Introduction
McArdle disease is a glycogen storage disease, a
rare genetic disorder caused by deficient myophosphorylase
activity. Myophosphorylase is a key enzyme in energy metabolism
because it catalyzes the rate-limiting step in glycogenolysis in
muscle (1). Glycogen phosphorylase
has three isoforms: PYGB (brain), PYGL (liver) and PYGM (muscle).
It has been reported that PYGM is present in skeletal muscle as
well as in other tissues and organs. The role of PYGM in tissues
other than muscle is unknown, but it is presumed to play a role in
not only McArdle disease but also in other diseases (2). McArdle disease is caused by genetic
defects of the myophosphorylase enzyme gene, PYGM, and the
disease has an autosomal recessive inheritance (3). More than 150 human PYGM
mutations have been identified, and the most common genetic defect
in Caucasians with McArdle disease is the nonsense mutation in
p.R50X of exon 1(4). Although
McArdle disease occurs in about 1 in 100,000 people in the United
States and Europe, it is very rare in Korea, showing racial
differences (5-7).
The p.R50X mutation accounts for more than 50% of PYGM mutations in
patients with McArdle disease in Europe, and only one case has been
reported in Japan (5). Therefore,
it has been suggested that there are ethnic differences in PYGM
variation between European and Asian individuals. Furthermore, 54%
of PYGM mutations were homozygous and the rest were heterozygous
allele (8,9). Patients with McArdle disease
experience progressive weakness and muscle wasting, and some
patients may not be diagnosed with this disease until they are over
50 years of age because they have no apparent symptoms (10). In patients with McArdle disease,
starting from the age of 50, symptoms of fixed muscle weakness
around the shoulder appear and may be accompanied by
cardiomyopathy, respiratory failure, and nervous system disorders
(1,11).
Recently, whole-exome sequencing (WES) has proved to
be a very effective method for the detection of familial disease
genes and a reliable methodology for detecting and quantifying
genetic defects clinically. Next-generation sequencing has made a
number of technological advances with minimal validation errors and
reliable levels of specificity and sensitivity (12). WES is a diagnostic approach for
detecting molecular defects in patients with suspected genetic
diseases (13). In the present
study, we attempted to analyze molecular defects through WES in a
13-year-old female patient who had not been diagnosed through a
conventional genetic approach. This study identified compound
heterozygous mutations of PYGM, which were found to be new
disease-causing missense mutations, in a Korean family by WES.
Subjects and methods
Subjects
A 13-year-old female patient visited the Eulji
University Hospital Growth Clinic because of her short stature (3rd
percentile). At first visit, she presented poor weight gain (1st
percentile) together and muscle weakness and dyspnea developed
immediately after short term exercise. At past history, she had
administered at hospital for 1 month a few years before because of
general ache and muscle weakness developed by excessive physical
activity. At first, we performed routine laboratory tests including
growth hormone stimulation to investigate the etiology of short
stature and poor weight gain but there were no remarkable findings.
We planned a genetic test for the patient, and the PYGM
mutation was discovered through WES, which brought attention to the
clinical features associated with myophosphorylase deficiency.
During the period of regular follow up, the patient showed exercise
intolerance and the second wind phenomenon. She was in the fifth
grade of elementary school, and her intelligence was normal. The
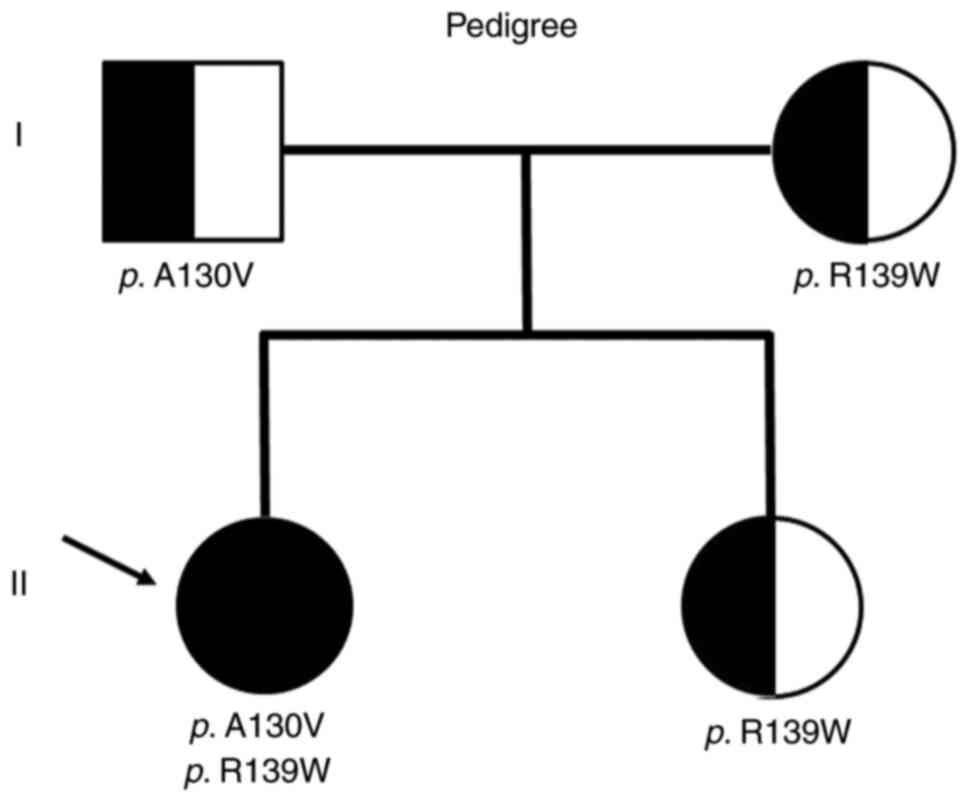
patient's parents and the younger sister, who was one year younger
than the patient, had no medically specific findings (Fig. 1). The present study was approved by
the Institutional Review Board of Eulji University (approval no.
