uptake experiment and the insulin-related receptor protein
expression levels, it was indicated that the insulin resistance
model of macrophages induced in the present study was successfully
established.
Identifying the key factors that mediate the
production of insulin resistance in macrophages is essential for
developing effective therapeutic targets. Macrophage-derived
cytokines have been considered the key regulators of the
transformation of obesity-related inflammation into insulin
resistance in rodents and humans. For example, Medina et al
(27) reported a key role of
macrophage-derived TNF-α in disrupting AKT-dependent insulin
signaling in adipocytes by reducing AKT levels. Orliaguet et
al (28) demonstrated that
IL-1β released by macrophages affected insulin signaling and
proinflammatory responses in human primary adipocytes via the
insulin signaling pathway. Macrophages also impair the insulin
signal transduction pathway by inhibiting the transcription of IRS1
and GLUT4, and can prompt adipocytes to activate peroxisome
proliferator-activated receptor γ and to secrete IL-6(29). A previous study (30) demonstrated that insulin resistance
is closely related to chronic inflammation and lipid metabolism.
Therefore, saturated fatty acid PA, the levels of which are
abnormally increased in patients with T2DM, was used in the present
study to induce insulin resistance and inflammation in
macrophages.
TLRs have been regarded as sensors that mediate
inflammation by identifying pathogens and activating downstream
signaling pathways that lead to the upregulation of inflammatory
gene expression (31). Conversely,
the activation of KLF4 serves a protective role in the inflammatory
response (32). The activation of
the TLR4-mediated inflammatory pathway in the saturated fatty acid
response has been confirmed in different types of cells (33-35).
Previous studies reported that the activation of KLF4 can inhibit
the phosphorylation of NF-κB, and that NF-κB is downstream of
TLR4(36). The degree of
phosphorylation NF-κB has an essential relationship with
inflammation (37,38). It has been reported that KLF4 can
induce the release of inflammatory factors in cells and serves a
proinflammatory role. For example, MARIE demonstrated in the
research on esophageal squamous cell carcinoma that KLF4 is an
epithelial-specific mediator of inflammation, which can activate
one or several proinflammatory cytokines, including TNF-α, C-X-C
motif chemokine 5, G-CSF and IL-1α (39). Luo et al (40) revealed that over-expression of KLF4
strongly induces the production of IL-6 in human fibroblast-like
synoviocytes of rheumatoid arthritis. In ischemic hemispheres, the
high expression of KLF4 is always related to relatively few
cerebrovascular endothelial inflammatory reactions (41). KLF4 overexpression can therefore
reduce inflammation. KLF4 protein expression levels were examined
in the present study. The relative protein expression levels of
KLF4 decreased significantly in PA-treated cells compared with
control cells, indicating that PA downregulated the expression of
KLF4. In addition, the content of inflammatory factors also
revealed that insulin-resistant macrophages were associated with
increased levels of inflammatory factors, which suggested that the
decrease in KLF4 expression may promote an increase in the M1
phenotype of macrophages, intensifying the inflammatory reaction.
It needs further study, and no similar report has been found so
far. The inflammatory response of macrophages serves an important
role in its phenotype, and the presence of KLF4 serves an important
role in inhibiting the inflammatory response of macrophages
(42-44).
These aforementioned studies indicate that KLF4 protein may serve
different inflammatory roles in different cells.
Gal-3 is a lectin secreted by macrophages, which is
involved in inflammation (44). The
expression of Gal-3 increased significantly in patients with T2DM
compared with the healthy controls. In addition, the expression of
Gal-3 increased in macrophages treated with PA compared with the
control group. Li et al (17) showed that the circulating Gal-3
levels in obese mice and humans were elevated; Gal-3 is a
macrophage-derived factor that could cause systemic insulin
resistance in the body. In addition, Gal-3 treatment inhibited the
activity of insulin receptors, resulting in a decrease in insulin
action in fat cells, muscle cells and liver cells in vitro,
thus leading to a decrease in downstream signal transmission to the
insulin action cascade (17).
Therefore, in obese individuals, the increased levels of immune
cell infiltration and inflammatory factor expression in adipose
tissue are considered to be the cause of insulin resistance, in
which Gal-3 serves an important role.
