Introduction
Persistent and progressive liver injury results in
the inability of the liver to regenerate and leads to liver
fibrosis (1). Liver fibrosis is a
wound healing response that is closely associated with hepatic
stellate cells (HSCs) and Kupffer cells (2). The activation and accumulation of a
large number of HSCs is a pivotal step in the formation and
progression of liver fibrosis (3).
Initiation and perpetuation are two stages that constitute the
activation of HSCs (4). Persistent
activation of HSCs can contribute to cellular events, including
extracellular matrix degradation and cytokine release, which result
in the production and accumulation of fibrotic extracellular matrix
components, including α-smooth muscle actin (α-SMA) and collagen
type I α1 (COL1A1) (5).
Additionally, HSCs can synthesize and secrete TIMP metallopeptidase
inhibitor 1 (TIMP1), which is a powerful inhibitor of enzymes that
degrade matrix molecules and serves a critical role in fibrosis
(6). Overall, liver fibrosis has
become an obstacle in clinical trials or during treatment of
patients with chronic liver diseases (7). Therefore, there is an urgent need to
identify novel and effective treatment strategies for this
disease.
Interleukin (IL)-22 was originally identified as an
IL-10-related T cell-derived inducible factor (8). IL-22 is an actively secreted protein
of 146 amino acids, which belongs to the IL-10 family of cytokines
(9,10). Despite the differences noted in
primary sequences among IL-10 members, they share a common gene
structure containing six helices (11). IL-22 expression is downregulated in
the serum of mice and in diabetic nephropathy patients (12). By binding to the IL-22 receptor
subunit α-1 (IL-22RA1) and IL-10R2, IL-22 exerts protective effects
on hepatocytes (13). It has been
shown that the binding of IL-22 to IL-22RA1 can induce senescence
of HSCs and ameliorate liver fibrosis (14). However, the activation of several
signaling pathways, such as STAT3, SOCS3, p53 and Notch, induced by
IL-22 inhibits the progression of liver damage (15).
Oxidative stress serves a crucial role in inducing
HSC activation and fibrogenic potential (16). Superoxide dismutase (SOD), one of
the most important protective enzymes, provides the first
antioxidant defense system in various organs and tissues, including
the liver (17). Glutathione (GSH)
is a key reactive oxygen species scavenger to protect liver cells
from oxidative stress (18). SOD
and GSH serve an important role in the antioxidant defense system,
while malondialdehyde (MDA) is considered to be the main marker of
lipid peroxidation in tissues (19). Inflammation serves a critical role
in the progression of liver injury and fibrosis. NOD-like receptor
protein 3 (NLRP3) is a well-established inflammasome that consists
of apoptosis-associated speck-like protein and the effector
molecule pro-caspase-1(20). Recent
studies have confirmed the role of NLRP3 in a variety of liver
diseases, such as alcoholic and non-alcoholic fatty liver diseases
and non-alcoholic steatohepatitis (21,22).
Inhibition of the NLRP3 inflammasome can alleviate renal injury and
fibrosis (23). NLRP3 is aberrantly
activated in vivo and in vitro following exposure to
aristolochic acid, and inhibition of NLRP3 ameliorates renal
fibrosis and renal failure (24).
However, whether IL-22 ameliorates liver fibrosis by directly
inhibiting the activation of NLRP3 has not been previously
investigated.
In the present study, the role of IL-22 was
investigated in transforming growth factor β (TGF-β)-induced HSCs.
Whether IL-22 exerted its effects on liver fibrosis via the
regulation of NLRP3 inflammasome signaling was also explored. The
current study may guide the future exploration of the pathogenesis
of liver fibrosis.
Materials and methods
Cell culture and treatment
HSCs were purchased from the American Type Culture
Collection and Nigericin (cat. no. N7143) was purchased from
Sigma-Aldrich (Merck KGaA). HSCs were cultured in DMEM supplemented
with 10% FBS (both Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml
penicillin and 100 mg/ml streptomycin in a PBS buffer (R&D
Systems, Inc.) at 37˚C in an incubator with 95% air/5%
CO2. The cells were treated with 5 ng/ml TGF-β
(Sigma-Aldrich; Merck KGaA) at 37˚C for 24 h and subsequently
cultured in the presence of 250, 500, 750 and 1,000 pg/ml IL-22
(Sigma-Aldrich; Merck KGaA) for 48 h at 37˚C (25,26).
Finally, the cells were treated with Nigericin (40 µM) for 30 min
at 37˚C to activate NLRP3 for subsequent experiments (27).
Cell Counting Kit-8 (CCK-8) assay
The CCK-8 assay was conducted for the determination
of cell viability. Briefly, HSCs (3x104 cells/well) were
inoculated into a 96-well plate at 37˚C. At 48 h after IL-22
treatment, 10 µl CCK-8 reagent (Dojindo Molecular Technologies,
Inc.) was added to each well and incubated for 3 h. The absorbance
at 450 nm was read using a microplate reader (Bio-Rad Laboratories,
Inc.). Three replicates were conducted for each sample.
Apoptosis assay
Apoptosis was evaluated via flow cytometry. HSCs
were collected and then re-suspended in 1X binding buffer to a
concentration of 1x104 cells/well. An Annexin V-FITC
Staining Cell Apoptosis Detection kit (Nanjing KeyGen Biotech Co.,
Ltd.) was employed to determine apoptosis in accordance with the
manufacturer's protocol. Briefly, cells were stained with Annexin
V-FITC and PI, and incubated in the dark for 15 min at 37˚C. Cell
apoptosis of each sample was assessed by flow cytometry (BD Accuri™
C6; BD Biosciences). Apoptosis was analyzed using FlowJo software
(version 7.6.3; FlowJo LLC).