EU17-42; approval date, December 14, 2017), and written informed
consent was obtained from all individual participants included in
this study; for participants <18 years old, the patients'
parents provided written informed consent. The patient's family has
provided written informed consent for publication.
Genomic DNA extraction
Genomic DNA was extracted from the blood of the
patient and all of her family members, and the gDNA was
quantitatively analyzed and confirmed by agarose gel
electrophoresis.
WES analysis
The entire exome sequencing process was performed in
three stages: Sample preprocessing and sequencing, primary data
processing, and secondary data processing. First, genomic DNA was
fragmented and reconnected to produce a library for sequencing.
Exome capture and enrichment were performed using a human exome
capture kit (Illumina). WES was performed using high-throughput
next-generation sequencing (NGS) technology in the Illumina
GA/HiSeq 2000 sequencing system (Illumina).
Genomic DNA was extracted using AccuPrep®
Genomic DNA Extraction kit (Bioneer) to prepare DNA samples for
sequencing. To generate standard exome capture libraries,
SureSelectXT Reagent kits (Agilent) were used. The DNA quantity and
quality were measured by Quant-iT™ PicoGreen™ dsDNA assay kit,
(Cat. # P7589, Thermo Fisher Scientific, Inc.) and agarose gel
electrophoresis, respectively. One microgram of each cell line's
genomic DNA was diluted with EB Buffer and sheared to a target peak
size of 150-200 bp with the Covaris LE220 focused-ultrasonicator
(Covaris) according to the manufacturer's recommendations.
Fragmentation was followed by end-repair and the addition of an ‘A’
tail. Then Agilent adapters were ligated to the fragments. After
evaluating the efficiency of ligation, the adapter ligated product
was polymerase chain reaction (PCR) amplified. The final purified
product was quantified by TapeStation DNA screentape D1000
(Agilent). For exome capture, 250 ng of DNA library was mixed with
hybridization buffers, blocking mixes, RNase block and 5 µl of
SureSelect all exon capture library, according to the standard
Agilent SureSelect Target Enrichment protocol. The captured DNA was
washed and amplified. Then final purified product was quantified by
qPCR according to the qPCR Quantification Protocol Guide (KAPA
Library Quantification kits for Illumina Sequencing platforms) and
qualified by the TapeStation DNA screentape D1000 (Agilent). The
concentration of the final library was 0.228 to 0.304 pmol, using
PicoGreen dsDNA Quantitation Reagent (Promega). NovaSeq 6000 S4
Reagent Kit v1.5 (Cat. # 20028312, Illumina) was used for
sequencing. When analyzing the data obtained through WES, BWA
(http://biobwa.sourceforge.net/bwa.shtml), Picard
(http://broadinstitute.github.io/picard), SnpEff, and
GATK (https://www.broadinstitute.org/gatk) software were
used.
An average of thirty times was considered sufficient
for accurate mutation detection. The data obtained from the NGS
platform were passed to raw sequence reads and processed. These
reads were aligned to the reference genome using the alignment
tool. When noise was generated in variant calling, PCR copies were
removed. In the subsequent analysis, variant calling and filtering
were performed to eliminate false positives according to quality
control standards. Finally, before testing to identify mutations
that cause disease, mutations were identified to obtain information
on gene location and action effects.
PCR and Sanger sequencing
PCR was performed using AccuPower® PreMix
(Bioneer) for sequence analysis of mutations in the 20 exons of the
PYGM gene. Two sets of primers were prepared, and nested PCR
was performed to obtain a clean PCR product without nonspecific
reactions. The amplified PCR products were purified and subjected
to dideoxy-sequencing PCR and purification, followed by sequencing
using a 3730XL DNA analyzer (Thermo Fisher Scientific Inc.).
