Introduction
Respiratory syncytial virus (RSV) is the most common
cause of pneumonia and bronchiolitis in children worldwide
(1). Although RSV has a significant
clinical impact on global human health, no vaccines have yet been
approved and effective antiviral treatments are lacking.
Furthermore, while RSV infection induces humoral and cellular
immune responses to clear the infection, these responses do not
provide strong protection against subsequent RSV infection. The 11
proteins encoded by the RSV genome include non-structural protein 1
(NS1) and NS2, which are key in mediating the ability of the virus
to evade immune surveillance (2,3). It
has been reported that a series of antiviral mechanisms evoked by
RSV infection in the host may be hampered by the NS proteins
(4). RSV infection is severely
attenuated in vitro and in vivo when either NS1 or
NS2 is deleted, indicating that the NS proteins serve a key role in
viral replication (4,5). In addition, it has been reported that
the expression of inflammatory cytokines IL-6 and IL-8 in the lung
is associated with the severity of disease in infants and adults
infected with RSV (6,7). The silencing of NS1 provides
substantial protection against RSV infection-induced inflammation
and airway reactivity (2). RNA
interference targeting the RSV nucleocapsid (8) and NS1 has been investigated as a new
approach for RSV antiviral therapy (2,9,10). The
NS proteins also prevent cells from undergoing apoptosis and
prolong cell life, thus increasing the amount of viral replication
(3). NS1 has been shown to inhibit
the interaction between mitochondrial antiviral signaling protein
(MAVS) and retinoic acid-inducible gene-I by binding to MAVS, and
thereby decrease the production of type-I interferon (IFN)
(11). RSV viruses lacking NS1
or/and NS2 are more sensitive to IFN, which leads to increased
apoptosis and decreased viral replication efficiency in cell and
animal models (12,13). We hypothesize that in the absence of
RSV infection, the overexpression of NS1 alone may induce cellular
inflammation and aim to explore the underlying mechanisms.
Autophagy is a highly conserved process that
involves the encapsulation of cytoplasmic contents in a bilayer
membrane, fusion with a lysosome and degradation. Autophagy plays
important role in antigen presentation, bacterial and viral
infection, and cell death (14-16).
During autophagy, intracellular components, including organelles
such as mitochondria, are phagocytized in double-membraned
autophagosomes, which fuse with lysosomes to form autophagosomes.
The components within the autophagosomes are subsequently degraded
by lysosomal hydrolases. In the process of autophagosome membrane
formation, the cytoplasmic form of microtubule-associated protein
1A/1B-light chain 3 (LC3-I) conjugates with
phosphatidylethanolamine to form LC3-II, a lipid-modified form of
LC3-I, via two consecutive ubiquitylation-like reactions. This
intra-autophagosomal LC3-II is degraded by lysosomal proteases when
autophagosomes and lysosomes fuse. Therefore, the cellular level of
LC3-II can be regarded as a marker of autophagic activity.
Monitoring LC3-I and LC3-II using immunofluorescence and western
blotting is important when investigating the mechanism of autophagy
(17). Beclin1, the first-described
mammalian autophagy protein, serves an key role in the initiation
of autophagy. The abundance of Beclin1 can also act as a
determinant of autophagic activity (18).
Mammalian target of rapamycin (mTOR) is a
serine/threonine protein kinase, which serves as a sensor of
cellular nutritional status, stress and growth factor signals and
thereby serves a key role in the occurrence of autophagy (19). mTOR pathway-mediated autophagy is
closely associated with apoptosis and inflammation. Reed et
al (20) reported that the
production of innate cytokines was decreased upon the blockade of
autophagy, while RSV infection induced inflammasome activation and
IL-1β secretion in autophagy-deficient cells. At present, the
majority of studies have indicated that NS1 suppresses the IFN
response signal to reinforce RSV infection. However, there have
been few reports concerning the relationship between NS1 and
autophagy or the mechanism of autophagy in NS1-induced
inflammation. Therefore, the present study aimed to explore the
role of autophagy in NS1-transfected cells with the aim of gaining
an improved understanding of the role of NS1 and its relationship
with autophagy. The findings of this study also may assist in the
identification of strategies for viral vaccine development.
Materials and methods
Cell culture and treatment
The A549 type II human lung epithelial cell line was
obtained from the American Type Culture Collection and cultivated
with high-glucose DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin and 100 mg/ml streptomycin
at 37˚C in an incubator containing 5% CO2. The cells
were passaged when they had grown to a dense monolayer. NS1 plasmid
tagged with Flag (pNS1-Flag) was donated by the laboratory of
Professor Zhao at Wuhan University Zhongnan Hospital (21). When the A549 cells reached a density
of 60-70% they were transfected with pNS1 for 24 h at 37˚C. For the
pNS1 transfection, a liposomal formulation
(Lipofectamine® 2000; Invitrogen; Thermo Fisher
Scientific, Inc.) was used according to the manufacturer's
instructions. Briefly, 2.5 µg plasmid, 7.5 µl Lipofectamine 2000
and 5 µl p3000 (Invitrogen; Thermo Fisher Scientific, Inc.) complex
were used to transfect the cells in each well of 6-well plates.
Cells were collected for subsequent experiments after 24 h. The
autophagy inhibitor 3-methyladenine (3MA; MedChemExpress) dissolved
in normal culture medium at a final concentration of 5 mM was used
to treat the cells for 12 h prior to transfection with pNS1. The
mTOR-specific inhibitor Torin-1 (Selleck Chemicals) dissolved in
DMSO and diluted to a final concentration of 1 µM in the medium was
used to treat the cells for 3 h prior to transfection with pNS1 as
previously described (22).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
The MTT (Sigma-Aldrich; Merck KGaA) colorimetric
assay was used to determine cell viability. Briefly, cells were
seeded in 96-well plates (5x103 cells per well), and
then subjected to different treatments (transfected with pNS1 for
24 h at 37˚C, treated with 3MA for 12 h and then transfected with
pNS1 for 24 h at 37˚C, or treated with Torin-1 for 3 h at 37˚C).