Detection of oxidative stress
The contents of MDA and GSH, as well as the activity
of SOD, were respectively detected using the MDA assay kit (cat.
no. A003-4-1), GSH assay kit (cat. no. A006-2-1) and SOD assay kit
(cat. no. A001-3-2; all from Nanjing Jiancheng Bioengineering
Institute) in accordance with the manufacturer's instructions.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from cells using
TRIzol® reagent (Sigma-Aldrich; Merck KGaA). cDNA was
synthesized using a Sensiscript RT kit (Qiagen GmbH) following
manufacturer's recommendations. According to the manufacturer's
recommendations, PCR assays were conducted using
SYBR-Green® Realtime PCR Master Mix and Premix Ex Taq
(Takara Bio., Inc.) on ViiA™ 7 Real-Time PCR System (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The following
thermocycling conditions were used: 5 min at 95˚C, followed by 35
cycles at 95˚C for 15 sec, 40 sec at 55˚C and 72˚C for 1 min.
Relative quantification of gene expression was performed using the
2−ΔΔCq method (28). The
relative gene expression levels were normalized to those of GAPDH.
Three independent experiments were conducted for each target gene.
The sequences of the gene-specific primers used in the present
study were as follows: α-SMA forward, 5'-TCCAGAGTC
CAGCACAATACCAG-3' and reverse, 5'-AATGACCCAGAT TATGTTTGAGACC-3';
COL1A1 forward, 5'-TCAGGGGCG AAGGCAACAGT-3' and reverse,
5'-TTGGGATGGAGG GAGTTTACACGA-3'; TIMP1 forward, 5'-ACCACCTTA
TACCAGCGTTATGA-3' and reverse, 5'-GGTGTAGACGAA CCGGATGTC-3'; GAPDH
forward, 5'-CTCACCGGATGC ACCAATGTT-3' and reverse,
5'-CGCGTTGCTCACAAT GTTCAT-3'.
Western blot analysis
HSCs were lysed in radioimmunoprecipitation (RIPA)
lysis buffer (Beyotime Institute of Biotechnology) and the
concentrations of the proteins were measured using a BCA protein
assay (Shanghai Yeasen Biotechnology Co., Ltd.). Proteins (40
µg/lane) were separated via 10% SDS-PAGE and were transferred to
PVDF membranes, which were then blocked with 5% skimmed milk for 1
h at room temperature. Subsequently, the membranes were incubated
at 4˚C overnight with primary antibodies (all 1:1,000) against
NLRP3 (cat. no. 15101S), IL-1β (cat. no. 12703S), caspase-1 (cat.
no. 3866T), α-SMA (cat. no. 19245T), COL1A1 (cat. no. 72026T),
TIMP1 (cat. no. 8946S) and GAPDH (cat. no. 5174T; all from Cell
Signaling Technology, Inc.). The membranes were washed three times
with TBS-0.2% Tween 20 before incubation with goat anti-rabbit
HRP-conjugated secondary antibody (1:3,000; cat. no. 7074S; Cell
Signaling Technology, Inc.) for 1 h at room temperature. The
expression levels of the targeted proteins were determined using a
Bio-Rad ChemiDoc MP imaging system (Bio-Rad Laboratories, Inc.).
The bands were detected using an enhanced chemiluminescence reagent
(EMD Millipore). The grayscale values of the membranes were
semi-quantified using ImageJ software (version 1.52r; National
Institutes of Health). GAPDH was used as an internal control.
Statistical analysis
Data were analyzed using GraphPad Prism version 6.0
(GraphPad Software, Inc.). Data were expressed as the mean ± SD.
All experiments were performed three times. Comparisons between two
groups were conducted by unpaired Student's t-test, while
comparisons among multiple groups were performed by one-way ANOVA
followed by Turkey's post-hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
IL-22 attenuates HSC viability and
oxidative stress activated by TGF-β
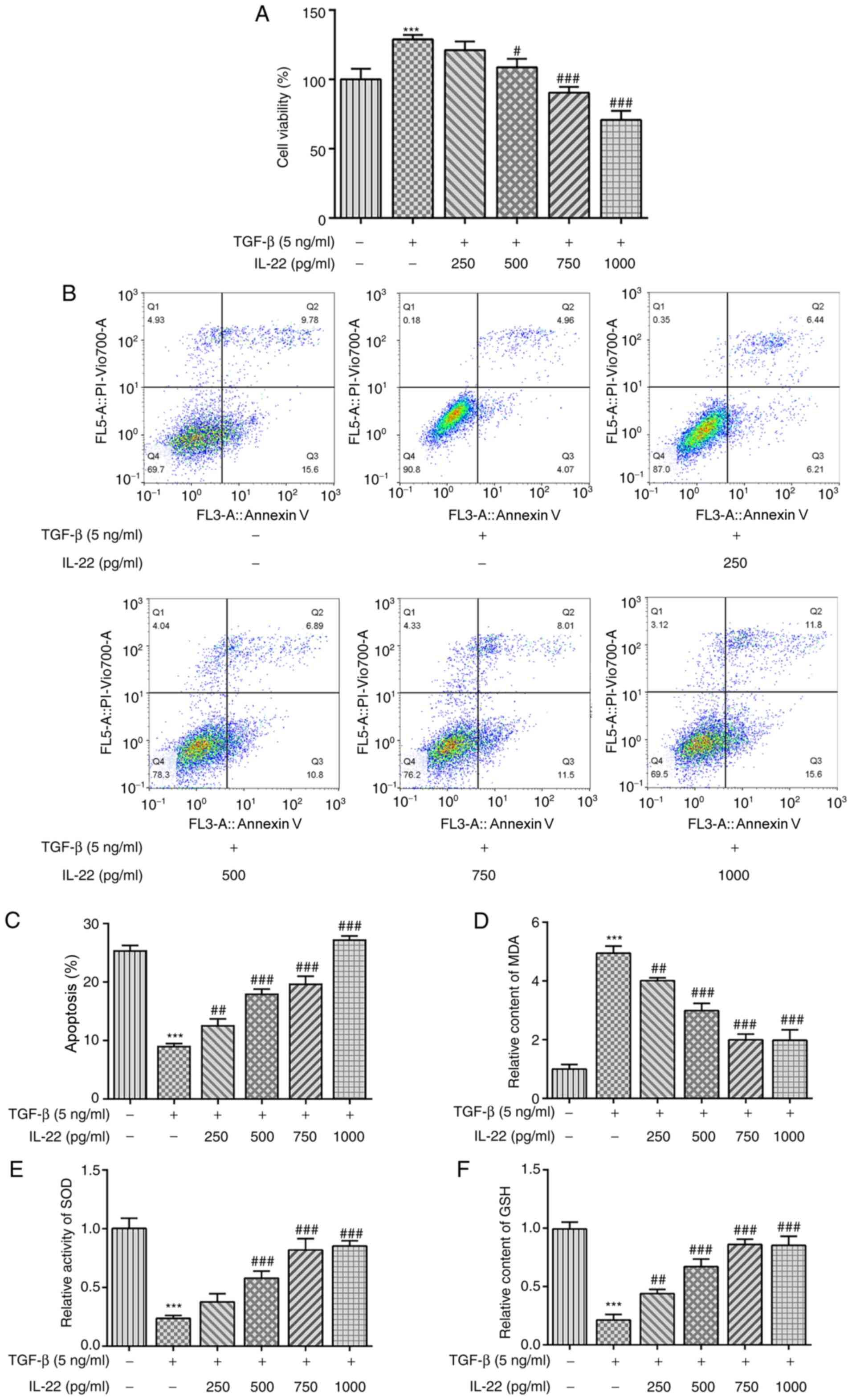
Following incubation of HSCs with TGF-β, their
viability was significantly increased, while concomitant IL-22
treatment led to significant decreases in HSC viability in a
dose-dependent manner (Fig. 1A). As
shown in Fig. 1B and C, TGF-β treatment significantly inhibited
the apoptosis of HSCs compared with the untreated group. By
contrast, IL-22 significantly increased TGF-β-induced apoptosis in
a dose-dependent manner (Fig. 1B
and C). Subsequently, factors
associated with oxidative stress were measured using commercially
available kits. As shown in Fig.