Gal-3 can induce macrophage chemotaxis in the
adipose tissue of obese mice and promote the accumulation of
inflammatory macrophages to mediate the inflammatory response in
adipose tissue. Li et al (17) determined that in vitro use of
Gal-3 directly enhanced the chemotaxis of macrophages and reduced
glucose uptake by insulin-stimulated muscle cells. That study also
demonstrated that Gal-3 directly bound to the IR and inhibited
downstream IR signal transduction to inhibit the glucose output of
mouse liver cells (17). This is
consistent with the results of the current study. Compared with the
control group, the expression of Gal-3 was significantly increased
in PA-induced insulin-resistant macrophages. The results also
showed that the addition of the Gal-3 inhibitor GB1107 could
relieve the PA-induced inhibition of glucose uptake and utilization
by macrophages, and that Gal-3 was closely related to insulin
resistance. Further analysis of bioinformatics revealed that Gal-3
had an interactive relationship with TLR4 and KLF4. TLR4 may
promote the expression of Gal-3 by activating the p-NF-κB in its
downstream pathway, and KLF4 reduced the effect of PA on NF-κB. The
inhibitor of TLR4 (TAK-242) was added to verify the predicted
results. The results showed that TAK-242 could reduce the
expression of TLR4, thereby affecting the level of p-NF-κB in its
downstream pathway and decreasing the expression level of
Gal-3.
In summary, the current study demonstrated that PA
treatment reduced the inhibitory effect on the TLR4/NF-κB pathway
by inhibiting KLF4, thereby promoting the high expression of Gal-3
and ultimately leading to the development of insulin resistance.
The findings may provide a theoretical basis for developing
treatment strategies against T2DM.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by The Medical Health
Science and Technology Project of Zhejiang Provincial Health
Commission (grant no. 2019KY192).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JL, YSM and FC designed and performed the
experiments. DXX and TQZ performed the literature search, research
design and manuscript editing. JL performed manuscript editing. JL
and YSM confirm the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
All protocols and the use of human peripheral blood
were approved by The Affiliated Hospital of Medical School of
Ningbo University (Ningbo, China; approval no. KY20190102). All
subjects were provided Oral informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang Y, Yan A, Li S, Liu B, Li H and Yan
Y: Efficacy and safety of berberine in the treatment of type 2
diabetes with insulin resistance: Protocol for a systematic review.
Medicine (Baltimore). 98(e16947)2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Nanditha A, Ma RC, Ramachandran A,
Snehalatha C, Chan JC, Chia KS, Shaw JE and Zimmet PZ: Diabetes in
Asia and the Pacific: Implications for the global epidemic.
Diabetes Care. 39:472–485. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Hu C and Jia W: Diabetes in China:
Epidemiology and genetic risk factors and their clinical utility in
personalized medication. Diabetes. 67:3–11. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wang YF, Sun MX, Xue H, Zhao WH, Yang XG,
Zhu XY, Zhao L and Yang YX: Understanding the China blue paper on
obesity prevention and control and policy implications and
recommendations for obesity prevention and control in China.
Zhonghua Yu Fang Yi Xue Za Zhi. 53:875–884. 2019.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
5
|
Qin X, Zhao Y, Gong J, Huang W, Su H, Yuan
F, Fang K, Wang D, Li J, Zou X, et al: Berberine protects
glomerular podocytes via inhibiting drp1-mediated mitochondrial
fission and dysfunction. Theranostics. 9:1698–1713. 2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lee YS, Kim JW, Osborne O, Oh DY, Sasik R,
Schenk S, Chen A, Chung H, Murphy A, Watkins SM, et al: Increased
adipocyte O2 consumption triggers HIF-1α, causing inflammation and
insulin resistance in obesity. Cell. 157:1339–1352. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Siwicki M, Engblom C and Pittet MJ: Gal3
links inflammation and insulin resistance. Cell Metab. 24:655–656.
2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Cimini FA, Barchetta I, Ciccarelli G,
Leonetti F, Silecchia G, Chiappetta C, Di Cristofano C, Capoccia D,
Bertoccini L, Ceccarelli V, et al: Adipose tissue remodelling in
obese subjects is a determinant of presence and severity of fatty
liver disease. Diabetes Metab Res Rev. 37(e3358)2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hotamisligil GS, Shargill NS and
Spiegelman BM: Adipose expression of tumor necrosis factor-alpha:
Direct role in obesity-linked insulin resistance. Science.
