Introduction
Osteonecrosis of the femoral head is a relatively
common and refractory osteoarthropathy. Glucocorticoid is one of
the leading causes of non-traumatic necrosis of the femoral head.
Its major feature is that the necrosis and regeneration of bone and
marrow cells are caused by disorders of the blood supply of the
femoral head. Eventually, the femoral head collapses and is
deformed and severe hip joint lesions occur (1). The exact pathogenesis of avascular
necrosis of the femoral head remains to be elucidated. There are
several hypotheses, including the theory of lipid metabolism
disorder, that of intravascular coagulation and that of
microvascular injury caused by immune complex deposition. However,
the obstacle in the bone tissue blood supply is known to be the
fundamental factor of femoral head necrosis. Microcirculation
disorder is speculated to be the most common pathway underlying
this disease. Bone microvascular endothelial cells (BMECs),
involved in the formation of microcirculation, have quickly become
a research hotspot. Previous studies have pointed out that BMECs in
the necrosed femoral head of patients exhibited different degrees
of injury (2). The damage caused by
glucocorticoids to BMECs was determined (3,4), while
the specific mechanisms remain to be studied.
Microfluidic technologies comprise the control,
operation and detection of complex fluids at microscopic sizes. It
is able to manipulate the fluids at the submillimeter level.
Microfluidic technologies range from simple cell culture chips to
organ chips and their application prospect is infinite. Urbaczek
et al (5) introduced a
microfluidic device that is able to simulate blood vessels. This
device may be used for cell perfusion culture, as well as
cardiovascular and toxicological studies. van Engeland et al
(6) cultured arterial endothelial
and smooth muscle cells in a 3-dimensional microfluidic chip,
achieving in vitro simulation of arterial wall composition,
interaction and mechanical environment. With the development of
microfluidic technology, the construction of organ chips has been
realized. It is known that the blood-brain barrier is one of the
important protective mechanisms of the human body. Through
microfluidics, organ-on-a-chip models of the blood-brain barrier
are becoming a reality. van der Helm et al (7) summarized the recent developments and
challenges of this chip model.
The use of multi-disciplinary strategies have now
become a trend. The continuous development of microfluidic
technology was bound to drive the development of medicine. In the
present study, microfluidic technology was used to generate a model
of femoral head necrosis by culturing BMEC in a microfluidic
organ-on-a-chip. The microfluidic organ-on-a-chip was used to
culture BMECs and apply relevant interventions to explore the
pathogenesis of osteonecrosis.
Materials and methods
Isolation of BMECs
The femoral head was donated voluntarily by patients
undergoing total hip arthroplasty at China-Japan Friendship
Hospital (Beijing, China) for femoral neck fractures during
November and December 2019. A total of 10 male patients were
selected and it was confirmed that their femoral head was normal
except from the femoral neck fracture. The patients were aged
between 55 and 60 years old and were in a generally good condition.
The experiment was approved by the Ethics Committee of China-Japan
Friendship Hospital (Beijing, China) and all patients donated their
bones voluntarily with written informed consent. Normal cancellous
bones from resected femoral heads were cut into 1-2-mm2
particles and washed three times with PBS to remove the adipose
tissue, blood cells and other debris. These bone granules were
digested with 5 ml of 0.2% collagenase I (Gibco; Thermo Fisher
Scientific, Inc.) for 30 min at 37˚C, followed by trypsinization
with 3 ml of 0.25% trypsin-EDTA for 5 min at 37˚C. The process was
stopped by adding 2 ml DMEM (Gibco; Thermo Fisher Scientific, Inc.)
containing 10% FBS (Shanghai Yeasen Biotechnology Co., Ltd.). The
supernatant was filtered through a 70-µM cell strainer and
subjected to centrifugation at 107 x g for 5 min at room
temperature (RT). The pellet consisted of the harvested
microvessels and the cells were resuspended with 80 µl MACS running
buffer (Miltenyi Biotec GmbH) and 20 µl of CD31 microbeads (cat.
no. 130-091-935; Miltenyi Biotec GmbH). Following incubation for 15
min at 4˚C, the cell suspension was mixed with 400 µl MACS running
buffer and added onto a MACS column fixed in the magnetic field.
Subsequently, the column was removed and 1 ml DMEM buffer was
pipetted onto the column to flush out the labeled cells into a
suitable collection tube. Following centrifugation, the pellet was
resuspended in 15 ml endothelial cell medium (ECM; ScienCell
Research Laboratories, Inc.) and cultivated in a cell incubator at
37˚C with 5% CO2. After 24 h, the unattached cells were
removed by washing with PBS. The BMECs were passaged at 80%
confluency and used for subsequent experiments.
Identification of BMECs
The purity of the isolated BMECs was evaluated by
immunofluorescence detection of the endothelial markers. The
isolated cells were grown on sterile adhesive microscopic slides
for 12 h, fixed in 4% paraformaldehyde for 20 min at 4˚C,
permeabilized with 0.5% Triton X-100 for 15 min at room
temperature, blocked with 10% FBS for 30 min at 37˚C and probed
overnight with primary antibody to CD31 (cat. no. ab28364;
dilution, 1:300; Abcam) and von Willebrand factor (vWF; cat. no.
ab6994; dilution, 1:300; Abcam) at 37˚C. Subsequently, the cells
were incubated with secondary antibody conjugated to FITC (cat. no.
ab6717; 1:200; Abcam) and Alexa 555 (cat. no. ab150078; 1:200;
Abcam) and counterstained with DAPI at RT for 1 h at room
temperature. The cell morphology was observed under a fluorescence
microscope and 10 fields were randomly selected. The number of
immunofluorescence-positive cells was counted and cell purity was
calculated.