After the treatments, the cells were washed with PBS and MTT
solution was added to the cells, which were then incubated for 2.5
h. DMSO was used to dissolve the purple formazan. The absorbance
was measured at 490 nm with a microplate reader (BioTek
Instruments, Inc.) to determine the cell viability.
Reverse transcription-quantitative PCR
(RT-qPCR)
The total RNA of A549 cells was separated using
RNAiso Plus (Takara Biotechnology Co., Ltd.) following the
manufacturer's protocol. A RevertAid First Strand cDNA Synthesis
kit (Thermo Fisher Scientific, Inc.) was used to reverse transcribe
the purified RNA into cDNA according to the manufacturer's
protocol. The levels of IL-6, IL-8 and β-actin were examined using
SYBR™ Green PCR Master Mix (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The thermocycling conditions for PCR was
as follows: 50˚C for 2 min, 95˚C for 2 min, followed by 40 cycles
of denaturation at 95˚C for 15 sec, followed by annealing and
extension at 60˚C for 30 sec. The primer sequences for IL-6, IL-8
and β-actin were purchased from Tianyi Huiyuan Biotech Co., Ltd.
The sequences of the primers were as follows: Human IL-6 forward,
5'-GAGGATACCACTCCCAACAGACC-3' and reverse,
5'-AAGTGCATCATCGTTGTTCATAC-3'; IL-8 forward,
5'-CTTGGCAGCCTTCCTGA-3' and reverse, 5'-TTCTTTAGCACTCCTTGGCAAAA-3';
β-actin forward, 5'-CCGACAGGATGCAGAAGGAG-3' and reverse,
5'-GTGGGGTGGCTTTTAGGATG-'3. The cycle threshold or quantitation
cycle (Cq) was detected, and the 2-ΔΔCq method (23) was used to determine the relative
expression of the target RNAs with normalization to β-actin
expression.
Western blot analysis
Cells were washed with ice-cold PBS and then
lysed for 30 min on ice with RIPA lysis buffer (Beyotime Institute
of Biotechnology) containing a protease inhibitor cocktail. A BCA
Assay kit (Beyotime Institute of Biotechnology) was then used for
protein quantification. Total proteins (30-40 µg/lane) were loaded
and isolated by 12.5 or 10.0% SDS-PAGE and blotted onto PVDF
membranes (EMD Millipore). The membranes were then blocked with 5%
non-fat milk for 1 h at room temperature (26˚C) and incubated
overnight at 4˚C with primary antibody. The primary antibodies used
were as follows: Rabbit anti-Flag (1:1,000; ##14793; Cell Signaling
Technology, Inc.), rabbit anti-IL-6 (1:1,000; #12153, Cell
Signaling Technology, Inc.), mouse anti-IL-8 (1:400; sc-376750;
Santa Cruz Biotechnology, Inc.), rabbit anti-LC3 (1:1,000; #4108;
Cell Signaling Technology, Inc.), mouse anti-Bcl-2 (1:500; sc-7382;
Santa Cruz Biotechnology, Inc.), mouse anti-Bax (1:500; sc-7480;
Santa Cruz Biotechnology, Inc.), mouse anti-Beclin1 (1:500,
sc-48381; Santa Cruz Biotechnology, Inc), anti-mTOR (1:1,000;
#2983, Cell Signaling Technology, Inc.), anti-phosphorylated
(p-)mTOR (1:1,000; #5536; Cell Signaling Technology, Inc.),
anti-p70 S6 kinase (S6KP70) (1:1,000; #2708; Cell Signaling
Technology, Inc.), anti-p-S6KP70 (1:1,000; #9234; Cell Signaling
Technology, Inc.) and rabbit anti-β-actin (1:10,000; AC026;
ABclonal Biotech Co., Ltd.). Then the membranes were incubated with
horseradish peroxidase (HRP)-conjugated secondary antibody (goat
anti-rabbit IgG, 1:5,000, cat. no. AS014; goat anti-mouse IgG,
1:5,000; cat. no. AS003; both ABclonal Biotech Co., Ltd.) for 1 h
at room temperature and the protein bands were visualized using ECL
HRP substrate (PerkinElmer, Inc.). A ChemiDoc™ Image Analyzer
(Bio-Rad Laboratories, Inc.) was used to detect the antibody
binding signals. The results were quantified using ImageJ (version
7.0; National Institutes of Health) based on the analysis of three
independent bands.
Immunofluorescence staining
A549 cells cultivated in a Nunc™ Petri dish (Thermo
Fisher Scientific, Inc.) were washed three times with 1X PBS, fixed
in 4% paraformaldehyde for 20 min at 4˚C and washed again three
times with PBS. The cells were then permeabilized with 0.2% Triton
X-100 in PBS and blocked for 30 min at 4˚C with 3% bovine serum
albumin (Thermo Fisher Scientific, Inc.). The cells were then
incubated overnight at 4˚C with anti-LC3 primary antibodies (1:400
dilution; SAB4300571, Sigma-Aldrich; Merck KGaA) in blocking
buffer. After washing with PBS, the cells were incubated with Alexa
Fluor 555 fluorescent secondary antibody (1:100; cat. no.
bs-0295G-AF555; BIOSS) for 60 min at room temperature. The nuclei
were then stained with 4',6-diamidino-2-phenylindole at 1:5,000
dilution for 5 min at room temperature. The cells were washed twice
with PBS and fluorescence images were captured using a confocal
microscope (Smartproof 5; Carl Zeiss AG). The intensity of staining
was determined by measuring the integrated optical density (IOD) in
10 different fields for each sample.
Apoptosis detection using flow
cytometry
The apoptosis of cells was detected using an Annexin
V-FITC Apoptosis Detection kit (Beyotime Institute of
Biotechnology) according to the manufacturer's protocol and
detected by flow cytometry (BD FACSAria™ III; BD Biosciences).
Briefly, the cells were washed with PBS and resuspended; then
Annexin V-FITC and PI binding solution was added to the
resuspension and the cells were incubated at room temperature for
20 min in the dark. Cells were then placed in an ice bath prior to
detected by flow cytometry. The results were analyzed using FlowJo
v.8.0 software (Tree Star, Inc.).