259:87–91. 1993.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Nieto-Vazquez I, Fernandez-Veledo S,
Krämer DK, Vila-Bedmar R, Garcia-Guerra L and Lorenzo M: Insulin
resistance associated to obesity: The link TNF-alpha. Arch Physiol
Biochem. 114:183–194. 2008.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Tesz GJ, Guilherme A, Guntur KV, Hubbard
AC, Tang X, Chawla A and Czech MP: Tumor necrosis factor alpha
(TNFalpha) stimulates Map4k4 expression through TNFalpha receptor 1
signaling to c-Jun and activating transcription factor 2. J Biol
Chem. 282:19302–19312. 2007.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Brady NJ, Chuntova P and Schwertfeger KL:
Macrophages: Regulators of the inflammatory microenvironment during
mammary gland development and breast cancer. Mediators Inflamm.
2016(4549676)2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Nicholas DA, Zhang K, Hung C, Glasgow S,
Aruni AW, Unternaehrer J, Payne KJ, Langridge W and De Leon M:
Palmitic acid is a toll-like receptor 4 ligand that induces human
dendritic cell secretion of IL-1β. PLoS One.
12(e0176793)2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Loh K, Deng H, Fukushima A, Cai X, Boivin
B, Galic S, Bruce C, Shields BJ, Skiba B, Ooms LM, et al: Reactive
oxygen species enhance insulin sensitivity. Cell Metab. 10:260–272.
2009.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Bes-Houtmann S, Roche R, Hoareau L,
Gonthier MP, Festy F, Caillens H, Gasque P, d'Hellencourt CL and
Cesari M: Presence of functional TLR2 and TLR4 on human adipocytes.
Histochem Cell Biol. 127:131–137. 2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Shi H, Kokoeva MV, Inouye K, Tzameli I,
Yin H and Flier JS: TLR4 links innate immunity and fatty
acid-induced insulin resistance. J Clin Invest. 116:3015–3025.
2006.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
Li P, Liu S, Lu M, Bandyopadhyay G, Oh D,
Imamura T, Johnson A, Sears D, Shen Z, Cui B, et al:
Hematopoietic-derived galectin-3 causes cellular and systemic
insulin resistance. Cell. 167:973–984. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Johnson A, Hou S and Li P: Inflammation
and insulin resistance: New targets encourage new thinking:
Galectin-3 and LTB4 are pro-inflammatory molecules that
can be targeted to restore insulin sensitivity. Bioessays.
39(10.1002)2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ren Z, Liang W, Sheng J, Xun C, Xu T, Cao
R and Sheng W: Gal-3 is a potential biomarker for spinal cord
injury and Gal-3 deficiency attenuates neuroinflammation through
ROS/TXNIP/NLRP3 signaling pathway. Biosci Rep.
39(BSR20192368)2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Huh JY, Park YJ, Ham M and Kim JB:
Crosstalk between adipocytes and immune cells in adipose tissue
inflammation and metabolic dysregulation in obesity. Mol Cells.
37:365–371. 2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhang M, Zhou Z, Wang J and Li S: MiR-130b
promotes obesity associated adipose tissue inflammation and insulin
resistance in diabetes mice through alleviating M2 macrophage
polarization via repression of PPAR-γ. Immunol Lett. 180:1–8.
2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Odegaard JI and Chawla A: Mechanisms of
macrophage activation in obesity-induced insulin resistance. Nat
Clin Pract Endocrinol Metab. 4:619–626. 2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sasidhar MV, Chevooru SK, Eickelberg O,
Hartung HP and Neuhaus O: Downregulation of monocytic
differentiation via modulation of CD147 by
3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors. PLoS
One. 12(e0189701)2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kapellos TS, Taylor L, Lee H, Cowley SA,
James WS, Iqbal AJ and Greaves DR: A novel real time imaging
platform to quantify macrophage phagocytosis. Biochem Pharmacol.
116:107–119. 2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Talbot NA, Wheeler-Jones CP and Cleasby
ME: Palmitoleic acid prevents palmitic acid-induced macrophage
activation and consequent p38 MAPK-mediated skeletal muscle insulin
resistance. Mol Cell Endocrinol. 393:129–142. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Medina EA, Morris IR and Berton MT:
Phosphatidylinositol 3-kinase activation attenuates the
TLR2-mediated macrophage proinflammatory cytokine response to
Francisella tularensis live vaccine strain. J Immunol.