Forearm exercise test
The forearm exercise test was performed using the
method developed by Kazemi-Esfarjani et al (14). Before the test, 0.5 ml of venous
blood was sampled to determine the amounts of ammonia and lactic
acid. A blood pressure measurement cuff was wrapped around the
right upper arm, and the patient was held at an interval of 3 sec
while air was injected into the cuff over the systolic period. At
this time, hand-grasping exercises were performed just until muscle
pain appeared, and the fingers could be completely bent and
unfolded. Antecubital venous blood samples were taken at 1, 2, 3,
and 5 min after the cuff air was removed. After that, the patient
had difficulty in hand movement for about 2 min, remarkable muscle
buildup was observed in the forearm, and the fingers could not be
spread. The concentrations of ammonia and lactic acid were measured
in the patient's blood taken at each time point.
Results
Variant filtering and gene
classification
The patient's whole-exome DNA was analyzed by
library production, sequencing, and data sorting. The quality score
and nucleotide distribution for NGS were in the normal range, the
DNA fragment inserted into the library was 100-200 bp in size, and
the depth distribution was in the normal range. The raw data
resulting from WES are summarized in Fig. 2. A total of 106,728 variations were
detected in the patient by WES. As a result of limiting the
mutation sites to exon and splicing sites, variation was reduced to
28,315, and the number was further reduced to 14,093 by filtering
out synonymous SNVs. As a result of applying the recessive,
compound heterozygous, and de novo conditions to the
remaining 2,033 variations by filtering under the conditions of
dbSNP135_common 1000G (freq > 0.01), 78 de novo
mutations, 12 simple recessive mutations, and 9 compound
heterozygous mutations were isolated. Twelve genes with homozygous
simple recessive mutations were analyzed. A detailed analysis of
the function of the genes encoding the proteins and related
diseases revealed that the three genes, HYAL1, LRRC17, and
ZFHX3, were associated with the patient, while the remaining
nine genes were found to be unrelated (Table I). Next, nine genes showing compound
heterozygous mutations were analyzed. As a result, mutations in two
genes, PYGM and MED15, were associated with the
patient (Table II). Finally,
through additional in-depth analysis, the patient's muscle weakness
symptoms were predicted to be most associated with myophosphorylase
dysfunction due to the PYGM mutation.
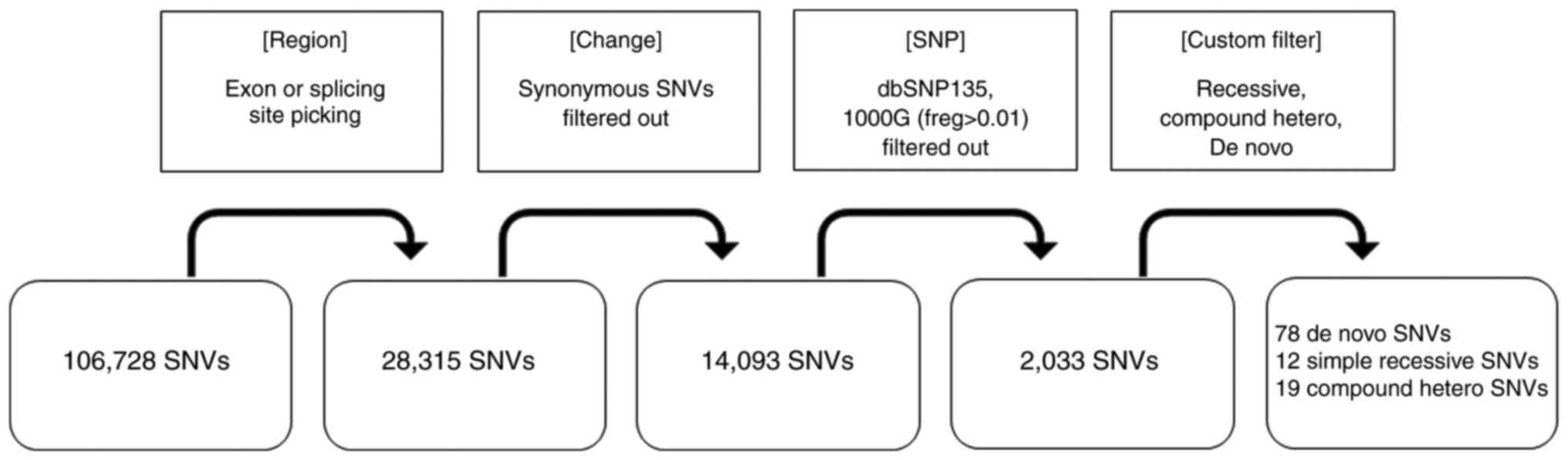 | Figure 2Workflow of data analysis. The raw
data resulting from Whole-Exome Sequencing was summarized. A total
of 106,728 variations were detected in the patient by WES. As a
result of limiting the mutation sites to exon and splicing sites,
variation was reduced to 28,315, and the number was further reduced
to 14,093 by filtering out synonymous SNVs. As a result of applying
the recessive, compound heterozygous, and de novo conditions
to the remaining 2,033 variations by filtering under the conditions
of dbSNP135_common 1000 G (freq. > 0.01), 78 de novo
mutations, 12 simple recessive mutations, and 9 compound
heterozygous mutations were isolated. SNV, single-nucleotide
variant; SNP, single-nucleotide polymorphism. |
 | Table IList of 12 homozygous recessive genes
of the patient. |
Table I
List of 12 homozygous recessive genes
of the patient.