Measurement of IFN-α level
The concentration of IFN-α in the culture
supernatants was measured by enzyme-linked immunosorbent assay
(ELISA). A commercially available human IFN-α ELISA kit (cat. no.
#41100; R&D Systems, Inc.) was used according to the
manufacturer's instructions. The results were normalized to the
amount of IFN-α in conditioned media and expressed in units of
pg/ml.
Statistical analysis
All data were analyzed using GraphPadPrism 8
(GraphPad Software, Inc.). Quantitative data are expressed as the
mean ± standard error of the mean. One-way ANOVA followed by
Bonferroni's multiple comparisons post hoc test was used for
comparisons among more than two groups; two-tailed Student's
t-test was used for comparisons of two groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
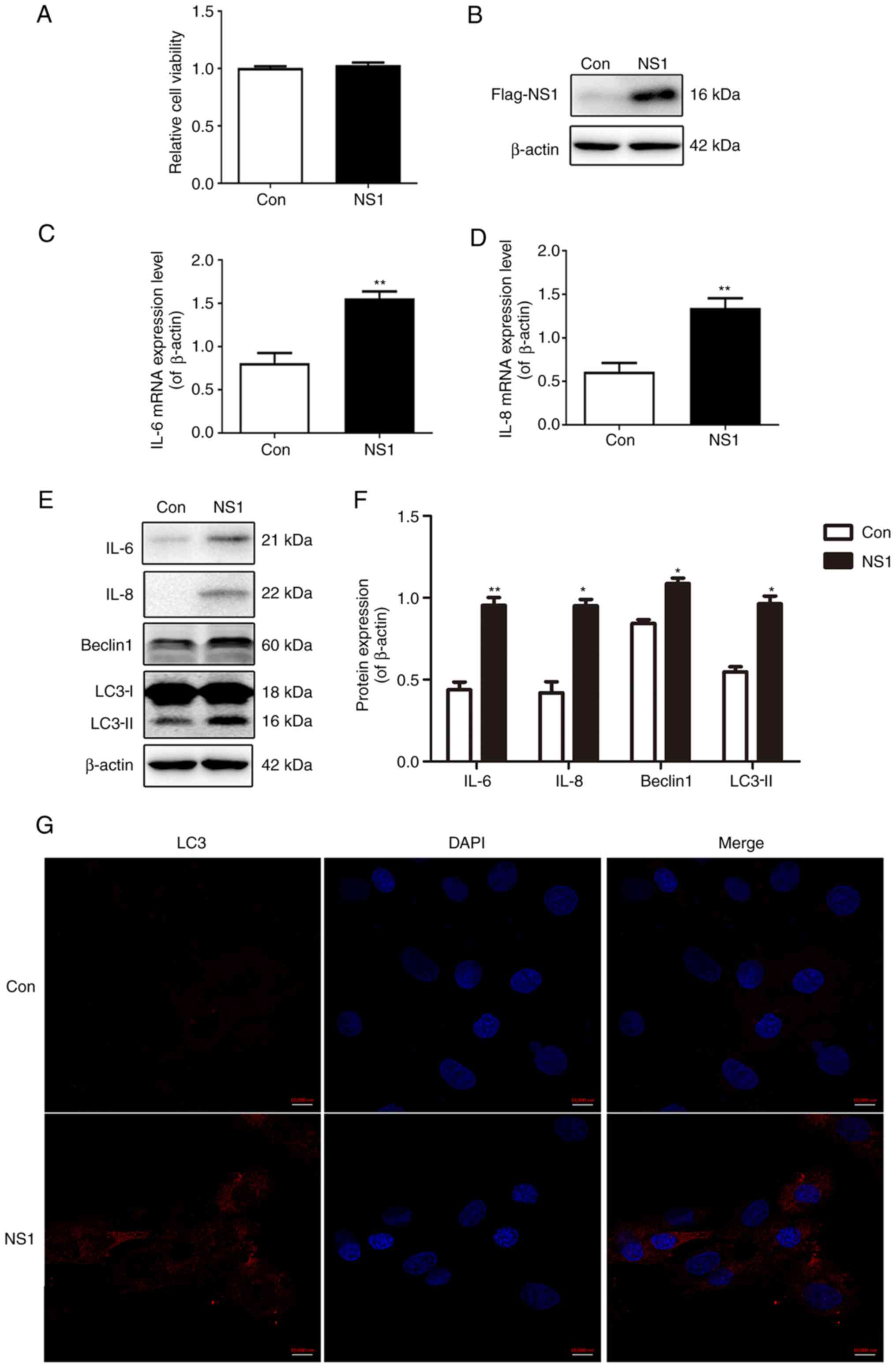
NS1 induces inflammatory cytokine
production and autophagy in A549 cells
The viability and expression of NS1 in A549 cells
transfected with the plasmid pNS1-Flag were examined. As shown in
Fig. 1A, NS1 transfection did not
affect the viability of the cells (Fig.
1A). In addition, the expression level of NS1 protein in the
cells 24 h after transfection with pNS1-Flag was higher than that
in the control cells transfected with empty plasmid (Fig. 1B). The mRNA expression levels of the
inflammatory cytokines IL-6 and IL-8 were increased significantly
following transfection with NS1 (Fig.
1C and D), as were the
respective protein expression levels (Fig. 1E and F). Furthermore, it was observed that
following transfection with NS1, the protein levels of the
autophagy markers LC3-II and Beclin1 were significantly increased
(Fig. 1E and F). The results of immunofluorescence
staining also showed that the fluorescence intensity of LC3
increased markedly after NS1 transfection (Fig. 1G). These results indicate that NS1
transfection increases the production of inflammatory cytokines and
autophagy levels in A549 cells.