1D, TGF-β stimulation caused a significant increase in the
relative content of MDA, whereas increasing concentrations of IL-22
led to a gradual decrease in MDA levels. Conversely, the levels of
SOD and GSH were significantly inhibited following TGF-β treatment,
whereas IL-22 restored their levels in a dose-dependent manner
(Fig. 1E and F). Overall, these findings revealed that
IL-22 inhibited viability and oxidative stress in HSCs induced by
TGF-β.
IL-22 alleviates fibrosis of HSCs
activated by TGF-β
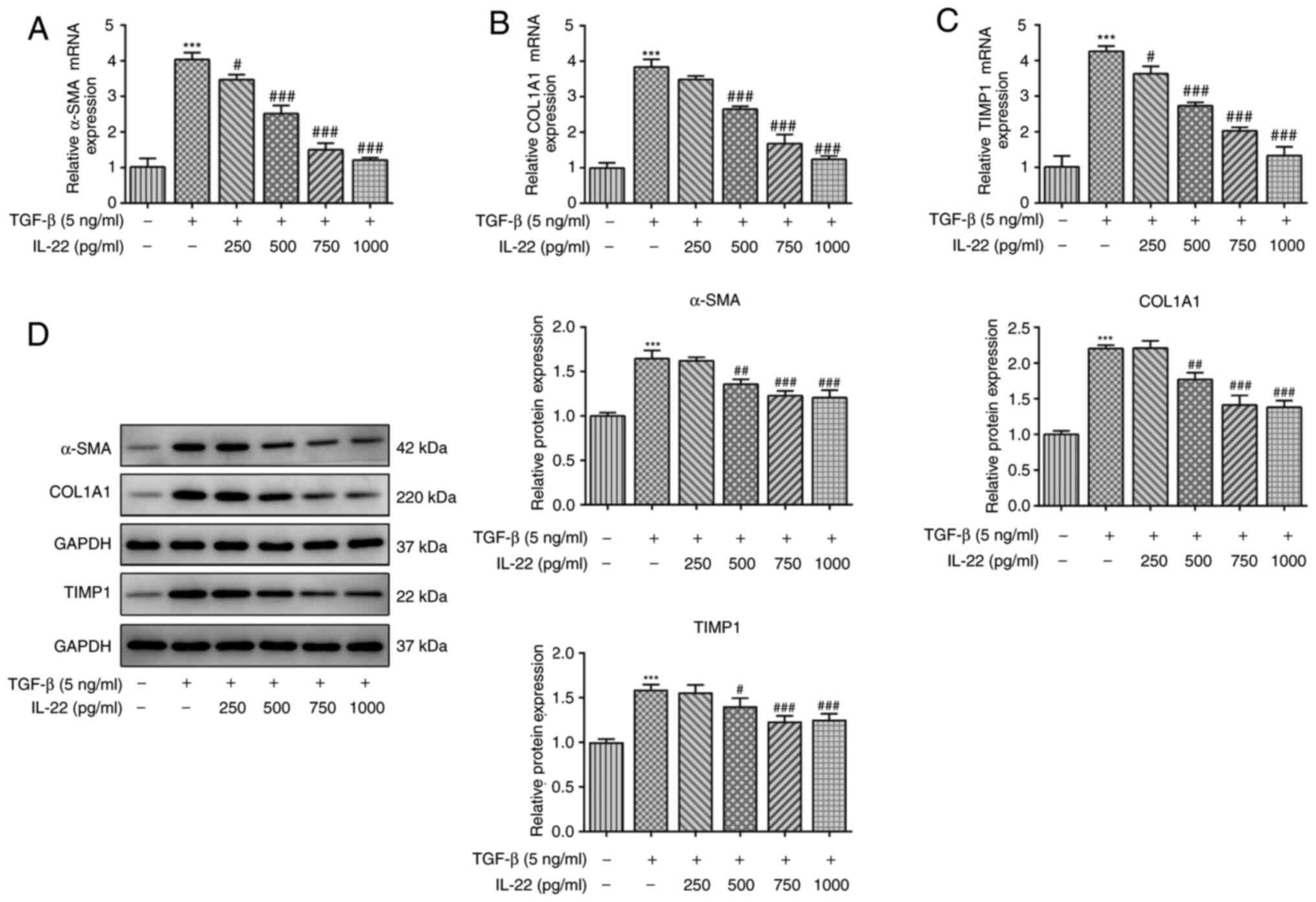
Subsequently, the ability of IL-22 to inhibit HSC
fibrosis activated by TGF-β was assessed. To confirm this
hypothesis, RT-qPCR and western blot analyses were conducted to
assess the expression levels of fibrosis-associated genes. TGF-β
activation in HSCs significantly enhanced the mRNA and protein
expression levels of α-SMA, COL1A1 and TIMP1 compared with the
levels in untreated HSCs (Fig.