| Gene | Polymorphic
change | Function | Related disease |
|---|
| ALLC | Non-frameshift
deletion | Purine
metabolism | Lymphatic
granuloma |
| FAM171B | Non-frameshift
insertion | Expression of
polyglutamine region | Polyglutamine
disease |
| HYAL1 | Nonsynonymous
SNV | Hyaluronic acid
(HA) | HA deficiency |
| TCP11 | Frameshift
deletion | Spermatogenesis | Abnormal sperm |
| TMEM184A | Non-frameshift
insertion | Expression in
testis | Abnormal sperm |
| AHR | Nonsynonymous
SNV | Regulation of
metabolic enzymes | Abnormal xenobiotics
metabolism |
| LRRC17 | Frameshift
deletion | Regulation of
osteoclast | Overdifferentiation
of osteoclast |
| PTCHD3 | Frameshift
insertion | Spermatozoa
function | Abnormal sperm |
| SPRN | Non-frameshift
deletion | Neuroprotection | Neuroprotection
associated disease |
| OR4L1 | Frameshift
deletion | Olfaction | Olfactory
weakness |
| ZFHX3 | Non-frameshift
deletion | Differentiation of
muscle and nerve | Abnormal muscle and
nerve differentiation |
| KRTAP17-1 | Non-frameshift
deletion | High
sulfur-associated protein | High Sulfur
associated |
| Table IIList of nine compound heterozygous
recessive genes of the patient. |
Table II
List of nine compound heterozygous
recessive genes of the patient.
| Gene | Polymorphic
change | Function | Related disease |
|---|
| CYP4A22 | Nonsynonymous
SNV | Cytochrome
P450 | Abnormal cyt
P450 |
| ZNF644 | Nonsynonymous
SNV | Eye
development | High myopia |
| SYNJ2 | Nonsynonymous
SNV | PIP activity | Charcot-Marie-Tooth
disease |
| NOL8 | Nonsynonymous
SNV | Cell growth | Abnormal
growth |
| AHNAK | Nonsynonymous
SNV | Cell migration | Miyoshi muscle
dystrophy |
| PYGM | Nonsynonymous
SNV |
Myophosphorylase | McArdle
disease |
| EXOC3L4 | Nonsynonymous
SNV | Glucose and GGT
activity connection | Seckel
syndrome |
| SPIRE2 | Non-frameshift
insertion | Intracellular
transport | Osteogenesis
imperfecta |
| MED15 | Nonsynonymous
SNV | RNA pol II
transcription | DiGeorge
syndrome |
Sanger sequencing of PYGM
The exon 3 region of the PYGM gene was
amplified by PCR. Primer sequences were as listed below: The first
PCR, forward 5'-AAAGAACTTCAACCGGCACCC-3' and reverse
5'-TAGAAGTGCACAGGTAGCGT-3'; nested PCR, forward
5'-TGGGCCTGGCTGAGTGTTGG-3' and reverse
5'-CCAGAGATGATAAACAAGTGGG-3'. PYGM gene sequences of the patient
and family were compared with the GenBank reference (AH006714.1) to
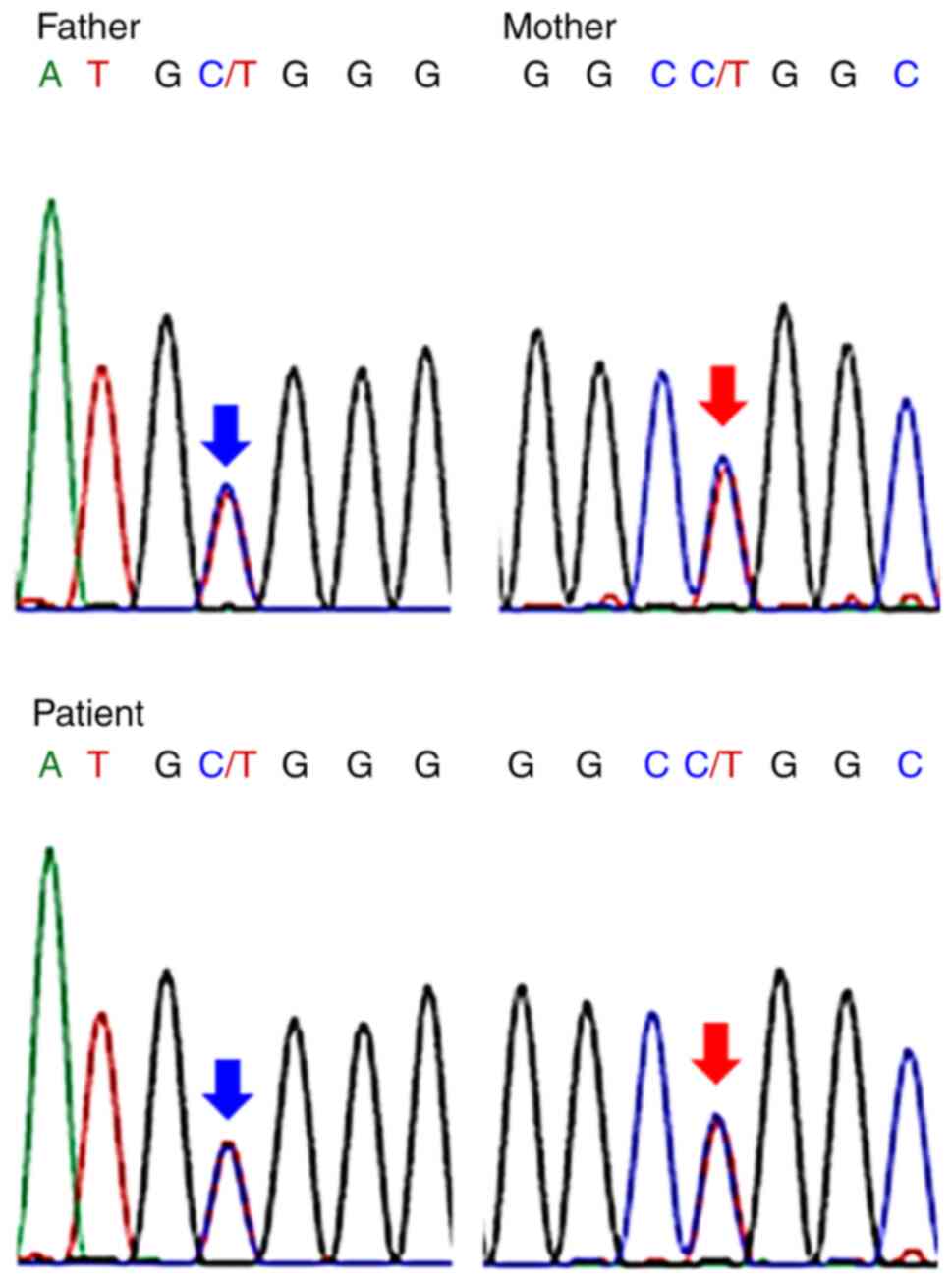
confirm the mutation. The patient's father had a heterozygous