Inhibition of autophagy increases
inflammatory factors and IFN-α production in NS1-transfected
cells
To explore the role of autophagy in the expression
of inflammatory factors induced by NS1, cells were pretreated with
the autophagy inhibitor 3MA for 12 h prior to transfection with the
NS1-expressing vector. Firstly, the MTT assay results demonstrated
that cell viability was not affected by transfection or the 3MA
treatment (Fig. 2A). Western
blotting results showed that 3MA further elevated the increase in
the protein expression levels of IL-6 and IL-8 induced by NS1,
while the cells treated with 3MA alone exhibited no significant
difference in the expression of inflammatory factors compared with
the control group (Fig. 2B-D). The
pretreatment with 3MA also attenuated the NS1-induced increase in
the expression of autophagy markers LC3-II and Beclin1, while 3MA
treatment alone had no significant effect on cell autophagy markers
or NS1 expression (Fig. 2B and
E-G). A significant weakening of
the NS1-induced increase in LC3-II fluorescence intensity was also
observed following 3MA treatment in the immunofluorescence assay
(Fig. 2H and I). The levels of IFN-α were also measured.
The results showed that transfection with NS1 downregulated the
IFN-α level and this affect was attenuated by 3MA, while 3MA alone
failed to induce IFN-α (Fig. 2J).
These results suggest that the inhibition of autophagy increased
inflammatory factors and IFN-α production in the cells transfected
with NS1.
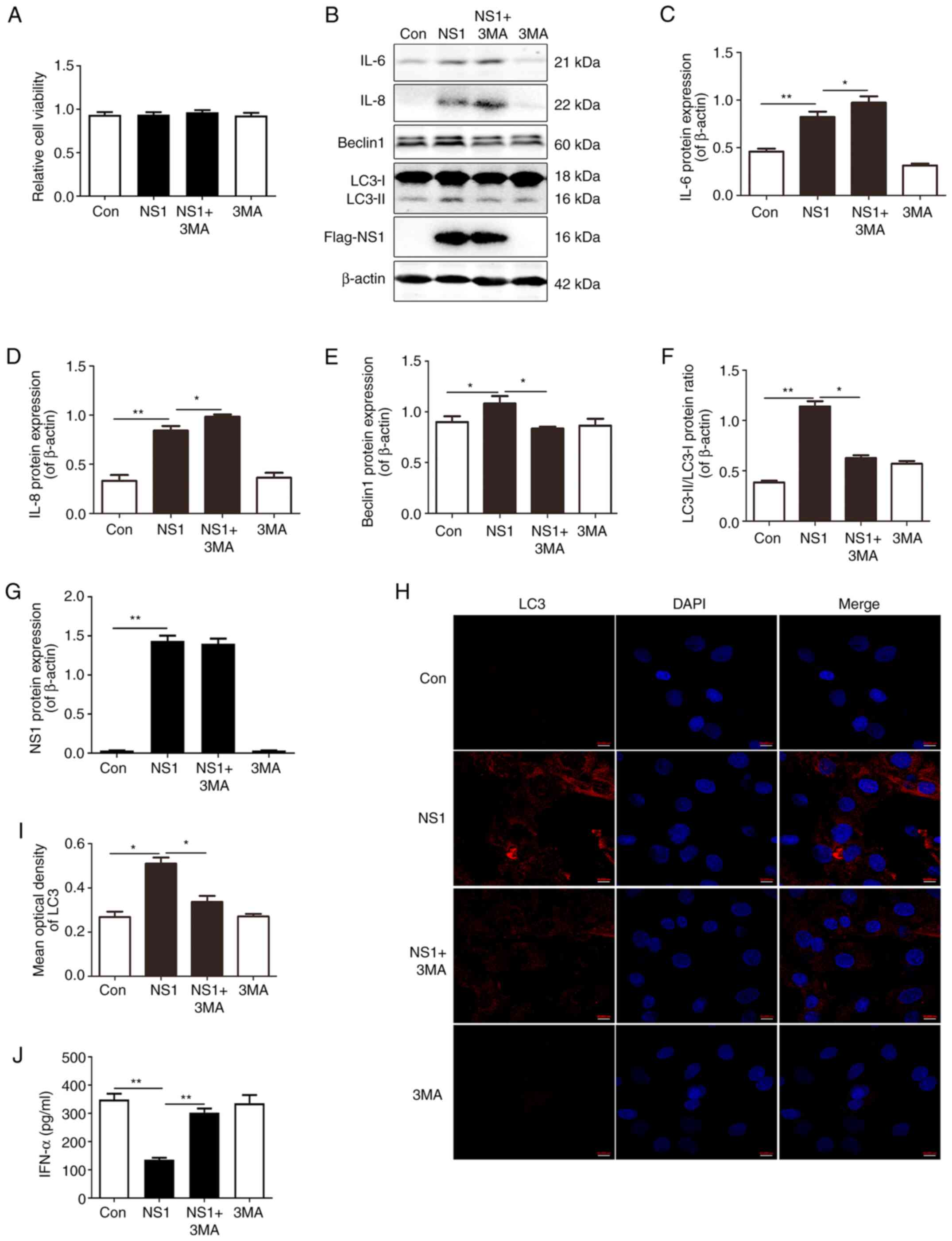 | Figure 2Inhibition of autophagy increases the
production of inflammatory factors and IFN-α in NS1-transfected
cells. A549 cells were treated with 3MA for 12 h prior to
transfection with plasmid pNS1-Flag. (A) Cell viability of various
treatment groups measured by MTT assay. (B) Representative
immunoblots and quantification of (C) IL-6, (D) IL-8, (E) Beclin1
and (F) the LC3-II/LC-I ratio, (G) NS1 in the cells. (H) Cell
immunofluorescence staining and (I) quantification of the optical
density value of LC3 in the cells. Scale bar, 10 µm. (J) IFN-α
production in the cell supernatant was determined by enzyme-linked
immunosorbent assay. Values are presented as the mean ± SEM and are
derived from three independent experiments. *P<0.05,
**P<0.01. IFN-α, interferon-α; NS1, non-structural
protein 1; 3MA, 3-methyladenine; LC3-I, cytoplasmic
microtubule-associated protein 1A/1B-light chain 3; LC3-II,
lipid-modified LC3-I; Con, control. |
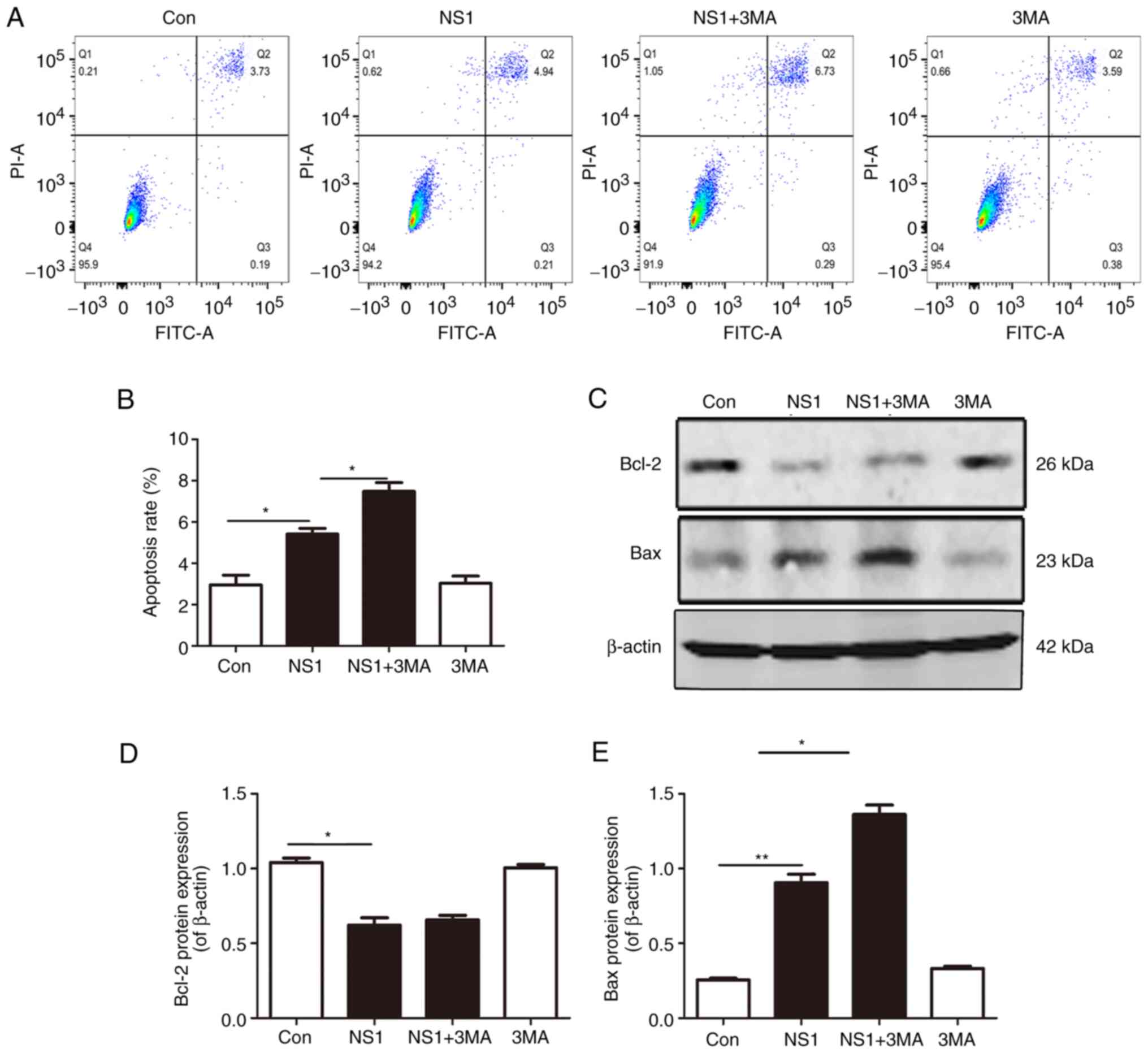
Inhibition of autophagy increases cell
apoptosis in NS1-transfected cells