2A-D). In addition, IL-22 treatment of HSCs activated by TGF-β
caused a downregulation in the expression levels of the
aforementioned fibrosis-associated factors (Fig. 2A-D). Therefore, the results
indicated that IL-22 suppressed HSC fibrosis activated by
TGF-β.
IL-22 inhibits NLRP3 inflammasome
signaling in TGF-β-induced HSCs
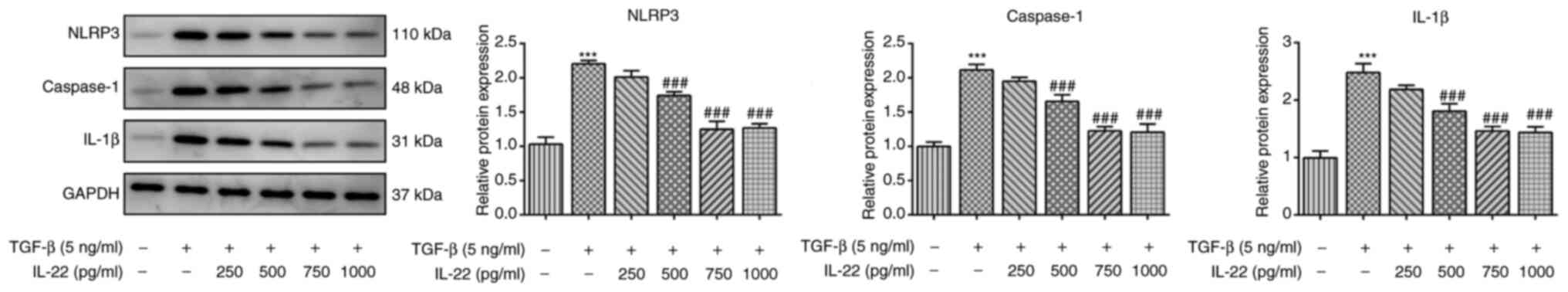
Subsequently, the expression levels of key proteins
involved in the NLRP3 inflammasome signaling pathway were detected
by western blot analysis in TGF-β-induced HSCs in order to confirm
whether IL-22 could alleviate inflammation of TGF-β-induced HSCs.
It was observed that TGF-β caused a significant increase in the
expression levels of NLRP3, caspase-1 and IL-1β, and this effect
was reversed by IL-22 (Fig. 3).
Therefore, the results suggested that IL-22 inhibited inflammation
of TGF-β-induced HSCs. Treatment of the cells with 500, 750 and
1,000 ng/ml IL-22 resulted in a significant decrease of the
expression levels of the inflammatory factors. Therefore, IL-22 was
used at a concentration of 750 ng/ml in subsequent experiments.
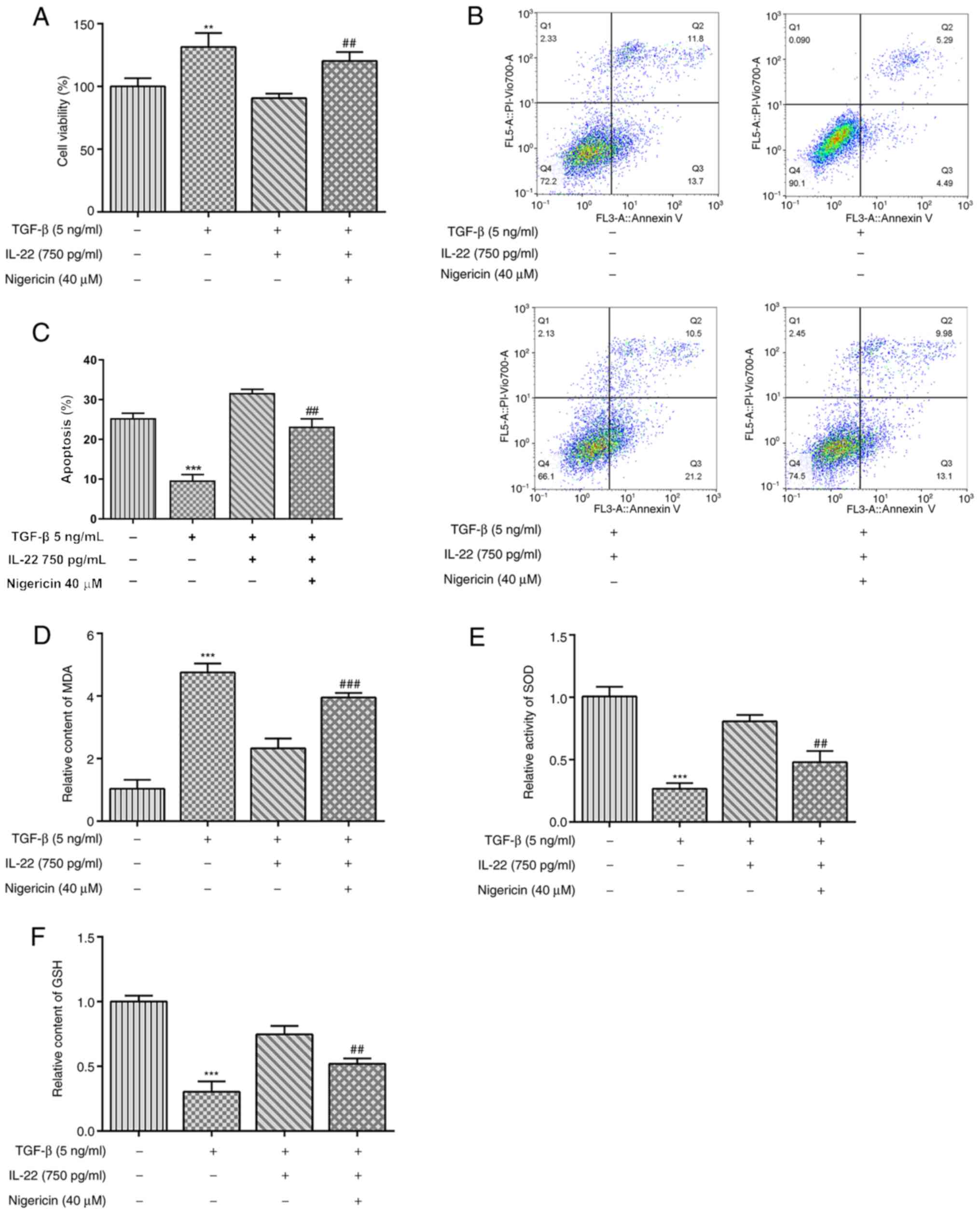
