Introduction
Chronic heart failure is a critical end-complication
and comorbidity of various cardiovascular diseases, such as
hypertension and coronary heart disease (1). Cardiac fibrosis is an inevitable
intermediate pathological process during the development of heart
failure (2). Cardiac fibrosis is
characterized by a net accumulation of extracellular matrix
proteins (including fibrillar collagen types I and III, Elastin and
Fibronectin) in the cardiac interstitium and contributes to both
systolic and diastolic dysfunction in myocardial remodeling and
heart failure (3,4). Cardiac fibroblasts (CFbs), the most
prevalent cell type in the heart, serve a key role in the adverse
cardiac fibrosis that occurs with heart failure (5,6). CFbs
have been recognized to have a fundamental contribution to the
cardiac response of a variety of injuries (7). For example, when ischemia-reperfusion
injury occurs in the myocardium, triggering an inflammatory
response, the inflammasome is activated in cardiac fibroblasts but
not in cardiac myocytes (8).
Furthermore, the inflammatory response generated by this injury is
the key pathophysiological process in myocardial remodeling and the
progression of heart failure (9).
In the fibrotic heart, excessive cardiac fibroblasts impair the
electromechanical coupling of cardiomyocytes, hence increasing the
risk of arrhythmogenesis and mortality (7). Hence, CFbs are regarded as a
therapeutic target for heart failure (10).
Recently, a study demonstrated genetic variation in
cardiac fibrosis by characterizing the response of CFbs from
multiple inbred mouse strains to isoproterenol (ISO) treatment
(8). In addition, the study
identified latent-transforming growth factor β-binding protein 2
(LTBP2) as a marker for cardiac fibrosis (11). LTBP2 is a protein expressed in
elastic tissues, such as heart and muscle (12), interact with a number of matrix
components, including collagen and fibronectin (13) and is a member of a superfamily of
proteins comprising extracellular proteins fibrillins and
latent-transforming growth factor β-binding proteins (13), however whether LTBP2 serves a role
in cardiac fibrosis remains unclear. Recent studies have shown that
LTBP2 is associated with apoptosis (14,15);
however, whether the upregulation of LTBP2 expression promotes or
inhibits apoptosis remains controversial. Liang et al
(14) demonstrated that LTBP2
knockdown alleviated apoptosis in osteosarcoma cells in
vitro, while Suri et al (15) demonstrated that LTBP2 deficiency
induced apoptosis in trabecular meshwork cells in vitro
(16). Hence, whether LTBP2 affects
cells apoptosis in cardiac fibrosis and its role in CFb apoptosis
are important to understand in the context of cardiac fibrosis as
well as heart failure development.
More importantly, the mechanism through which LTBP2
affects cell apoptosis has not been elucidated.
Mitochondria-related apoptosis is a key process in cardiac fibrosis
(17,18), and caspase-3, the execution protein
of mitochondrial-induced cell apoptosis serves a pivotal role in
heart failure (19). A previous
study illustrated that transforming growth factor β (TGF-β) induces
apoptosis by targeting caspase-3 activity and LTBP2 has been
identified to form latent complexes with TGFβ (20). Based on these aforementioned
results, it was hypothesized that LTBP2 may promote CFb apoptosis
by activating caspase-3. The current study was conducted to explore
the role and mechanism of LTBP2 in ISO-induced apoptosis in CFb to
provide a potential therapeutic target for heart failure.
Materials and methods
Mouse cell fibroblasts (MCFs) culture
and treatment
MCFs (cat. no. CP-M074) were obtained from Procell
Life Science & Technology Co. Ltd. Cells were cultured in 30 ml
of DMEM (Gibco; Thermo Fisher Scientific, Inc.) containing 10% FBS
(Gibco; Thermo Fisher Scientific, Inc.) and placed in a 37˚C, 5%
CO2 incubator for 90 min of absolute static incubation.