The apoptosis of the cells was tested following
transfection with the NS1 overexpression vector by flow cytometry
using Annexin V-FITC and PI. In Fig.
3A, cells in Q1 (Annexin V-FITC)-/PI+
were necrotic, in Q2 (Annexin V-FITC)+/PI+
were late apoptotic, in Q3 (Annexin
V-FITC)+/PI- were early apoptotic and in Q4
(Annexin V-FITC)-/PI- were living. The
results showed that the apoptosis of NS1-transfected cells was
increased compared with that in the control group and 3MA increased
the apoptosis induced by NS1, whereas 3MA treatment alone had no
significant effect on apoptosis (Fig.
3A and B). The detection of
apoptosis-associated proteins by western blotting showed that the
expression of anti-apoptotic protein Bcl-2 decreased and
pro-apoptotic protein Bax increased significantly following NS1
transfection. When compared with the NS1 transfection group, the
Bcl-2 expression level was not significantly changed in the
3MA-pretreated NS1-transfected cells, while Bax expression was
significantly upregulated. 3MA treatment alone had no significant
effect on these two proteins (Fig.
3B-D). These results demonstrate that transfection with NS1
increases apoptosis and the inhibition of autophagy exacerbated
this phenomenon.
NS1 activates autophagy through the
mTOR pathway
To further explore the mechanism by which autophagy
occurs in NS1-transfected cells, the classic mTOR pathway of
autophagy was examined. Western blotting results showed that
compared with cells that were not transfected with NS1, the
p-mTOR/mTOR and p-S6KP70/S6KP70 protein ratios were significantly
reduced in the NS1-transfected cells. In addition, pretreatment
with 3MA blocked the NS1-induced reduction in these protein ratios,
while the administration of 3MA alone did not change the
p-mTOR/mTOR and p-S6KP70/S6KP70 protein ratios significantly
(Fig. 4A-C). The cells were
pretreated with the mTOR-specific inhibitor Torin-1 to further
confirm the links between autophagy and the mTOR pathway. Firstly,
the results of an MTT assay showed that treatment with 1 µM Torin-1
alone had no significant effect on cell viability (Fig. 4D), and Torin-1 significantly
inhibited the activation of mTOR by phosphorylation (Fig. 4E and F). When mTOR activity was inhibited by
Torin-1, the LC3-II level was upregulated in the NS1-transfected
cells compared with the control and untreated NS1-transfected
cells. Treatment with Torin-1 alone also significantly increased
the level of LC3-II compared with that in the control group
(Fig. 4E and G). These results suggested that NS1 may
induce autophagy through the mTOR-S6KP70 pathway.