Microfluidic organ-on-a-chip
preparation
The SU-8 photolithography method was used to
manufacture a male mold. Silane treatment was performed after the
liquid polydimethylsiloxane (PDMS; Sylgard Dow Corning) (the ratio
of prepolymer to curing agent was 10:1) was poured on the positive
mold, followed by degassing, and after curing at 70˚C for 1 h, and
a PDMS microfluidic cover with a thickness of ~2.5 mm was obtained.
A 0.5-mm biopsy perforator (Harrick Plasma) was prepared for inlet
and outlet. Tape was used to remove dust from the surface, and
clean devices and coverslips were plasma-treated (PDC-002; Harrick
Plasma) and bonded together. The chip was sterilized with 75%
ethanol and ultraviolet light.
Matrix collagen manufacturing and chip
injection
A total of 3 mol/l collagen I (Discovery Labware,
Inc.) was prepared and placed on ice for later use. HUVECs
(BioCoat) and BMECs were trypsinized at 95% confluency and
harvested by centrifugation at 100 x g for 5 min at RT. Cells were
collected for counting and the concentration was adjusted to
2.5x106 cells/ml for later use.
Part I [co-culture experiment with
hydroxyapatite (HA)]
Three groups were designed: Group A (the HUVEC
group), Group B (the control group of BMECs) and group C (the
intervention group of BMECs with 0.2% HA), with 3 chips each.
In groups A and B, ECM (ScienCell Research
Laboratories, Inc.) and matrix collagen (Discovery Labware, Inc.)
were mixed in the equivalent volume of 20 µl each on ice and added
to 20 µl cell suspension. Next, 2.5 µl matrix collagen cell mixture
was injected into the second channel in the chip. The injection
hole was then sealed at both ends of the channel with PDMS pieces
to prevent the evaporation of the liquid in the channel. The
injected chip was placed in a Petri dish with 1 ml PBS to maintain
the moist environment and reduce the evaporation of the liquid in
the chip. The assembly was placed in an incubator for 30 min to
allow for solidification of the matrix collagen. Subsequently, 5 µl
ECM was added to the medium channel of the chip, the injection hole
was sealed with PDMS and the chip was placed into the culture dish
in the incubator (37˚C, 5% CO2) for culture. Following
incubation for 24 h, cell staining was performed.
In group C, ECM and HA were used to prepare a 0.6%
HA suspension. A volume of 20 µl HA suspension was mixed with an
equivalent volume of matrix collagen on ice and the same volume of
BMEC suspension was added. The remaining steps were the same as
above.
Part II (dexamethasone intervention
experiment on BMECs)
A total of four groups were established: Group D
(the blank group), group E (the 0.4 mg/ml dexamethasone
intervention group), group F (the 0.6 mg/ml dexamethasone
intervention group) and group G (the icariin protection group),
with 4 chips in each group (8-10).
The procedure for chip injection was the same as
described above. Following a 24-h culture, the growth of the
microvascular structure in each chip was observed under the
microscope. ECM containing 0.4 or 0.6 mg/ml dexamethasone were
prepared using 5 mg/ml dexamethasone (1 ml: 5 mg) mother liquor and
ECM. ECM in the medium channel was subsequently replaced with the
ECM containing dexamethasone, as aforementioned, for processing.
Group D was changed to normal ECM, Group E was altered with the ECM
containing 0.4 mg/ml dexamethasone, and group F was changed with
the ECM containing 0.6 mg/ml dexamethasone. In group G, the only
difference from group F was that medium containing
icariin(Solarbio) was first used (4x104 mol/l) to
culture for 24 h, followed by 0.6 mg/ml dexamethasone intervention.
Following incubation for 24 h, cell staining was performed.
Caspase 3/7, DAPI and phalloidin
staining
To analyze the chips from part I, the medium was
aspirated and the chips were washed with PBS twice, followed by
fixation with 4% formaldehyde at 4˚C for 10 min and treatment with
0.5% Triton X-100 for 10 min at RT. Subsequently, the chips were
probed with 496 nm FITC-labeled phalloidin (cat. no. F432; 300 nM;
Invitrogen; Thermo Fisher Scientific, Inc.) at RT for 1 h in the
dark. Finally, the nuclei were counterstained with 364 nm DAPI
(cat. no. 10236276001; 200 nM; Sigma-Aldrich; Merck KGaA) for 5 min
and the film was sealed. Samples could be kept for 3 months at 4˚C
in the dark.
In part II, since the fluorescence wavelength of the
label for caspase 3/7 (Ex/Em=502/530 nm) was close to that of the
496 nm FITC-labeled phalloidin, a fluorophore with another
wavelength, 647 nm FITC-labeled phalloidin, was selected. After the
medium was aspirated, the chips were washed with PBS and incubated
in 15 µl Caspase-3/7 Green Detection Reagent (cat. no. C10723; 8
µM; Invitrogen; Thermo Fisher Scientific, Inc.) for 1 h to remove
extra caspase-3/7 reagent. Subsequently, 4% formaldehyde was used
for fixation at 4˚C for 10 min, followed by treatment with 0.5%
Triton X-100 for 10 min at RT. Next, 647 nm FITC-labeled phalloidin
(cat. no. 40762ES75; 200 nM; Shanghai Yeasen Biotechnology Co.,
Ltd.) was added and samples were incubated at RT for 1 h in the
dark. The nuclei were counterstained with 364 nm DAPI for 5 min
prior to sealing and storing at 4˚C in the dark.
Cell imaging detection
Staining images were captured by a confocal
microscope using the 10-fold lens or 40-fold oil lens. An interface
with optimal cell morphology, clear staining and no bubble
interference was selected. The number of cells was compared by
counting the number of nuclei, the area coverage of microfilament
skeletons was determined to compare the growth status of the cells
and the area coverage of cleaved caspase-3/7 was determined to
compare the cell damage among the groups. The imaging data were
analyzed using ImageJ software (https://imagej.nih.gov/ij/) (11,12).