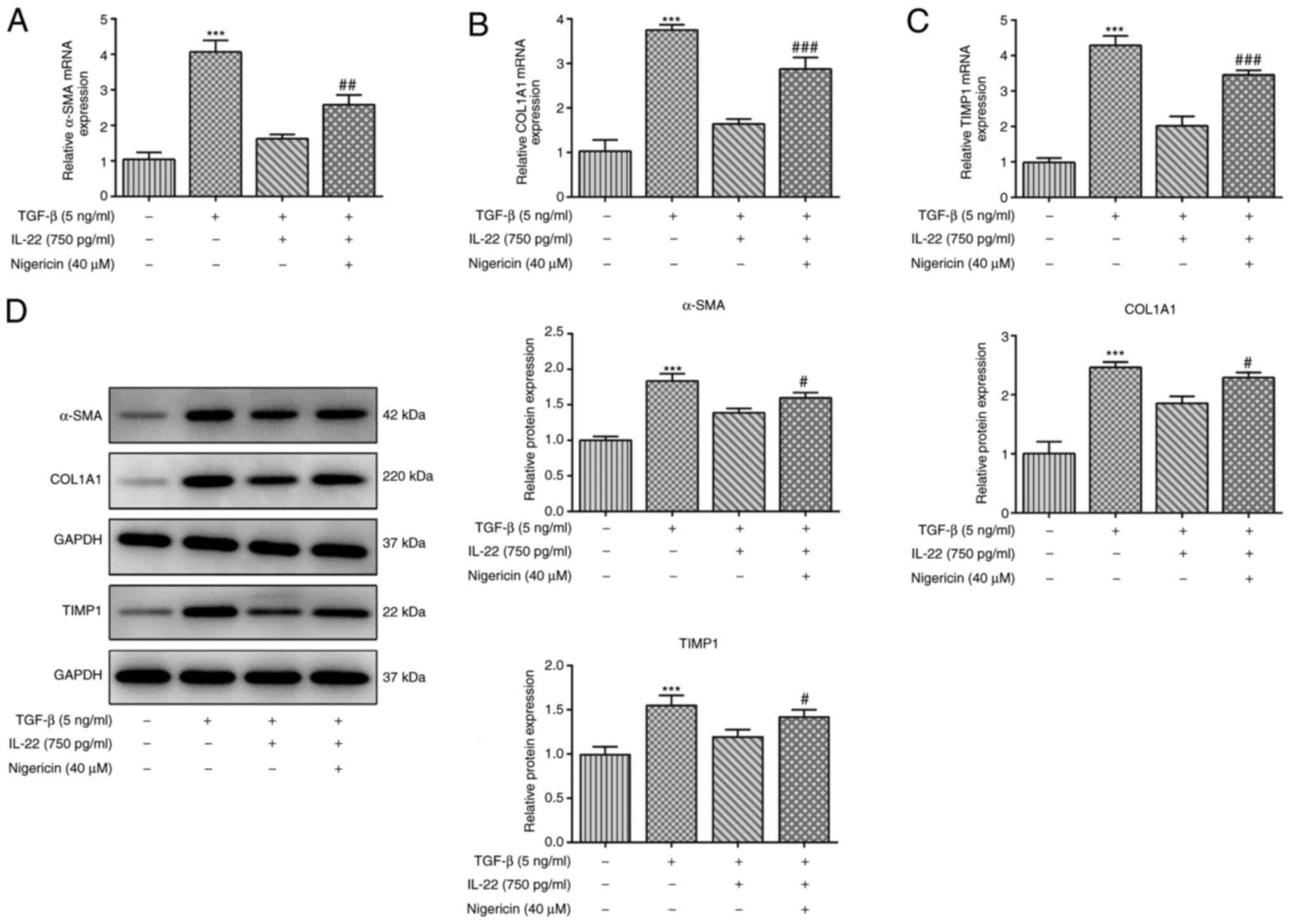
Nigericin reverses the inhibitory
effects of IL-22 on oxidative stress and fibrosis in HSCs
stimulated by TGF-β
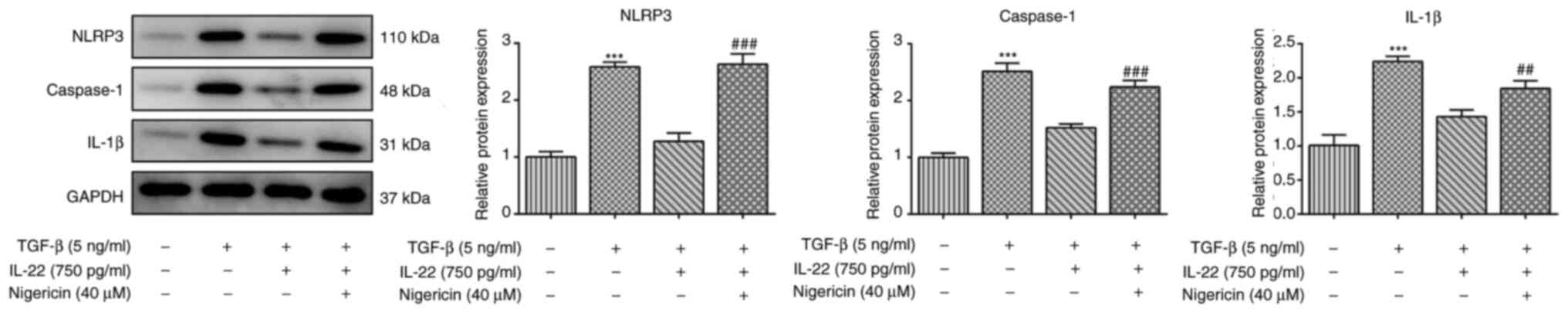
To further investigate the exact mechanism by which
IL-22 exerts its cytoprotective effects, the NLRP3 activator
Nigericin was used to treat HSCs. TGF-β significantly upregulated
the protein expression levels of NLRP3, caspase-1 and IL-1β, and
this effect was reversed by co-treatment with TGF-β and IL-22
(Fig. 4). However, the addition of
Nigericin in HSCs co-treated with TGF-β and IL-22 significantly
increased the expression levels of NLRP3, caspase-1 and IL-1β
compared with HSCs treated with TGF-β and IL-22 (Fig. 4). Similarly, CCK-8 and flow
cytometry assays indicated that Nigericin treatment reversed the
inhibition of IL-22 on cell viability and apoptosis of
TGF-β-induced HSCs (Fig. 5A-C). In
addition, the levels of the oxidative stress indices measured in
the present study were significantly reversed following Nigericin
treatment in TGF-β- and IL-22-induced HSCs compared with TGF-β and
IL-22 treatment (Fig. 5D-F).