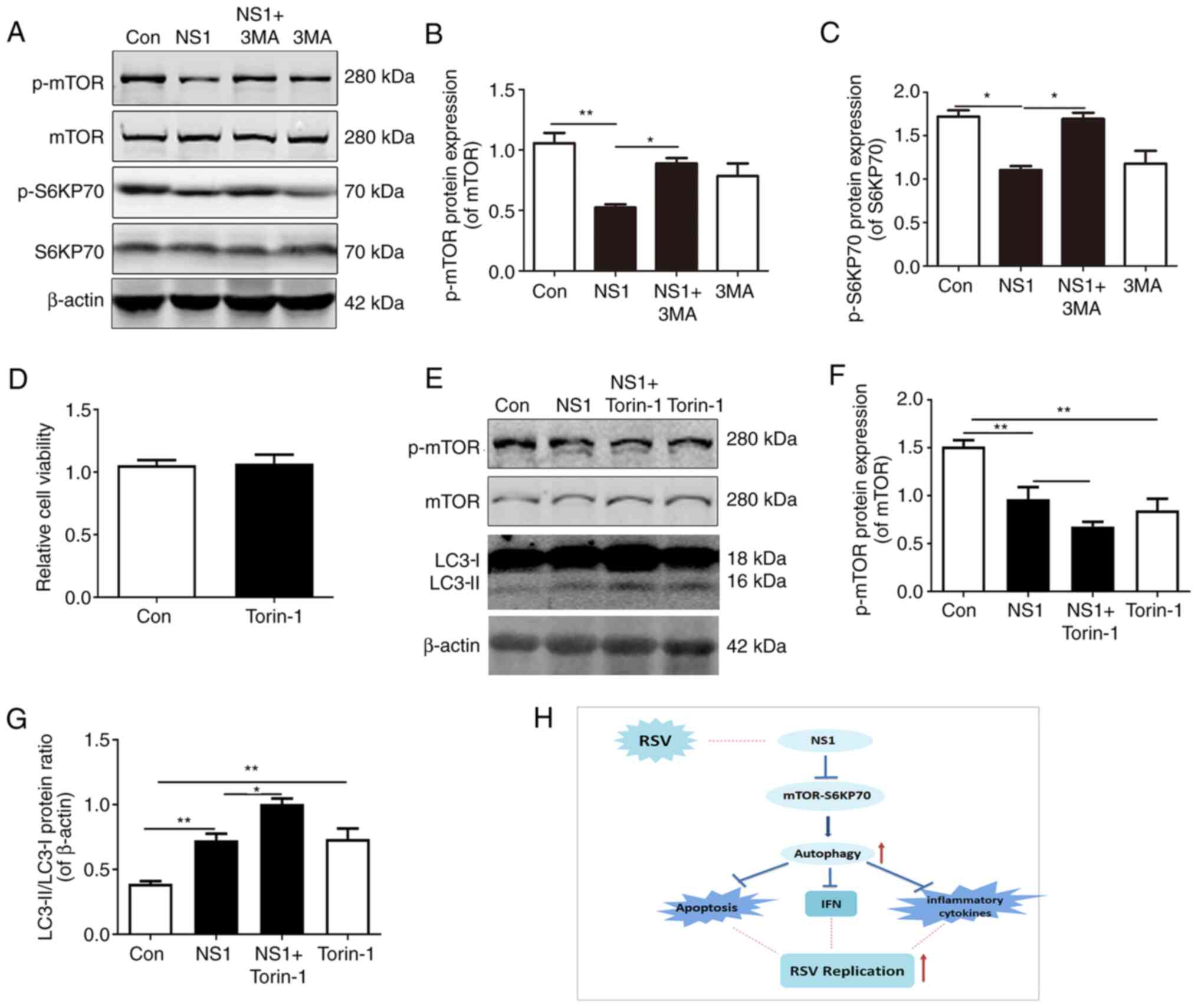 | Figure 4NS1 activates autophagy through the
mTOR pathway. (A-C) A549 cells were treated with 3MA for 12 h prior
to transfection with plasmid pNS1-Flag. (A) Representative
immunoblots and quantification of (B) p-mTOR/mTOR and (C)
p-S6KP70/S6KP70 ratios in cells. (D-G) Cells were treated with
Torin-1 for 3 h prior to transfection with plasmid pNS1-Flag, then
(D) an MTT assay and (E) western blot analysis of p-mTOR and LC3
levels were performed. (F) p-mTOR/mTOR and (G) LC3-II/LC3-I ratios
were quantified (H) Scheme of the potential mechanism for the
beneficial effect of NS1 in RSV-infected epithelial cells via
autophagy. Values are presented as the mean ± SEM and are derived
from three independent experiments. *P<0.05,
**P<0.01. NS1, non-structural protein 1; mTOR,
mammalian target of rapamycin; 3MA, 3-methyladenine; p-,
phosphorylated; S6KP70, p70 S6 kinase; LC3-I, cytoplasmic
microtubule-associated protein 1A/1B-light chain 3; LC3-II,
lipid-modified LC3-I; Con, control; RSV, respiratory syncytial
virus; IFN, interferon. |
Discussion
RNA and DNA viral infections can induce
non-identical autophagy responses, and a recent study demonstrated
that the inhibition of autophagy by 3MA decreased RSV replication
and ameliorated lung pathology in the lungs of mice following RSV
infection (24). It also has been
shown that the induction of autophagy is beneficial for the
replication of viruses, but the detailed mechanisms remain elusive
(25-27).