Statistical analysis
Values are expressed as the mean ± standard
deviation. All data were analyzed by SPSS 20.0 statistical software
(IBM Corp.). Differences among multiple groups were analyzed by
one-way ANOVA followed by Tukey's post-hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Cell culture and chip preparation
A microfluidic organ-on-a-chip and schematic diagram
of the top view is presented in Fig.
1A. It contained four channels, including those for infusion of
drugs and cell microchannels, infusion of VEGF-containing medium
microchannels and an ECM simulation region. The internal size
specification of each channel was identical and a micropillar
structure separated the channels. The cells were able to be
inoculated into either channel and the medium was injected into
adjacent channels. In the identification of BMECs, under a
fluorescence microscope, immunofluorescence demonstrated high
expression of CD31 and vWF (Fig.
1B).
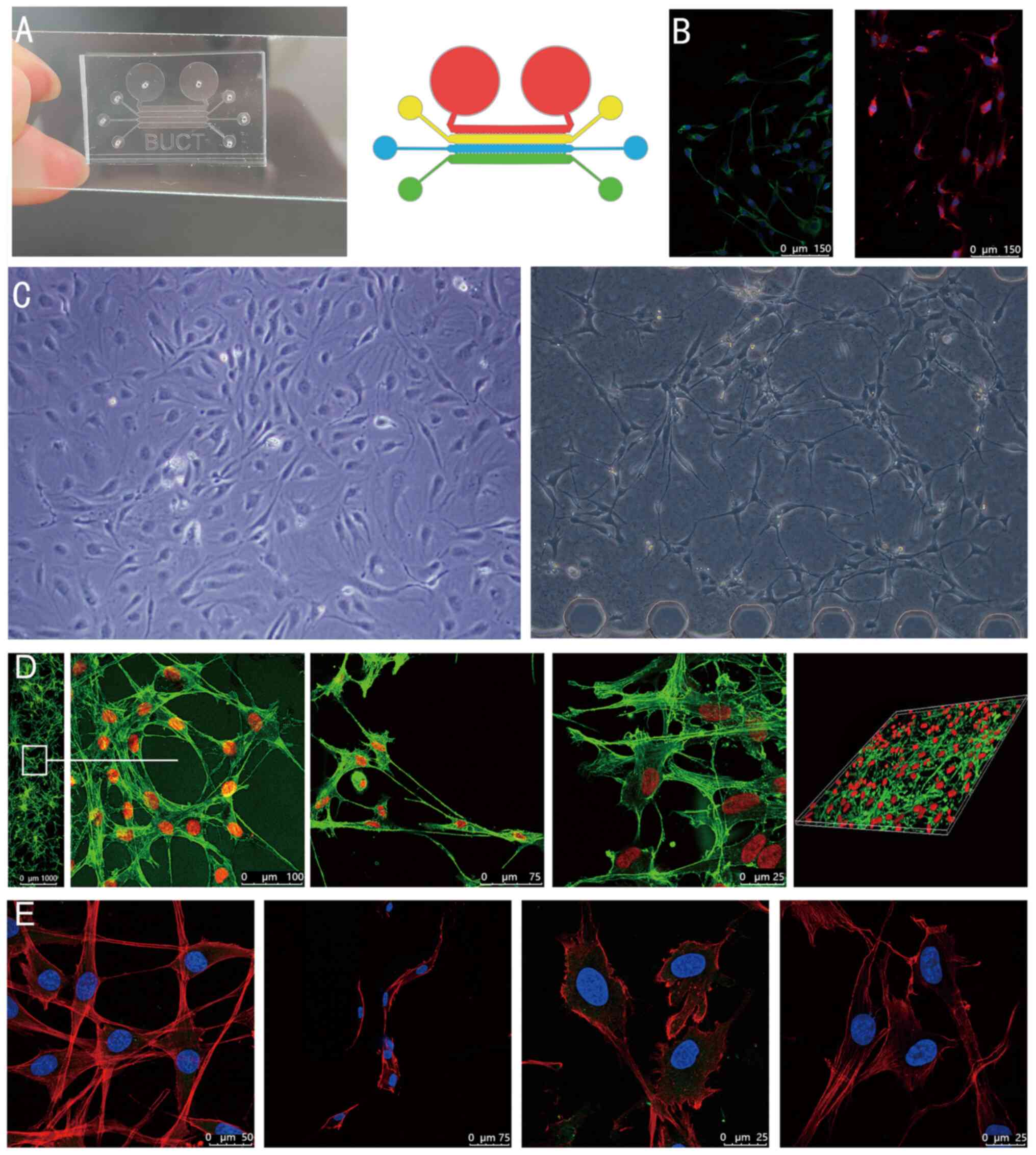 | Figure 1(A) Microfluidic chip and schematic
diagram of the top view. Four different coloured areas represent
its four channels. (B) Identification of bone microvascular
endothelial cells and immunofluorescence staining revealed high
expression of von Willebrand factor (green) on the left side and
CD31 (red) on the right side, and both nuclei were counterstained
with DAPI (scale bars, 150 µm). (C) Ordinary optical microscope
images. The left is taken from ordinary Petri dishes, and the right
is from the microfluidic chip (Magnification, x100). (D) Confocal
microscopy images. From left to right, the first image is the
microfluidic chip channel panorama taken by a confocal microscope
using the 10-fold lens and a partial enlargement (scale bar in
magnified window, 100 µm). The second and third images are
high-resolution images obtained by a confocal microscope using the
40-fold oil lens (scale bars, 75 and 25 µm, respectively). The last
image is a laminated and reconstructed solid image obtained using
confocal microscopy. (E) Cells in high-definition close-up (scale
bars, 50, 75, 25 and 25 µm, respectively, from left to right). |
Fig. 1C is the cell
image acquired by an ordinary optical microscope (magnification,
x100). The left is the BMECs cultured in ordinary Petri dishes, and
the right is the BMECs inoculated into the chip. In Petri dishes,
most cells clustered together, and there are few connective
structures between cells. And the microscopic connection between
BMECs in the chip was increased. The structure of the cells, with
the microscopic connections, with similar vascular structures, was
different from the common culture model.