Moreover, the results from the RT-qPCR and western blot analyses
suggested that the IL-22-induced decreases in the mRNA and protein
expression levels of α-SMA, COL1A and TIMP1 in TGF-β-induced HSCs
were significantly reversed by Nigericin (Fig. 6A-D). The present results indicated
that Nigericin reversed the inhibitory effects of IL-22 on the
induction of oxidative stress and fibrosis in HSCs stimulated by
TGF-β.
Discussion
HSCs are a type of non-parenchymal cells that are
important members in the occurrence and development of liver
fibrosis and cirrhosis (29). The
exposure of HSCs to TGF-β transforms their still state into an
activated cell form, where HSCs transdifferentiate into
proliferative myofibroblasts with pro-fibrogenic transcriptional
and secretory properties that are vital for the development of
liver fibrosis and collagen deposition (30,31).
Therefore, the present study used HSCs activated by TGF-β for the
simulation of liver fibrosis in vitro.
IL-22 is a factor that protects liver tissues from
the development of various chronic diseases, such as nonalcoholic
steatohepatitis, liver fibrosis and hepatocellular carcinoma
(15,32). IL-22 can promote liver repair and
tissue regeneration by stimulating the proliferation and survival
of hepatocytes (33). A previous
study has suggested that IL-22 exerts inhibitory effects on liver
fibrosis and that the attenuation of HSC activation and inhibition
of inflammatory factors by IL-22 can restrict liver fibrogenesis
(34). Overexpression of IL-22
protects mice from liver injury and liver apoptosis and necrosis
stimulated by various factors (35). In the present study, IL-22 inhibited
the proliferation of HSCs and the development of liver fibrosis
in vitro, which was consistent with the aforementioned
studies. Immune response and inflammation in the liver are
contributors of hepatocyte injury and activation of HSCs (29). The induction of oxidative stress and
inflammation activated by TGF-β in HSCs was attenuated by IL-22 in
the present study.
NLRP3 is an intracellular multi-protein complex, and
its activation, which occurs by various types of stimuli, such as
endogenous danger signals and environmental irritants, can lead to
host defense against microbial infections, activation of caspase-1
and further induction of maturation and secretion of IL-1β and
IL-18. This process initiates inflammation (36,37).
Inhibition or inactivation of the NLRP3 inflammasome is suggested
to enhance lipid metabolism and decrease the levels of
inflammation, pyroptosis and the infiltration of immune cells into
plaques, thereby alleviating inflammatory responses (38-40).
An increased number of studies have reported that the inflammasome
and its downstream effectors can trigger liver fibrosis (41-43).
The activation of the NLRP3 inflammasome leads to liver
inflammation, hepatocyte pyroptosis and liver tissue fibrosis in
mice, while pharmacological inhibition of NLRP3 decreases
hepatocyte injury and liver inflammation (42,44).
Moreover, activation of the NLRP3 signaling pathway triggers the
activation of HSCs, which is of great importance for the initiation
and development of liver fibrosis (45,46). A
previous study focused on the potential drug therapies of liver
fibrosis and revealed that the suppression of NLRP3 by oridonin
could effectively alleviate this process (47). The selective inhibitor of NLRP3,
MCC950, can markedly inhibit liver injury in mice induced by bile
duct ligation (41). In the present
study, the NLRP3 activator Nigericin was used to confirm the
specific role of NLRP3 in liver fibrosis. The addition of Nigericin
markedly reversed the inhibitory effect of IL-22 on liver fibrosis
of TGF-β-induced HSCs, which indirectly demonstrated that
inhibition of the NLRP3 inflammasome may be an effective and
potential therapeutic strategy used in the treatment of liver
injury and fibrosis.
In conclusion, the current data indicated that IL-22
may serve an important role in inhibiting liver fibrosis by
inactivation of NLRP3 inflammasome signaling, which may provide
further insight on the underlying mechanism by which IL-22 exerts
protective effects on liver fibrosis. However, in vivo experiments
were not performed and the effects of IL-22-knockdown on NLRP3
inflammasome signaling were not investigated in the present study.