When changing the culture medium, a cross was used to shake the
dish and the principle of differential cell adhesion was used to
wash and discard non-adherent cells
After repeated washing with PBS at 37˚C, 10 ml of
DMEM containing 10% FBS was added to the cells. Cells were then
placed in an incubator for static culturing and the medium was
changed every 3 days. Cells were passaged when 90% confluency was
reached. MCFs were then treated with 100 µM/l ISO (8) (Sigma-Aldrich; Merck KGaA) in a 37˚C,
5% CO2 incubator for 0, 24, 48 or 72 h, while the
control group was treated with an equivalent amount of PBS.
Small interfering (si) RNA
transfection
LTBP2 knockdown by siRNA reverses myocardial
oxidative stress injury, fibrosis and remodelling during dilated
cardiomyopathy. Therefore, LTBP2 and scrambled NC siRNAs were
obtained from Shanghai Shenggong Biology Engineering Technology
Service, Ltd. MCFs were grown in 12-well plates at a density of
6-8x105 and transfected in a 37˚C, 5% CO2
incubator for 12 h. Samples were then transfected with a mixture
containing 4 µl of Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific Inc.) and 3 µl of siRNA in 125 µl of
Opti-MEM (Invitrogen; Thermo Fisher Scientific Inc.). Cells were
then incubated at 37˚C in 5% CO2 for another 12 h, after
which the medium was changed to DMEM containing 100 µM/l ISO or
PBS. Knockdown efficiency was determined by western blotting. The
primer sequences used were as follows: siRNA LTBP2 forward,
5'-CCGGGUUAUAAGCGGGUUAUU-3' and reverse, GAACCAAACGUCUGCGCAAUU-3';
scrambled NC siRNA forward, 5'-UUCUCCGAACGUGUCACGUTT-3' and
reverse, 5'-ACGUGACACGUUCGGAGAATT-3'.
Reverse transcription-quantitative
(RT-q)PCR
The thermocycling conditios for OCR were as follows:
Pre-denaturation 95°C, 10 min, followed by 40 cycles at
95°C for 15 sec, 60°C for 60 sec and melting
curve analysis at 60-95°C, with a 0.3°C rise
every 15 sec. the 2-ΔΔCq method was used for
quantification (21). Experimental
methods and conditions were performed according to the instructions
of the PrimeScript RT Master Mix kit (Takara Bio, Inc.; cat. no.
RR036A). Experimental group MCFs were treated with 100 µM/l ISO at
37˚C in 5% CO2, while control group cells were treated
with an equivalent amount of PBS. After 72 h, all cells were
harvested. Total RNA was isolated from MCFs using a RNAiso reagent
(Takara Bio, Inc.). The PrimeScript RT reagent kit (Takara Bio
Inc.) was used to transcribe isolated RNA to cDNA according to the
manufacturer's protocol. qPCR was performed using a SYBR Premix Ex
Taq II kit (Takara Bio Inc.). The mRNA level of β-actin was used as
an internal control. The primer sequences used were: LTBP2 forward,
5'-TTACAAGCAGAGACTCAC-3'; reverse, 5'-ACAACAGAAGAGACCAGAT-3';
caspase-3 forward, 5'-ACAGCACCTGGTTACTATTC-3', reverse,
5'-CAGTTCTTTCGTGAGCAT-3'; and β-actin, forward,
5'-GCTGCGTGTGGCCCCTGAG-3', reverse, 5'-ACGCAGGATGGCATGAGGGA-3'.