Fig. 1D and E present high-resolution images of cells
captured with a confocal microscope following cell death. In
Fig. 1D, from left to right, the
first image is a microfluidic chip channel panorama obtained by
confocal microscopy using the 10-fold lens and a partial
enlargement. The second and third images are high-resolution images
acquired by a confocal microscope using the 40-fold oil lens. The
last image is a laminated and reconstructed solid image obtained
using confocal microscopy. Fig. 1E
presents selected cells in high-definition close-up during the
experiment in part II. Through these high-definition images, the
microscopic morphology and structure of the cells, the
microfilament connections between cells and the morphology of the
nucleus can be observed more clearly.
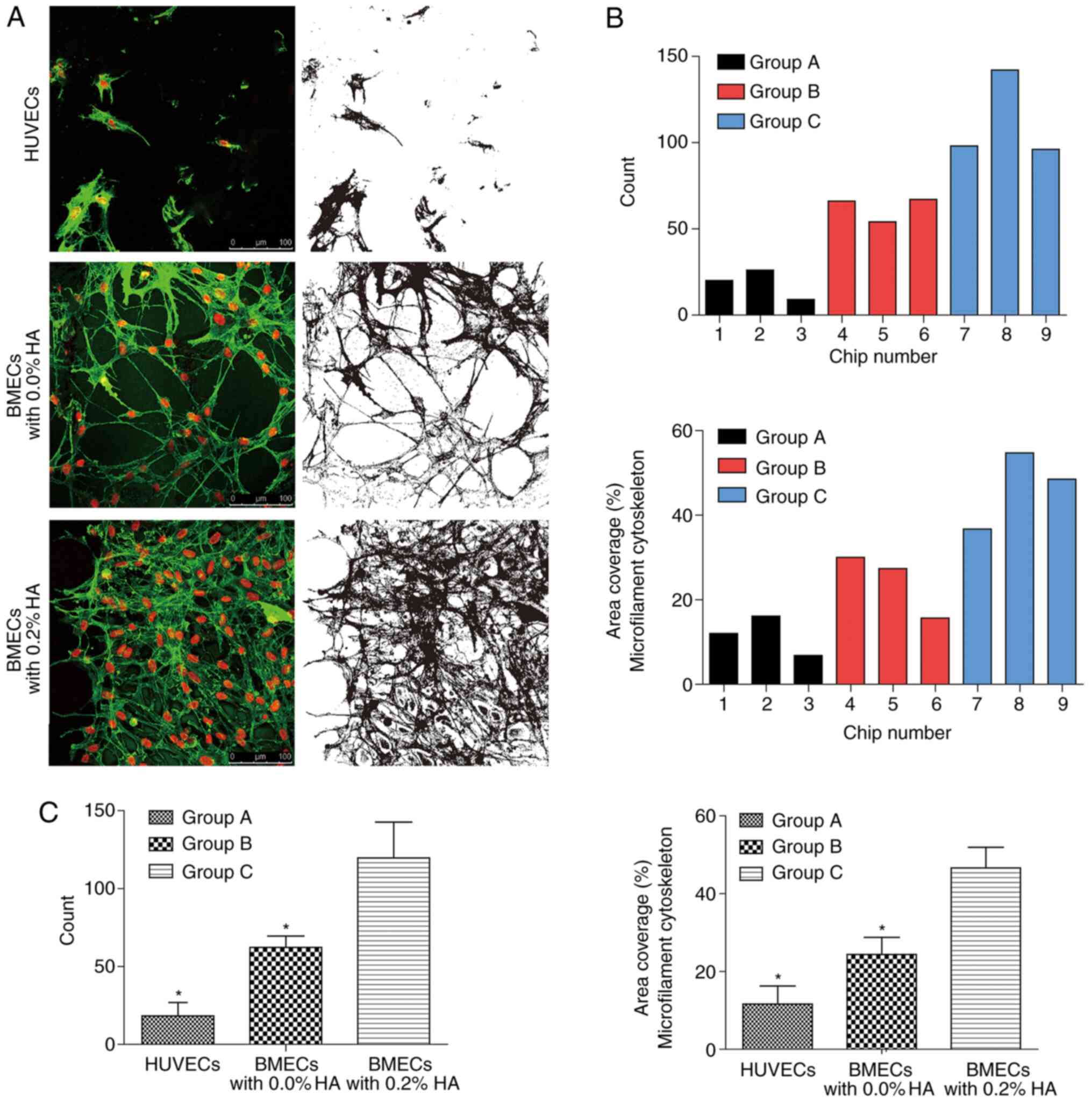
Part I (co-culture experiment of
HA)
The images and data were analyzed following
collection (Fig. 2). The results
were as follows: The number of cells that survived in group B was
significantly higher than that in group A (P<0.05), and that in
group C was also significantly higher compared with that in group B
(P<0.05). In addition, the area coverage percentage of the
microfilament cytoskeleton in group C was significantly higher than
that in group B (P<0.05). Morphologically, several microfilament
connections between cells and circular structures that resemble
blood vessels were observed, particularly in group B. In addition,
BMECs grew more vigorously compared with HUVECs, and BMECs
co-cultured with HA grew most vigorously.