Although it is known that NS1 attenuates the production of IFN
during RSV infection, the precise role of the RSV NS1 protein in
the early host-virus interaction during the infection is poorly
understood. Zhang et al (28) suggested that JNK activation
regulates virus replication via the induction of autophagy. On the
basis of these previous findings, it was hypothesized that
autophagy may play a role in RSV viral replication. This hypothesis
was supported by the observation of increased autophagic markers,
notably LC3-II and Beclin1, when cells were transfected with NS1,
and the increase of inflammatory cytokine and IFN-α production when
autophagy was inhibited. The present study suggests that when cells
are transfected with NS1, cell autophagy is activated to prevent
the production of cytokines and IFN-α and the activation of
apoptosis, which may be conducive to RSV viral replication.
Autophagy serves a dual role as it mediates both
cell survival and cell death. Autophagy limits the spread of a
virus from infected to healthy tissues and regulates the programmed
death of adjacent uninfected cells, thereby acting as a cell
survival mechanism during viral infection (29). The influenza A virus induces the
apoptosis of autophagy protein-deficient cells, which suggests that
autophagy is beneficial for the survival of infected cells
(30). Autophagy is also thought to
prolong the survival of erythroid cells infected with human
parvovirus B19(31). The impairment
of autophagy in cells may mediate apoptotic cell death. The results
from a study by Shrivastava et al (32) demonstrated that hepatitis C virus
infection in cells with autophagy-associated protein knockdown
induced apoptotic cell death. In the present study, the findings of
increased IL-6 and IL-8 secretion, decreased Bcl-2 and increased
Bax levels when autophagy was inhibited concur with a previous
report of RSV infection inducing cytokine secretion in dendritic
cells via an LC3-dependent mechanism (20). The current study also documented
increased apoptosis in NS1-transfected cells, which was further
increased by the inhibition of autophagy. This suggests that
autophagy may promote cell survival in response to NS1 by
preventing apoptosis.
mTOR is a key regulator of autophagy, and inhibition
of the mTOR-S6KP70 signaling pathway is one of the main mechanisms
by which autophagy is activated (33). Torin-1 is a specific mTOR inhibitor,
which has been shown to induce autophagy by inhibiting mTOR
activity (22). The results of the
present study showed that transfection with NS1 decreased the
levels of p-mTOR and p-S6KP70, and the effects of NS1 were blocked
by 3MA. Furthermore, LC3-II levels increased in cells with or
without NS1 transfection when mTOR activity was inhibited by
Torin-1. These results suggest that NS1 induced autophagy through
the mTOR-S6KP70 signaling pathway. However, the mechanism by which
NS1 modulates the mTOR-S6KP70 signaling pathway remains unknown. To
summarize, the postulated mechanism of the beneficial effect of
NS1-induced autophagy in RSV-infected epithelial cells mediated
through the mTOR pathway is illustrated by the scheme in Fig. 4H.
In conclusion, the present study demonstrated that
the induction of autophagy by NS1 transfection via the mTOR pathway
may hinder the production of inflammatory cytokines and IFN-α, and
inhibit cell apoptosis, which may help to explain why autophagy has
been found to be beneficial to viral replication in most studies.
The findings of the present study suggest that autophagy may be a
novel and effective therapeutic target against RSV infections.
Acknowledgements
The authors would like to thank Professor Zhao of
Wuhan University Zhongnan Hospital for providing the NS1 plasmids
and Medical Research Center for Structural Biology of Basic Medical
Sciences, Wuhan University for technical help with the confocal
fluorescence microscopy and flow cytometry.
Funding
Funding: No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
MZ co-designed the study, performed experiments,
analyzed data and co-wrote and revised the manuscript. BH
co-designed the study, analyzed data and co-wrote the manuscript.
YW performed experiments, analyzed data and co-wrote the
manuscript. BH and YW confirm the authenticity of all the raw data.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shi T, McAllister DA, O'Brien KL, Simoes
EAF, Madhi SA, Gessner BD, Polack FP, Balsells E, Acacio S, Aguayo
C, et al: Global, regional, and national disease burden estimates
of acute lower respiratory infections due to respiratory syncytial
virus in young children in 2015: A systematic review and modelling
study. Lancet. 390:946–958. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Zhang W, Yang H, Kong X, Mohapatra S, San
Juan-Vergara H, Hellermann G, Behera S, Singam R, Lockey RF and
Mohapatra SS: Inhibition of respiratory syncytial virus infection
with intranasal siRNA nanoparticles targeting the viral NS1 gene.
Nat Med. 11:56–62. 2005.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Lo MS, Brazas RM and Holtzman MJ:
Respiratory syncytial virus nonstructural proteins NS1 and NS2
mediate inhibition of Stat2 expression and alpha/beta interferon
responsiveness. J Virol. 79:9315–9319. 2005.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Sedeyn K, Schepens B and Saelens X:
Respiratory syncytial virus nonstructural proteins 1 and 2:
Exceptional disrupters of innate immune responses. PLoS Pathog.
15(e1007984)2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Murphy BR and Collins PL: Live-attenuated
virus vaccines for respiratory syncytial and parainfluenza viruses:
Applications of reverse genetics. J Clin Invest. 110:21–27.
2002.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Stoppelenburg AJ, Salimi V, Hennus M,
Plantinga M, Huis in 't Veld R, Walk J, Meerding J, Coenjaerts F,
Bont L and Boes M: Local IL-17A potentiates early neutrophil
recruitment to the respiratory tract during severe RSV infection.
PLoS One. 8(e78461)2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Faber TE, Groen H, Welfing M, Jansen KJ
and Bont LJ: Specific increase in local IL-17 production during
recovery from primary RSV bronchiolitis. J Med Virol. 84:1084–1088.