mutation in the c.389C>T (p.A130V), and her mother had a
heterozygous mutation at c.415C>T (p.R139W). On the other hand,
her sister had the same mutation as her mother. The patient had
complex heterozygous mutations inherited from the father and
mother, respectively (Fig. 3). As
described above, the region of the PYGM gene mutation
analyzed by the Sanger sequence analysis of the patient's family
was exactly the same as the region in the whole-exome sequencing
analysis.
Non-ischemic forearm exercise
test
A non-ischemic lower arm exercise test was performed
to check for changes in the intramuscular myophosphorylase enzyme
due to the PYGM mutation. Anaerobic exercise increases
lactic acid levels in normal subjects, but lactic acid does not
increase in glycogen storage disease patients because glycogen in
muscle cells cannot be used as an energy source. Following a
handgrip exercise, in which a tourniquet for blood pressure
measurement was closed on the hand, grasped by the hand, and then
decompressed, venous blood was collected, and lactate and ammonia
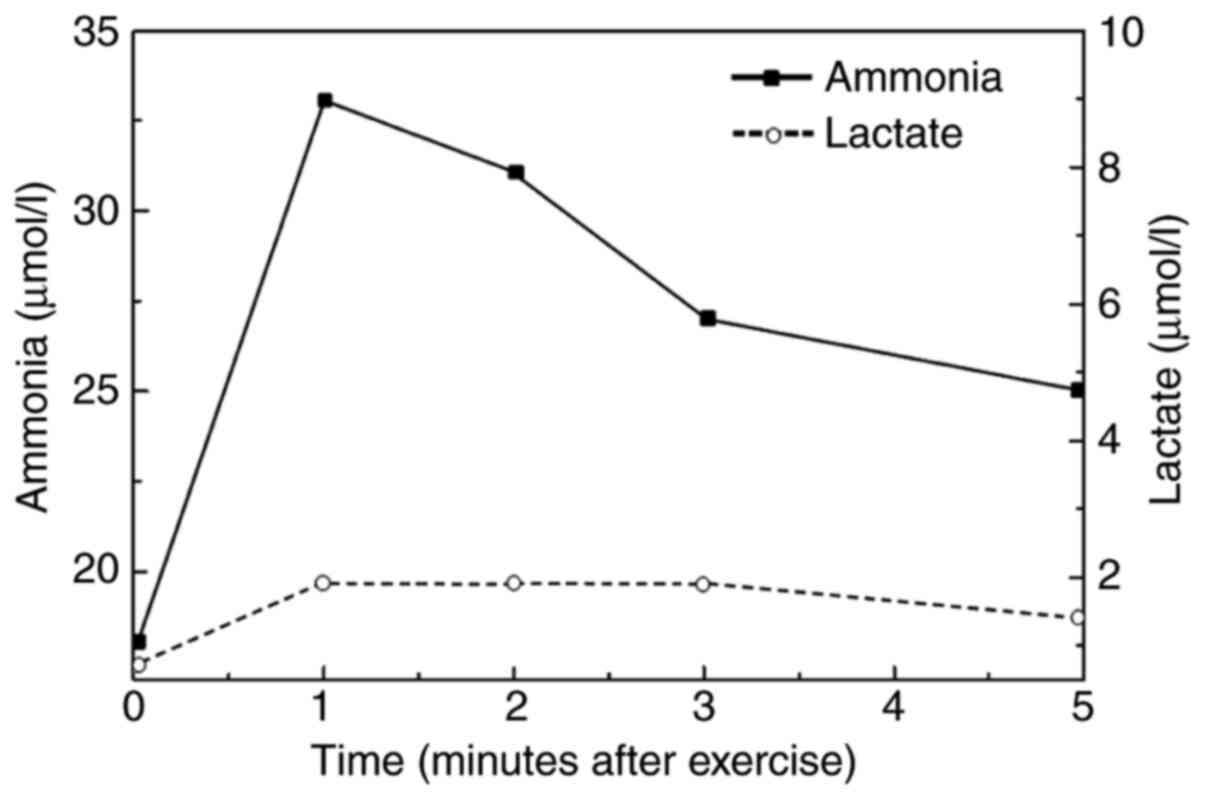
concentrations were measured. As a result, the concentration of
ammonia in the blood (0 min, 11 µmol/l; 1 min, 33 µmol/l; 2 min, 31
µmol/l) rapidly increased within 1 min, whereas the concentration
of lactate (0 min, 0.77 mmol/l; 1 min, 2.11 mmol/l; 2 min, 2.11
mmol/l; 3 min, 2.11 mmol/l; 5 min. 1.8 mmol/l) did not increase.
The ratio of ammonia to lactate was higher than normal, which is
consistent with the results of other McArdle patients (Fig. 4).
Discussion
A 13-year-old patient was diagnosed with McArdle
disease via sequencing of the whole exome. The patient initially
visited the growth clinic due to short stature and complained of
muscle weakness. WES analysis of the patient and her family members
revealed compound heterozygous mutations in the PYGM gene,
which consisted of a new missense mutation and a previously
reported mutation. In this patient, McArdle disease was caused by a
compound heterozygous mutation of the PYGM gene, including
one from her father and the other from her mother. The novel
PYGM mutation in the patient, c.389C>T, is a missense
mutation of p.A130V. Another mutation in the patient, c.415C>T,
substituted amino acid arginine for tryptophan at position 139
(R139W). In two variants, the patient inherited A130V from her
father and R139W from her mother, resulting in a myophosphorylase