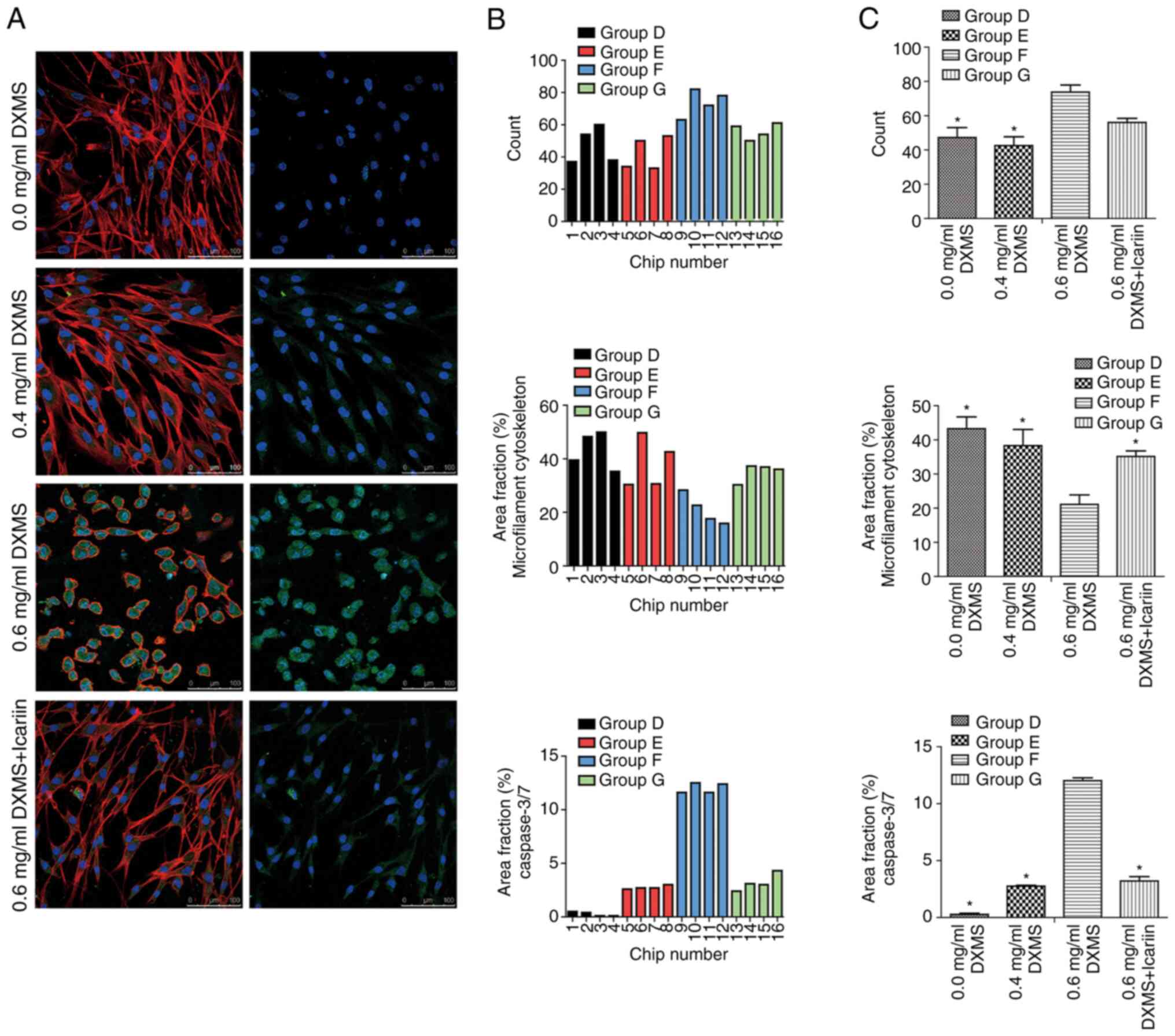
Part II (dexamethasone intervention
experiment of BMECs)
The images and data for the experimental Part II are
presented in Fig. 3. The results
demonstrated that there was no significant difference in the number
of cells between groups D, E and G (P>0.05); however, the number
of cells in group F was significantly higher than that in groups D
and E (P<0.05).
In addition, a significant difference was detected
in the content of cleaved caspase-3/7. The production of cleaved
caspase-3/7 was evident in group F and was significantly higher
than that in all other groups (P<0.05). There was no significant
difference in the content of cleaved caspase-3/7 between groups E
and G (P>0.05). Additionally, the content of cleaved caspase-3/7
was significantly lower compared with all other groups
(P<0.05).
Furthermore, the area coverage of the microfilament
cytoskeleton in group F was significantly lower than that in all
other groups (P<0.05). There was no significant difference among
the other three groups (P>0.05).
In addition to the quantitative results, there were
numerous observations regarding morphology and cellular location of
proteins. First, the localization of cleaved caspase-3/7 was
distinct in the cell, and these proteins were located around the
nucleus. Furthermore, in group F, the morphological structure of
the cells was altered, the microfilament structure between the
cells had disappeared and the cells changed from elongated to a
round shape. Finally, the cell morphology in the other three groups
was similar.
Discussion
HA has been widely used in bone tissue simulation
for a long time, due to its similar structure and osteoinductive
characteristics. It has also been recognized as the best bone graft
material. Bones consist mainly of collagen and calcium phosphate
(Cap) minerals and Cap minerals are similar to HA minerals in
composition and structure. The brittle texture of HA limits its
application as a load-bearing material, and therefore, it requires
to be synthesized with other polymers to improve its mechanical
properties (13,14). Jusoh et al (15) simulated the bone structure with HA
in a microfluidic chip, using HA to induce the growth of HUVECs
across channels. The results indicated that the group treated with
0.2% HA exhibited the best endothelial cell morphology and the most
reliable vasculature-forming ability. Based on the above
experience, in addition to the traditional cultivation of HUVECs,
BMECs extracted and cultured by our group were used for a similar
experiment and co-culture was also performed in the microfluidic
organ-on-a-chip. It was identified that the viability of HUVECs in
the chip was relatively weak. At the same concentration of injected
cells, HUVECs growth was not as vigorous as BMECs's. Although there
were also microfilaments between HUVECs, they did not form a
structure similar to blood vessels as BMECs in Group B did. This
may be due to the limited vitality of the purchased HUVECs. As
BMECs are primary cells extracted from cancellous bone, their
vitality may be higher than that of frozen HUVECs. In the
co-culture group, the microfilament connections between cells were
significantly increased (P<0.05) and the cells were dense. The
results were similar to those of the study by Jusoh et al
(15). In addition, the number of
BMECs co-cultured with HA also increased significantly (P<0.05).