Therefore, further experiments should be performed in future
studies to confirm the present conclusions.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZX and YW searched the literature, designed the
experiments and performed the experiments. NL analyzed, interpreted
the data and wrote the manuscript. ZX revised the manuscript. ZX
and YW confirmed the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Seki E and Brenner DA: Recent advancement
of molecular mechanisms of liver fibrosis. J Hepatobiliary Pancreat
Sci. 22:512–518. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
Matsuda M and Seki E: The liver fibrosis
niche: Novel insights into the interplay between fibrosis-composing
mesenchymal cells, immune cells, endothelial cells, and
extracellular matrix. Food Chem Toxicol. 143(111556)2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zhang CY, Yuan WG, He P, Lei JH and Wang
CX: Liver fibrosis and hepatic stellate cells: Etiology,
pathological hallmarks and therapeutic targets. World J
Gastroenterol. 22:10512–10522. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Friedman SL: Mechanisms of disease:
Mechanisms of hepatic fibrosis and therapeutic implications. Nat
Clin Pract Gastroenterol Hepatol. 1:98–105. 2004.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wu S, Liu L, Yang S, Kuang G, Yin X, Wang
Y, Xu F, Xiong L, Zhang M, Wan J, et al: Paeonol alleviates
CCl4-induced liver fibrosis through suppression of hepatic stellate
cells activation via inhibiting the TGF-β/Smad3 signaling.
Immunopharmacol Immunotoxicol. 41:438–445. 2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kumar P, Smith T, Rahman K, Thorn NE and
Anania FA: Adiponectin agonist ADP355 attenuates CCl4-induced liver
fibrosis in mice. PLoS One. 9(e110405)2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Schuppan D: Liver fibrosis: Common
mechanisms and antifibrotic therapies. Clin Res Hepatol
Gastroenterol. 39 (Suppl 1):S51–S59. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kunkl M, Amormino C, Frascolla S, Sambucci
M, De Bardi M, Caristi S, Arcieri S, Battistini L and Tuosto L:
CD28 autonomous signaling orchestrates IL-22 expression and
IL-22-regulated epithelial barrier functions in human T
lymphocytes. Front Immunol. 11(590964)2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Xie MH, Aggarwal S, Ho WH, Foster J, Zhang
Z, Stinson J, Wood WI, Goddard AD and Gurney AL: Interleukin
(IL)-22, a novel human cytokine that signals through the interferon
receptor-related proteins CRF2-4 and IL-22R. J Biol Chem.
275:31335–31339. 2000.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Nagem RA, Colau D, Dumoutier L, Renauld
JC, Ogata C and Polikarpov I: Crystal structure of recombinant
human interleukin-22. Structure. 10:1051–1062. 2002.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wolk K, Witte E, Witte K, Warszawska K and
Sabat R: Biology of interleukin-22. Semin Immunopathol. 32:17–31.
2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wang S, Li Y, Fan J, Zhang X, Luan J, Bian
Q, Ding T, Wang Y, Wang Z, Song P, et al: Interleukin-22
ameliorated renal injury and fibrosis in diabetic nephropathy
through inhibition of NLRP3 inflammasome activation. Cell Death
Dis. 8(e2937)2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Xing WW, Zou MJ, Liu S, Xu T, Gao J, Wang
JX and Xu DG: Hepatoprotective effects of IL-22 on fulminant
hepatic failure induced by d-galactosamine and lipopolysaccharide
in mice. Cytokine. 56:174–179. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kim DK, Jo A, Lim HS, Kim JY, Eun KM, Oh
J, Kim JK, Cho SH and Kim DW: Enhanced type 2 immune reactions by
increased IL-22/IL-22Ra1 signaling in chronic rhinosinusitis with
nasal polyps. Allergy Asthma Immunol Res. 12:980–993.
2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wu Y, Min J, Ge C, Shu J, Tian D, Yuan Y
and Zhou D: Interleukin-22 in liver injury, inflammation and
cancer. Int J Biol Sci. 16:2405–2413. 2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Que R, Shen Y, Ren J, Tao Z, Zhu X and Li
Y: Estrogen receptor β dependent effects of saikosaponin d on the
suppression of oxidative stress induced rat hepatic stellate cell
activation. Int J Mol Med. 41:1357–1364. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Pan X, Shao Y, Wang F, Cai Z, Liu S, Xi J,
He R, Zhao Y and Zhuang R: Protective effect of apigenin magnesium
complex on H2O2-induced oxidative stress and
inflammatory responses in rat hepatic stellate cells. Pharm Biol.
58:553–560. 2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Novo E, Busletta C, Bonzo LV, Povero D,
Paternostro C, Mareschi K, Ferrero I, David E, Bertolani C,
Caligiuri A, et al: Intracellular reactive oxygen species are
required for directional migration of resident and bone
marrow-derived hepatic pro-fibrogenic cells. J Hepatol. 54:964–974.
2011.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Li C, Meng M, Guo MZ, Wang MY, Ju AN and
Wang CL: The polysaccharides from Grifola frondosa attenuate
CCl4-induced hepatic fibrosis in rats via the TGF-beta/Smad
signaling pathway. RSC Advances. 9:33684–33692. 2019.
|
|
20
|
Ting JP, Lovering RC, Alnemri ES, Bertin
J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA,
et al: The NLR gene family: A standard nomenclature. Immunity.
28:285–287. 2008.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wu X, Dong L, Lin X and Li J: Relevance of
the NLRP3 inflammasome in the pathogenesis of chronic liver
disease. Front Immunol. 8(1728)2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Feng D, Mukhopadhyay P, Qiu J and Wang H:
Inflammation in liver diseases. Mediators Inflamm.
2018(3927134)2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wang S, Li Y, Fan J, Zhang X, Luan J, Bian
Q, Ding T, Wang Y, Wang Z, Song P, et al: Interleukin-22
ameliorated renal injury and fibrosis in diabetic nephropathy
through inhibition of NLRP3 inflammasome activation. Cell Death
Dis. 8(e2937)2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wang S, Fan J, Mei X, Luan J, Li Y, Zhang
X, Chen W, Wang Y, Meng G and Ju D: Interleukin-22 attenuated renal
tubular injury in aristolochic acid nephropathy via suppressing
activation of NLRP3 inflammasome. Front Immunol.
10(2277)2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Chen E, Cen Y, Lu D, Luo W and Jiang H:
IL-22 inactivates hepatic stellate cells via downregulation of the
TGF-β1/Notch signaling pathway. Mol Med Rep. 17:5449–5453.