defect. The p.R139W missense mutation of the PYGM gene was
reported in two Dutch male patients (15). However, the p.A130V missense
mutation of the patient and her father was a new mutation that has
not been reported at all.
To date, 167 variants of PYGM genes (OMIM
#608455) have been reported, and all variants can be retrieved from
the LOVD database (http://database.lovd.nl/shared/variants/PYGM).
Approximately 93.4% of the PYGM mutations are in exons, and
6.6% are located in the intron regions. Most PYGM mutations are
missense mutations, accounting for 50% of all mutations. Exon 1 of
the PYGM gene contains the p.R50X mutation, the most common
mutation among Caucasians (16,17).
Exon 17 is the second most variable, and the most common variant in
Japanese is c.2128_2130delTTC (p.F710del) (5). The mutations found in the patient in
this study, c.389 C> T and c.415 C> T, are located in exon
3.
The p.R50X mutation account for more than 50% of
PYGM mutations in patients with McArdle disease in Europeans, and
only one case has been reported in Japanese (5). Therefore, it was suggested that there
are ethnic differences between Europeans and Asians in PYGM
variation. In addition, 54% of PYGM mutations were
homozygous and the rest were heterozygous allele (8,9). In
one study, out of a total of fifty-four Spanish McArdle patients,
78% of the PYGM mutations were R50X and G205S (18). Further, Santalla et al
(7) analyzed forty-five patients
with McArdle disease in Caucasians. Among mutated alleles of
PYGM, p.R50X was 55%, whereas p.W798R and p.G205S were 10
and 9%, respectively.
McArdle disease, caused by myophosphorylase
deficiency, affects one in 100,000 people. Beginning in their
fifties, symptoms of fixed muscle weakness appear around the
shoulder and may be accompanied by cardiomyopathy, respiratory
failure, and nervous system disorders (1,11). In
patients with McArdle disease, biopsy can be performed because of
the accumulation of glycogen in skeletal muscle cells when viewed
under an electron microscope. Recently, muscle biopsies have not
been used to diagnose McArdle disease in children because of the
burden of biopsy after general anesthesia. The exercise load test
showed no false positive results, could sufficiently replace muscle
biopsy, and was confirmed through sequencing of PYGM gene
mutations (19). Further, de Luna
et al (20) suggested that
McArdle disease can be successfully diagnosed by assaying
myophosphorylase in leukocytes.