Considering the space in the channel and the concentration of
injected cells, the physical structure of HA may provide a more
favorable growth environment for cells. HA was considered to be
able to keep BMECs alive rather than to promote cell division. The
reason for the higher number of cells in the HA group was that more
cells in the control group died. In the presence of HA, Whisler
et al (16) also co-cultured
the HUVECs and pulmonary fibroblasts in a microfluidic chip and
concluded that pulmonary fibroblasts were useful for maintaining
the morphology of vascular cells and prolonging the survival time
of vascular cells in the chip for up to 4 days. A notable
phenomenon was identified in the present study as in that the
morphological structure of the HUVECs being not as healthy as that
of the cells in the study by Whisler et al (16), and no vascular lumen was observed as
it was in their study. However, the morphological structure of
BMECs was similar to that of the HUVECs in the study by Whisler
et al (16), particularly in
Group B. The cells formed a structure similar to that of the
circular lumen, which is similar to the image published in the
literature, and even the panoramic image. Given that both BMECs and
HUVECs are essentially vascular endothelial cells, just derived
from different parts of the human body, it is understandable that
their morphology and structure were similar.
In the present study, although there was not a very
typical three-dimensional structure, fluorescence staining
indicated that the cells were connected by microfilaments in the
matrix collagen. Furthermore, the morphology was somewhat different
from that of cells in ordinary Petri dishes. The major
characteristics of the present microfluidic organ-on-a-chip were as
follows: i) Fluidity. The cells were suspended in the matrix
collagen, were able to move slightly and grew in all directions,
not limited to a plane. ii) Independence. The microfluidic
organ-on-a-chip had four channels, each of which was able to be
used for liquid injection. However, due to the effect of surface
tension of the liquids, there would be no leakage between the empty
and liquid channels. iii) Feasibility of intervention. Adjacent
channels were available for the addition of different drugs and
other interventions, as well as for changes in the medium or the
final addition required for fluorescent staining.
Apoptosis is programmed cell death regulated by
apoptosis-associated genes in order to maintain the stability and
integrity of the genome. Apoptosis involves the activation,
expression and regulation of a series of genes and it differs from
cell necrosis in morphology and biochemistry. The caspase family is
a cysteine protease system with a specific aspartic cysteine that
is a key player in apoptosis (17,18).
To date, ≥14 caspase members have been implicated in apoptosis in
mammals. The stimulation of various proteases also activates
caspase-induced apoptosis through different pathways. However,
mammalian cell apoptosis is effectuated via caspase-3. The
increased expression and stimulation of caspase-3 is a central link
in the early initiation of the apoptotic signaling pathway and is
also one of the vital proteases involved in the regulation and
execution of apoptosis (19).
Microfluidic organ-on-a-chip provided a new
environment for the growth of cells. In the chip, it was indicated
that the microfilaments between cells not only increased but it was
also easier for the cells to form tubular structures between each
other. In some areas, the formation of tubular structures between
only three cells was observed. In petri dishes, cells attach
themselves to the flat surface of the dish and could only grow in
the directions on one surface to touch the surrounding cells. In
the chip, cells could grow in all directions in a three-dimensional
environment, touching cells all around rather than being confined
to a single plane. This increased the probability of contact
between cells and made it easier for them to form tubular
structures. The chip, combined with confocal microscopy, provides
high-resolution morphological results for cellular experiments. In
the experiment of the present study, the appearance of apoptotic
proteins (caspase-3/7) was able to be directly observed in the
microfluidic organ-on-a-chip and it was also identified that the
glucocorticoid caused the apoptosis of BMECs. The comparison not
only revealed a significant increase in the cleaved caspase-3/7
content (P<0.05), but its release from the nucleus was also
directly visible to the naked eye. The glucocorticoid-treated BMECs
produced more cleaved caspase-3/7, and with the increase of the
glucocorticoid concentration, cleaved caspase-3/7 also increased
significantly (P<0.05). Particularly in the high-concentration
glucocorticoid intervention group, in addition to high expression
of cleaved caspase-3/7, BMECs also exhibited obvious atrophy in
morphology; the proportional area of the whole cytoskeleton was
significantly smaller than that of the other three groups
(P<0.05). The microfilaments all disappeared, the cells became
round, and the whole cell appeared to be on the verge of death.
Furthermore, the number of cells in this group increased
significantly compared with that in the other groups (P<0.05).
Combined with cell morphology, high-concentration glucocorticoid
resulted in cell atrophy and volume reduction; therefore, the
number of cells increased in the same field area. Furthermore, the
concentration of dexamethasone used in the present study was
selected according to our previous experiment (8-10).
The concentration of drug used in the present study was slightly
higher than the previously used concentration of 0.1 or 0.2 mg/ml
for two main reasons. First, different from using a culture dish,
the ECM containing dexamethasone was added to the chip through
channels and the volume of the two channels of the ECM and the cell
directly diluted the concentration of dexamethasone by 50%.
Furthermore, in an ordinary culture dish, cells are in direct
contact with dexamethasone in the ECM, while in the microfluidic
chip, cells are suspended in the matrix collagen and dexamethasone
is required to permeate through the matrix collagen to be in
contact with the cells. Therefore, a higher concentration is
required to achieve the same effect as in an ordinary culture dish.
In addition, the concentrations of 0.4 and 0.6 mg/ml used in the
present study were low for the human body but were sufficiently
high to be lethal for the cells in vitro. The purpose of the
present study was to investigate the mechanism of cell damage in
order to guide clinical treatment, and it is difficult to directly
calculate the appropriate clinical drug concentration through cell
experiments. Therefore, further investigation may be required to
determine a more appropriate dose to be used in the clinic.