2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Mu M, Zuo S, Wu RM, Deng KS, Lu S, Zhu JJ,
Zou GL, Yang J, Cheng ML and Zhao XK: Ferulic acid attenuates liver
fibrosis and hepatic stellate cell activation via inhibition of
TGF-β/Smad signaling pathway. Drug Des Devel Ther. 12:4107–4115.
2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Martine P, Chevriaux A, Derangère V,
Apetoh L, Garrido C, Ghiringhelli F and Rébé C: HSP70 is a negative
regulator of NLRP3 inflammasome activation. Cell Death Dis.
10(256)2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Aslamazova EB, Stroganova EV and
Dzhinanian VL: Pathogenesis of liver cirrhosis. Ter Arkh. 47:30–34.
1975.PubMed/NCBI(In Russian).
|
|
30
|
Lakner AM, Steuerwald NM, Walling TL,
Ghosh S, Li T, McKillop IH, Russo MW, Bonkovsky HL and Schrum LW:
Inhibitory effects of microRNA 19b in hepatic stellate
cell-mediated fibrogenesis. Hepatology. 56:300–310. 2012.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Friedman SL: Hepatic stellate cells:
Protean, multifunctional, and enigmatic cells of the liver. Physiol
Rev. 88:125–172. 2008.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Hwang S, He Y, Xiang X, Seo W, Kim SJ, Ma
J, Ren T, Park SH, Zhou Z, Feng D, et al: Interleukin-22
ameliorates neutrophil-driven nonalcoholic steatohepatitis through
multiple targets. Hepatology. 72:412–429. 2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zenewicz LA, Yancopoulos GD, Valenzuela
DM, Murphy AJ, Karow M and Flavell RA: Interleukin-22 but not
interleukin-17 provides protection to hepatocytes during acute
liver inflammation. Immunity. 27:647–659. 2007.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Lu DH, Guo XY, Qin SY, Luo W, Huang XL,
Chen M, Wang JX, Ma SJ, Yang XW and Jiang HX: Interleukin-22
ameliorates liver fibrogenesis by attenuating hepatic stellate cell
activation and downregulating the levels of inflammatory cytokines.
World J Gastroenterol. 21:1531–1545. 2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Xu K, Sun J, Chen S and Li Y, Peng X, Li M
and Li Y: Hydrodynamic delivery of IL-38 gene alleviates
obesity-induced inflammation and insulin resistance. Biochem
Biophys Res Commun. 508:198–202. 2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Vandanmagsar B, Youm YH, Ravussin A,
Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM and Dixit
VD: The NLRP3 inflammasome instigates obesity-induced inflammation
and insulin resistance. Nat Med. 17:179–188. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
37
|
Swanson KV, Deng M and Ting JPY: The NLRP3
inflammasome: Molecular activation and regulation to therapeutics.
Nat Rev Immunol. 19:477–489. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Duewell P, Kono H, Rayner KJ, Sirois CM,
Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nuñez G, Schnurr
M, et al: NLRP3 inflammasomes are required for atherogenesis and
activated by cholesterol crystals. Nature. 464:1357–1361.
2010.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Wang R, Wang Y, Mu N, Lou X, Li W, Chen Y,
Fan D and Tan H: Activation of NLRP3 inflammasomes contributes to
hyperhomocysteinemia-aggravated inflammation and atherosclerosis in
apoE-deficient mice. Lab Invest. 97:922–934. 2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Abderrazak A, Couchie D, Mahmood DF,
Elhage R, Vindis C, Laffargue M, Matéo V, Büchele B, Ayala MR, El
Gaafary M, et al: Anti-inflammatory and antiatherogenic effects of
the NLRP3 inflammasome inhibitor arglabin in ApoE2.Ki mice fed a
high-fat diet. Circulation. 131:1061–1070. 2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Qu J, Yuan Z, Wang G, Wang X and Li K: The
selective NLRP3 inflammasome inhibitor MCC950 alleviates
cholestatic liver injury and fibrosis in mice. Int Immunopharmacol.
70:147–155. 2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Wree A, Eguchi A, McGeough MD, Pena CA,
Johnson CD, Canbay A, Hoffman HM and Feldstein AE: NLRP3
inflammasome activation results in hepatocyte pyroptosis, liver
inflammation, and fibrosis in mice. Hepatology. 59:898–910.
2014.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Wu X, Zhang F, Xiong X, Lu C, Lian N, Lu Y
and Zheng S: Tetramethylpyrazine reduces inflammation in liver
fibrosis and inhibits inflammatory cytokine expression in hepatic
stellate cells by modulating NLRP3 inflammasome pathway. IUBMB
Life. 67:312–321. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
44
|
Mridha AR, Wree A, Robertson AAB, Yeh MM,
Johnson CD, Van Rooyen DM, Haczeyni F, Teoh NC, Savard C, Ioannou
GN, et al: NLRP3 inflammasome blockade reduces liver inflammation
and fibrosis in experimental NASH in mice. J Hepatol. 66:1037–1046.
2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Watanabe A, Sohail MA, Gomes DA, Hashmi A,
Nagata J, Sutterwala FS, Mahmood S, Jhandier MN, Shi Y, Flavell RA,
et al: Inflammasome-mediated regulation of hepatic stellate cells.
Am J Physiol Gastrointest Liver Physiol. 296:G1248–G1257.
2009.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Inzaugarat ME, Johnson CD, Holtmann TM,
McGeough MD, Trautwein C, Papouchado BG, Schwabe R, Hoffman HM,
Wree A and Feldstein AE: NLR family pyrin domain-containing 3
inflammasome activation in hepatic stellate cells induces liver
fibrosis in mice. Hepatology. 69:845–859. 2019.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Liu D, Qin H, Yang B, Du B and Yun X:
Oridonin ameliorates carbon tetrachloride-induced liver fibrosis in
mice through inhibition of the NLRP3 inflammasome. Drug Dev Res.
81:526–533. 2020.PubMed/NCBI View Article : Google Scholar
|