2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zamora MR, Budev M, Rolfe M, Gottlieb J,
Humar A, Devincenzo J, Vaishnaw A, Cehelsky J, Albert G, Nochur S,
et al: RNA interference therapy in lung transplant patients
infected with respiratory syncytial virus. Am J Respir Crit Care
Med. 183:531–538. 2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Bitko V, Musiyenko A, Shulyayeva O and
Barik S: Inhibition of respiratory viruses by nasally administered
siRNA. Nat Med. 11:50–55. 2005.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Kong X, Zhang W, Lockey RF, Auais A,
Piedimonte G and Mohapatra SS: Respiratory syncytial virus
infection in Fischer 344 rats is attenuated by short interfering
RNA against the RSV-NS1 gene. Genet Vaccines Ther.
5(4)2007.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Boyapalle S, Wong T, Garay J, Teng M, San
Juan-Vergara H and Mohapatra S and Mohapatra S: Respiratory
syncytial virus NS1 protein colocalizes with mitochondrial
antiviral signaling protein MAVS following infection. PLoS One.
7(e29386)2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Rostad CA, Stobart CC, Gilbert BE, Pickles
RJ, Hotard AL, Meng J, Blanco JCG, Moin SM, Graham BS, Piedra PA
and Moore ML: A recombinant respiratory syncytial virus vaccine
candidate attenuated by a low-fusion F protein is immunogenic and
protective against challenge in cotton rats. J Virol. 90:7508–7518.
2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yang P, Zheng J, Wang S, Liu P, Xie M and
Zhao D: Respiratory syncytial virus nonstructural proteins 1 and 2
are crucial pathogenic factors that modulate interferon signaling
and Treg cell distribution in mice. Virology. 485:223–232.
2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Yan S, Yue YZ, Sun MM, Wu BS and Wang XP:
Suppressive effect of Aurantii Fructus Immaturus and Atractylodis
Macrocephalae Rhizoma on glutamic acid-induced autophagy of
interstitial cells of Cajal. J Integr Med. 18:334–343.
2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Huang XP, Ding H, Lu JD, Tang YH, Deng BX
and Deng CQ: Autophagy in cerebral ischemia and the effects of
traditional Chinese medicine. J Integr Med. 13:289–296.
2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Deretic V, Saitoh T and Akira S: Autophagy
in infection, inflammation and immunity. Nat Rev Immunol.
13:722–737. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
Tanida I, Ueno T and Kominami E: LC3 and
autophagy. Methods Mol Biol. 445:77–88. 2008.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Shi B, Ma M, Zheng Y, Pan Y and Lin X:
mTOR and Beclin1: Two key autophagy-related molecules and their
roles in myocardial ischemia/reperfusion injury. J Cell Physiol.
234:12562–12568. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Jung CH, Ro SH, Cao J, Otto NM and Kim DH:
mTOR regulation of autophagy. FEBS Lett. 584:1287–1295.
2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Reed M, Morris SH, Owczarczyk AB and
Lukacs NW: Deficiency of autophagy protein Map1-LC3b mediates
IL-17-dependent lung pathology during respiratory viral infection
via ER stress-associated IL-1. Mucosal Immunol. 8:1118–1130.
2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Xu X, Zheng J, Zheng K, Hou Y, Zhao F and
Zhao D: Respiratory syncytial virus NS1 protein degrades STAT2 by
inducing SOCS1 expression. Intervirology. 57:65–73. 2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zullo AJ, Jurcic Smith KL and Lee S:
Mammalian target of Rapamycin inhibition and mycobacterial survival
are uncoupled in murine macrophages. BMC Biochem.
15(4)2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Li M, Li J, Zeng R, Yang J, Liu J, Zhang
Z, Song X, Yao Z, Ma C, Li W, et al: Respiratory syncytial virus
replication is promoted by autophagy-mediated inhibition of
apoptosis. J Virol. 92:e02193–e02217. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
O'Donnell V, Pacheco JM, LaRocco M,
Burrage T, Jackson W, Rodriguez LL, Borca MV and Baxt B:
Foot-and-mouth disease virus utilizes an autophagic pathway during
viral replication. Virology. 410:142–150. 2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Pokharel SM, Shil NK and Bose S:
Autophagy, TGF-β, and SMAD-2/3 signaling regulates interferon-β
response in respiratory syncytial virus infected macrophages. Front
Cell Infect Microbiol. 6(174)2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Owczarczyk AB, Schaller MA, Reed M, Rasky
AJ, Lombard DB and Lukacs NW: Sirtuin 1 regulates dendritic cell
activation and autophagy during respiratory syncytial virus-induced
immune responses. J Immunol. 195:1637–1646. 2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhang J, Ruan T, Sheng T, Wang J, Sun J,
Wang J, Prinz RA, Peng D, Liu X and Xu X: Role of c-Jun terminal
kinase (JNK) activation in influenza A virus-induced autophagy and
replication. Virology. 526:1–12. 2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Pandey A, Ding SL, Qin QM, Gupta R, Gomez
G, Lin F, Feng X, Fachini da Costa L, Chaki SP, Katepalli M, et al:
Global reprogramming of host kinase signaling in response to fungal
infection. Cell Host Microbe. 21:637–649.e6. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Gannage M, Dormann D, Albrecht R, Dengjel
J, Torossi T, Ramer PC, Lee M, Strowig T, Arrey F, Conenello G, et
al: Matrix protein 2 of influenza A virus blocks autophagosome
fusion with lysosomes. Cell Host Microbe. 6:367–380.
2009.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Nakashima A, Tanaka N, Tamai K, Kyuuma M,
Ishikawa Y, Sato H, Yoshimori T, Saito S and Sugamura K: Survival
of parvovirus B19-infected cells by cellular autophagy. Virology.
349:254–263. 2006.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Shrivastava S, Raychoudhuri A, Steele R,
Ray R and Ray RB: Knockdown of autophagy enhances the innate immune
response in hepatitis C virus-infected hepatocytes. Hepatology.
53:406–414. 2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ghiglieri V, Pendolino V, Bagetta V,
Sgobio C, Calabresi P and Picconi B: mTOR inhibitor rapamycin
suppresses striatal post-ischemic LTP. Exp Neurol. 226:328–331.
2010.PubMed/NCBI View Article : Google Scholar
|