Several mechanisms are able to induce apoptosis, and
it has been well observed that glucocorticoid induces apoptosis of
BMECs (10). However, specific
mechanisms remain elusive and should be clarified to improve
treatment approaches. It has been speculated that oxidative stress
is the primary mechanism to trigger apoptosis. In the case of
oxidative stress, excessive reactive oxygen species (ROS) cause
cell damage and the body forms a complex oxidative stress response
system against ROS damage (20).
Tang et al (21) reported
that polychlorinated biphenyl 118 is able to elevate the ROS levels
in HUVECs to initiate apoptosis. In addition to directly triggering
apoptosis, ROS accumulation may activate a series of signaling
pathways, leading to the dysregulation of apoptosis and excessive
cell death. Excessive apoptosis leads to autoimmune diseases and
inflammation, which are associated with various acute and chronic
diseases (22). ROS generated by
oxidative stress causes cell damage, which in turn leads to
excessive accumulation of ROS. The accumulated ROS induces
excessive apoptosis, which further aggravates damage (23).
Icariin is a flavonoid compound that has biological
activities, including immune regulation, anti-tumor, anti-aging and
cardiovascular effects. Previously, Zhao et al (10) studied the protective effect of
icariin on glucocorticoid-treated BMECs. Using the TUNEL method, it
was identified that icariin inhibited apoptosis of BMECs by
glucocorticoids. In addition, Hu et al (24) revealed that icariin prevented the
injury and apoptosis of HUVECs by regulating caspase-3 and B-cell
lymphoma 2. In the icariin-protected group of the present study, it
was observed that although a considerable amount of cleaved
caspase-3/7 was produced in the cytoplasm, the content was
significantly lower than that in the high-concentration
glucocorticoid group (P<0.05). In addition, the microfilamentous
structural connections between the cells were similar to those in
the other groups and the cells did not shrink. Furthermore, the
area of the whole cytoskeleton was not significantly different
compared with that in the blank group (P>0.05). This suggested
that advanced protection with icariin reduced the damaging effect
of glucocorticoid on the cells and protected the microvascular
structure. Chen et al (25)
investigated the protective mechanism of icariin by using
endothelial cells from rat femurs. It was identified that icariin
had a protective effect on the high glucose-induced endothelial
cells by inhibiting p38/cyclic AMP response element-binding protein
and activating the Akt/eNOS/NO pathway. Xiao et al (26) also reported that icariin decreased
ROS production in HUVECs. Wang et al (27) summarized the various functions of
icariin, including attenuation of hydrogen peroxide impairment,
improvement of cell viability, inhibition of cell apoptosis and
increase of superoxide dismutase and glutathione peroxidase
activity. It may be suggested that further studies are required in
the future to demonstrate the inhibitory effect of icariin on cell
apoptosis, as well as its cellular protective mechanisms.
The advantage of the microfluidic organ-on-a-chip
used in the present study was that it was easy to operate. Through
adjacent channels, it was possible to add various substances used
to directly stain the cells in situ and target the exact
position in the cell. As for caspase-3, in the present experiment,
it is well known that western blot analysis is able to analyze
intracellular proteins and detect the types and content of
proteins. A microfluidic organ-on-a-chip can be an excellent
supplement to western blot analysis. The target protein may be
observed directly with the naked eye by staining, and at the same
time, a simple content comparison may be performed. It is also
possible to determine whether the location of the target protein is
consistent with the theory and it provides a high-resolution
morphological visualization for cell experiments.
In conclusion, from the perspective of morphology,
BMECs were more likely to survive compared with HUVECs in the chip
used in the present study and HA was able to promote the growth of
BMECs through the microfluidic platform. Glucocorticoids were able
to damage cells through the production of cleaved caspase 3/7 and
icariin protected the cells to a certain extent and reduced the
damage of glucocorticoids to cells.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by the Beijing Natural Science
Foundation (grant no. 7182146), the Biomedical Translational
Engineering Research Center of BUCT-CJFH (grant no. RZ2020-02) and
the National Natural Science Foundation of China (grant nos.
81672236, 81802224 and 81871830).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YD and WS were responsible for designing and
directing the whole experiment. TL was responsible for performing
the cell experiment, collecting the data and writing the initial
manuscript. YL was responsible for designing and making
microfluidic chips, and writing the initial manuscript. QZ was
responsible for isolating cells, analyzing the data, and reviewing
the manuscript. TL, YL, QZ, YD and WS confirmed the authenticity of
all the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The experiment was approved by the Ethics Committee
of China-Japan Friendship Hospital (Beijing, China) and all
patients donated voluntarily with written informed consent.
Patient consent for publication
The patients provided consent for publication.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Zhao D, Liu Y, Ma C, Gu G and Han DF: A
mini review: Stem cell therapy for osteonecrosis of the femoral
head and pharmacological aspects. Curr Pharm Des. 25:1099–1104.
2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Maruyama M, Lin T, Pan CC, Moeinzadeh S,
Takagi M, Yang YP and Goodman SB: Cell-Based and scaffold-based
therapies for joint preservation in early-stage osteonecrosis of
the femoral head: A review of basic research. JBJS Rev.
7(e5)2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Yu Q, Guo W, Shen J and Lv Y: Effect of
glucocorticoids on lncRNA and mRNA expression profiles of the bone
microcirculatory endothelial cells from femur head of Homo sapiens.
Genom Data. 4:140–142. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Yu QS, Guo WS, Cheng LM, Lu YF, Shen JY
and Li P: Glucocorticoids significantly influence the transcriptome
of bone microvascular endothelial cells of human femoral head. Chin
Med J (Engl). 128:1956–1963. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Urbaczek AC, Leão PAGC, Souza FZR, Afonso
A, Vieira Alberice J, Cappelini LTD, Carlos IZ and Carrilho E:
Endothelial cell culture under perfusion on A polyester-toner
microfluidic device. Sci Rep. 7(10466)2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
van Engeland NCA, Pollet AMAO, den Toonder
JMJ, Bouten CVC, Stassen OMJA and Sahlgren CM: A biomimetic
microfluidic model to study signalling between endothelial and
vascular smooth muscle cells under hemodynamic conditions. Lab
Chip. 18:1607–1620. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
van der Helm MW, van der Meer AD, Eijkel
JC, van den Berg A and Segerink LI: Microfluidic organ-on-chip
technology for blood-brain barrier research. Tissue Barriers.