Glycogen phosphorylase has three isoforms: Brain
(PYGB), liver (PYGL), and muscle (PYGM). In a
recent study, Llavero et al (2) showed that PYGM is present in
skeletal muscle as well as in other tissues and organs, such as
astrocytes, kidneys, and retinal pigment epithelium (RPE). The role
of PYGM in tissues other than muscle is still unknown, but suggests
the possibility of McArdle disease and comorbidities (2). There were no other comorbidities
associated with PYGM mutations at the patient and family
members. However, we are closely following and observing the
patient and the family.
The forearm exercise test is widely used because it
is useful and sensitive to diagnose muscle glycogen accumulation
disorders such as McArdle disease. Diagnostic forearm exercise
protocol includes ischemic and non-ischemic tests, and has high
diagnostic value. In McArdle patients, myophosphorylase activity is
low, resulting in a flat or blunt lactate response. In the forearm
exercise test, ammonia level was used as a control, and the
variation of the lactate to ammonia ratio is measured (14). As shown the results of this study,
the initial increase in lactate (0-1 min) was presumed to be due to
the consumption of existing glucose present in the blood. However,
during the forearm exercise between 1 min and 5 min, there was no
increase in lactate because glycogen stored in the muscle was not
used. In this study, the results of the forearm exercise test were
consistent with the diagnostic results of McArdle disease
patients.
Anaerobic loading does not increase blood lactate
compared with a rapid increase in blood ammonia, which is often
observed in patients with McArdle disease (21). In the present study, the forearm
exercise test demonstrated no increase in lactate levels compared
to a normal increase in ammonia level. Further exercise tests were
not performed after five minutes to prevent muscle injury because
the patient complained of muscle pain.
Next-generation sequencing (NGS) has made a number
of technological advancements, with minimal validation errors as
well as specificity and sensitivity at reliable levels (12). WES is a NGS method and a diagnostic
approach for identifying molecular defects in patients with
suspected genetic diseases. WES is used to determine the cause of
genetic diseases and diagnose 25% of genetic-disease patients
(13). This study is expected to
contribute to the development of precision medicine in the future
because NGS can be used to diagnose genetic diseases and provide
accurate treatment opportunities in patients who have not been
diagnosed clinically.
Acknowledgements
Not applicable.
Funding
Funding: This research was supported by the Basic Science
Research Program through the National Research Foundation of Korea
funded by the Ministry of Education (grant number
2017R1D1A1B03035276).
Availability of data and materials
The datasets used and/or analyzed during the current
study are not publicly available due to difficulties in the
complete disclosure of human genome data under Korean law but are
available from the corresponding author on reasonable request.
Authors' contributions
JHK, JHP and HWB contributed to study conception and
design. JSP, JHP and JHK acquired data. SKL, SHL, JHP, JHK and HWB
analyzed and interpretated data. JHK, JHP and HWB drafted the
manuscript. JHP, JHK and HWB critically revised the manuscript.
JHK, JSP and JHP performed statistical analysis. HWB obtained
funding. JHK, JHP, JSP, SKL, SHL and HWB provided administrative,
technical or material support. JHK, JHP and HWB supervised the
study. JHK, JHP and HWB confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board of Eulji University (approval no. EU17-42; approval
date, December 14, 2017). Written informed consent was obtained
from all individual participants included in this study; for
participants <18 years old, the patients' parents provided
written informed consent.
Patient consent for publication
The patient's family has provided written informed
consent for publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Quinlivan R, Buckley J, James M, Twist A,
Ball S, Duno M, Vissing J, Bruno C, Cassandrini D, Roberts M, et
al: McArdle disease: A clinical review. J Neurol Neurosurg
Psychiatry. 81:1182–1188. 2010.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Llavero F, Arrazola Sastre A, Luque
Montoro M, Gálvez P, Lacerda HM, Parada LA and Zugaza JL: McArdle
disease: New insights into its underlying molecular mechanisms. Int
J Mol Sci. 20(5919)2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lebo RV, Gorin F, Fletterick RJ, Kao FT,
Cheung MC, Bruce BD and Kan YW: High-resolution chromosome sorting
and DNA spot-blot analysis assign McArdle's syndrome to chromosome
11. Science. 225:57–59. 1984.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Nogales-Gadea G, Santalla A, Brull A, de
Luna N, Lucia A and Pinós T: The pathogenomics of McArdle disease -
genes, enzymes, models, and therapeutic implications. J Inherit
Metab Dis. 38:221–230. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sugie H, Sugie Y, Ito M, Fukuda T, Nonaka
I and Igarashi Y: Genetic analysis of Japanese patients with
myophosphorylase deficiency (McArdle's disease): Single-codon
deletion in exon 17 is the predominant mutation. Clin Chim Acta.