185:7562–7572. 2010.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Orliaguet L, Dalmas E, Drareni K,
Venteclef N and Alzaid F: Mechanisms of macrophage polarization in
insulin signaling and sensitivity. Front Endocrinol (Lausanne).
11(62)2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Cipolletta D, Feuerer M, Li A, Kamei N,
Lee J, Shoelson SE, Benoist C and Mathis D: PPAR-γ is a major
driver of the accumulation and phenotype of adipose tissue treg
cells. Nature. 486:549–553. 2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Chen L, Chen R, Wang H and Liang F:
Mechanisms linking inflammation to insulin resistance. Int J
Endocrinol. 2015(508409)2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Zhu Y, Deng J, Nan ML, Zhang J, Okekunle
A, Li JY, Yu XQ and Wang PH: The interplay between pattern
recognition receptors and autophagy in inflammation. Adv Exp Med
Biol. 1209:79–108. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Jin L, Ye H, Pan M, Chen Y, Ye B, Zheng Y,
Huang W, Pan S, Shi Z and Zhang J: Kruppel-like factor 4 improves
obesity-related nephropathy through increasing mitochondrial
biogenesis and activities. J Cell Mol Med. 24:1200–1207.
2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Wang Z, Liu D, Wang F, Liu S, Zhao S, Ling
EA and Hao A: Saturated fatty acids activate microglia via
toll-like receptor 4/NF-κB signalling. Br J Nutr. 107:229–241.
2012.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Tian H, Liu C, Zou X, Wu W, Zhang C and
Yuan D: miRNA-194 regulates palmitic acid-induced toll-like
receptor 4 inflammatory responses in THP-1 cells. Nutrients.
7:3483–3496. 2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Xu D, Liang J, Cui M, Zhang L, Ren S,
Zheng W, Dong X and Zhang B: Saturated fatty acids activate the
inflammatory signalling pathway in schwann cells: Implication in
sciatic nerve injury. Scand J Immunol. 92(e12896)2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Kaushik DK, Mukhopadhyay R, Kumawat KL,
Gupta M and Basu A: Therapeutic targeting of krüppel-like factor 4
abrogates microglial activation. J Neuroinflammation.
9(57)2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Allen KL, Hamik A, Jain MK and McCrae KR:
Endothelial cell activation by antiphospholipid antibodies is
modulated by kruppel-like transcription factors. Blood.
117:6383–6391. 2011.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Huang H, Wei L, Qin T, Yang N, Li Z and Xu
Z: Circular RNA ciRS-7 triggers the migration and invasion of
esophageal squamous cell carcinoma via miR-7/KLF4 and NF-κB
signals. Cancer Biol Ther. 20:73–80. 2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Tetreault MP, Wang ML, Yang Y, Travis J,
Yu QC, Klein-Szanto AJ and Katz JP: Klf4 overexpression activates
epithelial cytokines and inflammation-mediated esophageal squamous
cell cancer in mice. Gastroenterology. 139:2124–2134.
2010.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Luo X, Chen J, Ruan J, Chen Y, Mo X, Xie J
and Lv G: Krüppel-like factor 4 is a regulator of proinflammatory
signaling in fibroblast-like synoviocytes through increased IL-6
expression. Mediators Inflamm. 2016(1062586)2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhang X, Wang L, Han Z, Dong J, Pang D, Fu
Y and Li L: KLF4 alleviates cerebral vascular injury by
ameliorating vascular endothelial inflammation and regulating tight
junction protein expression following ischemic stroke. J
Neuroinflammation. 17(107)2020.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Wen Y, Lu X, Ren J, Privratsky JR, Yang B,
Rudemiller NP, Zhang J, Griffiths R, Jain MK, Nedospasov SA, et al:
KLF4 in macrophages attenuates TNFα-mediated kidney injury and
fibrosis. J Am Soc Nephrol. 30:1925–1938. 2019.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Liao X, Sharma N, Kapadia F, Zhou G, Lu Y,
Hong H, Paruchuri K, Mahabeleshwar GH, Dalmas E, Venteclef N, et
al: Krüppel-like factor 4 regulates macrophage polarization. J Clin
Invest. 121:2736–2749. 2011.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Henderson NC and Sethi T: The regulation
of inflammation by galectin-3. Immunol Rev. 230:160–171.
2009.PubMed/NCBI View Article : Google Scholar
|