4(e1142493)2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Yu Q, Guo W, Cheng L, Lu Y and Li P:
Preliminary study of impact of steroids on expression profile and
transcriptome of bone microvascular endothelial cells. Zhonghua Yi
Xue Za Zhi. 94:3817–3820. 2014.PubMed/NCBI(In Chinese).
|
|
9
|
Zhang Q, Gao F, Cheng L, Liu L, Sun W and
Li Z: Effects of icariin on autophagy and exosome production of
bone microvascular endothelial cells. Zhongguo Xiu Fu Chong Jian
Wai Ke Za Zhi. 33:568–577. 2019.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
10
|
Zhao DY, Yu QS, Guo WS and Cheng LM:
Effect of icariin on the proteomic expression profile of bone
microvascular endothelial cells of human femoral head against
steroids-induced lesion. Zhonghua Yi Xue Za Zhi. 96:1026–1030.
2016.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
11
|
Wang X, Sun Q and Pei J:
Microfluidic-Based 3D engineered microvascular networks and their
applications in vascularized microtumor models. Micromachines
(Basel). 9(493)2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhang Z, Chen YC, Cheng YH, Luan Y and
Yoon E: Microfluidics 3D gel-island chip for single cell isolation
and lineage-dependent drug responses study. Lab Chip. 16:2504–2512.
2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Rh Owen G, Dard M and Larjava H:
Hydoxyapatite/beta-tricalcium phosphate biphasic ceramics as
regenerative material for the repair of complex bone defects. J
Biomed Mater Res B Appl Biomater. 106:2493–2512. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ramesh N, Moratti SC and Dias GJ:
Hydroxyapatite-polymer biocomposites for bone regeneration: A
review of current trends. J Biomed Mater Res B Appl Biomater.
106:2046–2057. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Jusoh N, Oh S, Kim S, Kim J and Jeon NL:
Microfluidic vascularized bone tissue model with
hydroxyapatite-incorporated extracellular matrix. Lab Chip.
15:3984–3988. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Whisler JA, Chen MB and Kamm RD: Control
of perfusable microvascular network morphology using a multiculture
microfluidic system. Tissue Eng Part C Methods. 20:543–552.
2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Chen P, Zhang H, Zhang Q, Zhou W, Deng Y,
Hu X and Zhang L: Basic Fibroblast growth factor reduces
permeability and apoptosis of human brain microvascular endothelial
cells in response to oxygen and glucose deprivation followed by
reoxygenation via the fibroblast growth factor receptor 1
(FGFR1)/ERK pathway. Med Sci Monit. 25:7191–7201. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chen X, Jiang Y, Wang J, Liu Y, Xiao M,
Song C, Bai Y, Yinuo Han N and Han F: Synapse impairment associated
with enhanced apoptosis in post-traumatic stress disorder. Synapse.
74(e22134)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Gao X, Zhang Y, Zhang R, Zhao Z, Zhang H,
Wu J, Shen W and Zhong M: Cyclin-dependent kinase 1 disruption
inhibits angiogenesis by inducing cell cycle arrest and apoptosis.
Exp Ther Med. 18:3062–3070. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Yuan W, Chang H, Liu X, Wang S, Liu H and
Xuan H: Brazilian green propolis inhibits Ox-LDL-Stimulated
Oxidative Stress In Human Umbilical Vein Endothelial Cells Partly
through PI3K/Akt/mTOR-Mediated Nrf2/HO-1 Pathway. Evid Based
Complement Alternat Med. 2019(5789574)2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Tang L, Cheng JN, Long Y, He XM, Liang GN,
Tang XP, Jiang CX and Chen F: PCB 118-induced endothelial cell
apoptosis is partially mediated by excessive ROS production.
Toxicol Mech Methods. 27:394–399. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Munoz M, López-Oliva ME, Rodríguez C,
Martínez MP, Sáenz-Medina J, Sánchez A, Climent B, Benedito S,
García-Sacristán A, Rivera L, et al: Differential contribution of
Nox1, Nox2 and Nox4 to kidney vascular oxidative stress and
endothelial dysfunction in obesity. Redox Biol.
28(101330)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Fernandez-Bertolez N, Costa C, Bessa MJ,
Park M, Carriere M, Dussert F, Teixeira JP, Pásaro E, Laffon B and
Valdiglesias V: Assessment of oxidative damage induced by iron
oxide nanoparticles on different nervous system cells. Mutat Res.
845(402989)2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Hu Y, Li H, Liu K, Zhang Y, Ren L and Fan
Z: Protective effects of icariin on human vascular endothelial
cells induced by oxidized low-density lipoprotein via modulating
caspase-3 and Bcl-2. Mol Med Rep. 17:6835–6839. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Chen S, Wang Z, Zhou H, He B, Hu D and
Jiang H: Icariin reduces high glucose-induced endothelial
progenitor cell dysfunction via inhibiting the p38/CREB pathway and
activating the Akt/eNOS/NO pathway. Exp Ther Med. 18:4774–4780.
2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Xiao HB, Liu ZK, Lu XY, Deng CN and Luo
ZF: Icariin regulates PRMT/ADMA/DDAH pathway to improve endothelial
function. Pharmacol Rep. 67:1147–1154. 2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wang FY, Jia J, Song HH, Jia CM, Chen CB
and Ma J: Icariin protects vascular endothelial cells from
oxidative stress through inhibiting endoplasmic reticulum stress. J
Integr Med. 17:205–212. 2019.PubMed/NCBI View Article : Google Scholar
|