236:81–86. 1995.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lucia A, Ruiz JR, Santalla A,
Nogales-Gadea G, Rubio JC, García-Consuegra I, Cabello A, Pérez M,
Teijeira S, Vieitez I, et al: Genotypic and phenotypic features of
McArdle disease: Insights from the Spanish national registry. J
Neurol Neurosurg Psychiatry. 83:322–328. 2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Santalla A, Nogales-Gadea G, Encinar AB,
Vieitez I, González-Quintana A, Serrano-Lorenzo P, Consuegra IG,
Asensio S, Ballester-Lopez A, Pintos-Morell G, et al: Genotypic and
phenotypic features of all Spanish patients with McArdle disease: A
2016 update. BMC Genomics. 18 (Suppl 8)(819)2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
García-Consuegra I, Rubio JC,
Nogales-Gadea G, Bautista J, Jiménez S, Cabello A, Lucía A, Andreu
AL, Arenas J and Martin MA: Novel mutations in patients with
McArdle disease by analysis of skeletal muscle mRNA. J Med Genet.
46:198–202. 2009.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Nogales-Gadea G, Brull A, Santalla A,
Andreu AL, Arenas J, Martín MA, Lucia A, de Luna N and Pinós T:
McArdle disease: Update of reported mutations and polymorphisms in
the PYGM Gene. Hum Mutat. 36:669–678. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Aquaron R, Bergé-Lefranc JL, Pellissier
JF, Montfort MF, Mayan M, Figarella-Branger D, Coquet M, Serratrice
G and Pouget J: Molecular characterization of myophosphorylase
deficiency (McArdle disease) in 34 patients from Southern France:
Identification of 10 new mutations. Absence of genotype-phenotype
correlation. Neuromuscul Disord. 17:235–241. 2007.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Burr ML, Roos JC and Ostör AJ: Metabolic
myopathies: A guide and update for clinicians. Curr Opin Rheumatol.
20:639–647. 2008.PubMed/NCBI View Article : Google Scholar
|
|
12
|
De Castro M, Johnston J and Biesecker L:
Determining the prevalence of McArdle disease from gene frequency
by analysis of next-generation sequencing data. Genet Med.
17:1002–1006. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yang Y, Muzny DM, Reid JG, Bainbridge MN,
Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al:
Clinical whole-exome sequencing for the diagnosis of mendelian
disorders. N Engl J Med. 369:1502–1511. 2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kazemi-Esfarjani P, Skomorowska E, Jensen
TD, Haller RG and Vissing J: A nonischemic forearm exercise test
for McArdle disease. Ann Neurol. 52:153–159. 2002.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Martín MA, Rubio JC, Wevers RA, Van
Engelen BG, Steenbergen GC, Van Diggelen OP, De Visser M, De
Die-Smulders C, Blázquez A, Andreu AL, et al: Molecular analysis of
myophosphorylase deficiency in Dutch patients with McArdle's
disease. Ann Hum Genet. 68:17–22. 2004.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Tsujino S, Shanske S and DiMauro S:
Molecular genetic heterogeneity of myophosphorylase deficiency
(McArdle's disease). N Engl J Med. 329:241–245. 1993.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Deschauer M, Morgenroth A, Joshi PR,
Gläser D, Chinnery PF, Aasly J, Schreiber H, Knape M, Zierz S and
Vorgerd M: Analysis of spectrum and frequencies of mutations in
McArdle disease. Identification of 13 novel mutations. J Neurol.
254:797–802. 2007.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Martín MA, Rubio JC, Buchbinder J,
Fernández-Hojas R, del Hoyo P, Teijeira S, Gámez J, Navarro C,
Fernández JM, Cabello A, et al: Molecular heterogeneity of
myophosphorylase deficiency (McArdle's disease): A
genotype-phenotype correlation study. Ann Neurol. 50:574–581.
2001.PubMed/NCBI
|
|
19
|
Hogrel JY, van den Bogaart F, Ledoux I,
Ollivier G, Petit F, Koujah N, Béhin A, Stojkovic T, Eymard B,
Voermans N, et al: Diagnostic power of the non-ischaemic forearm
exercise test in detecting glycogenosis type V. Eur J Neurol.
22:933–940. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
de Luna N, Brull A, Lucia A, Santalla A,
Garatachea N, Martí R, Andreu AL and Pinós T: PYGM expression
analysis in white blood cells: A complementary tool for diagnosing
McArdle disease? Neuromuscul Disord. 24:1079–1086. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Sinkeler SP, Daanen HA, Wevers RA, Oei TL,
Joosten EM and Binkhorst RA: The relation between blood lactate and
ammonia in ischemic handgrip exercise. Muscle Nerve. 8:523–527.
1985.PubMed/NCBI View Article : Google Scholar
|