Western blotting
5x106 cells MCFs were collected and
homogenized in lysis buffer (Beyotime Institute of Biotechnology)
containing protease and phosphatase inhibitors (Roche Diagnostics)
and incubated on ice for 30 min. Whole cell lysates were
centrifuged at 12,000 x g for 15 min at 25˚C and protein
concentrations were determined using the bicinchoninic acid (BCA)
assay (Beyotime Institute of Biotechnology). Proteins (20 µg) were
then separated by 8% SDS-PAGE and transferred onto PVDF membranes
(EMD Millipore). Membranes were blocked with 5% skimmed milk in
Tris-buffered saline solution with 0.1% Tween-20 (TBS/T) at room
temperature for 2 h and subsequently incubated with specific
primary antibodies (all Abcam) against LTBP2 (1:1,000; cat. no.
ab121193), caspase-3 (1:1,000; cat. no. ab184787), β-actin
(1:2,000; cat. no. ab8226) at 4˚C overnight. The membranes were
washed with TBS/T 3 times and then incubated with an HRP-conjugated
secondary antibody (goat anti-mouse IgG; 1:2,000, Beyotime
Institute of Biotechnology; cat. no. A0216) at 37˚C for 2 h.
Protein bands were visualized by chemiluminescence detection
(ChemiDoc MP; Bio-Rad Laboratories, Inc.) using Image Lab software
for densitometry (version 5.2; Bio-Rad Laboratories, Inc.).
Caspase-3 activity
Caspase-3 activity was determined using a
colorimetric Caspase-3 assay kit (Abcam). 5x106 MCFs
were collected and resuspended in 50 µl of lysis buffer and
incubated on ice for 15 min, subsequently 50 µl of the 2X reaction
buffer was added to each sample. DEVD-pNA substrate (1 mM) was
added, and samples were incubated for 2 h at 37˚C. Caspase-3
activity was measured at an optical density (OD) of 400 nm. Protein
concentration was assayed by BCA assay (Beyotime Institute of
Biotechnology), BCA assay and protein concentration were performed
as aforementioned. Caspase-3 activity was normalized to the protein
concentration.
Cell proliferation
CCK-8 (Beyotime Insitute of Biotechnology) assay was
used to determine cell proliferation. MCFs (2x104
cells/ml) were seeded onto 96-well plates at 100 µl per well (total
of 56 wells) and further supplemented with 100 µl of DMEM
containing 10% FBS at 37˚C. Experimental group cells were treated
with 100 µM/l ISO, while controls were treated with PBS. After 24,
48, and 72 h of incubation, 100 µl of culture medium was aspirated
and 10 µl of CCK-8 working solution (Beyotime Institute of
Biotechnology) was added for 1 h at room temperature. The
absorbance (A) values at 450 nm were measured by a microplate
reader and 0 was set using blank control wells. After continuous
detection for 7 days, by measuring absorbance at 450 nm, the
proliferation rate of MCFs was calculated.
Cell apoptosis
Cell Death Detection ELISA kit (Roche Diagnostics
GmBH) was used to evaluate the apoptotic rates according to the
manufacturer's instructions. Cells (2x104 cells/ml) were
treated with ISO or PBS and centrifuged at 11,000 x g at 25˚C for
10 min. After the supernatant was removed, cell pellets were
incubated with 200 µl lysis buffer for 30 min at room temperature.
Cytoplasmic lysates were transferred to a streptavidin-coated
plate, then a mixture of anti-DNA-POD and anti-histone-biotin was
added at 25˚C for 2 h. Absorbance at 405 nm was detected with a
reference wavelength at 490 nm (Synergy Mx; BioTek Instruments,
Inc.). Apoptotic rate was determined by measuring the absorbance at
405 nm.
Statistical analysis
SPSS 20.0 software (IBM Corp.) was used for
statistical analysis. Results are expressed as mean ± standard
error of mean (SEM). All experiments were repeated at least 3
times. An unpaired t-test was performed for comparison between 2
groups, multiple comparisons were compared using one-way analysis
of variance (ANOVA) followed by the post hoc Tukey's test.
P<0.05 was considered to indicate a statistically significant
difference, while P<0.01 was considered to indicate a highly
significant difference.
Results
Cell apoptosis and LTBP2 expression
increased in a time-dependent manner in MCFs treated with ISO
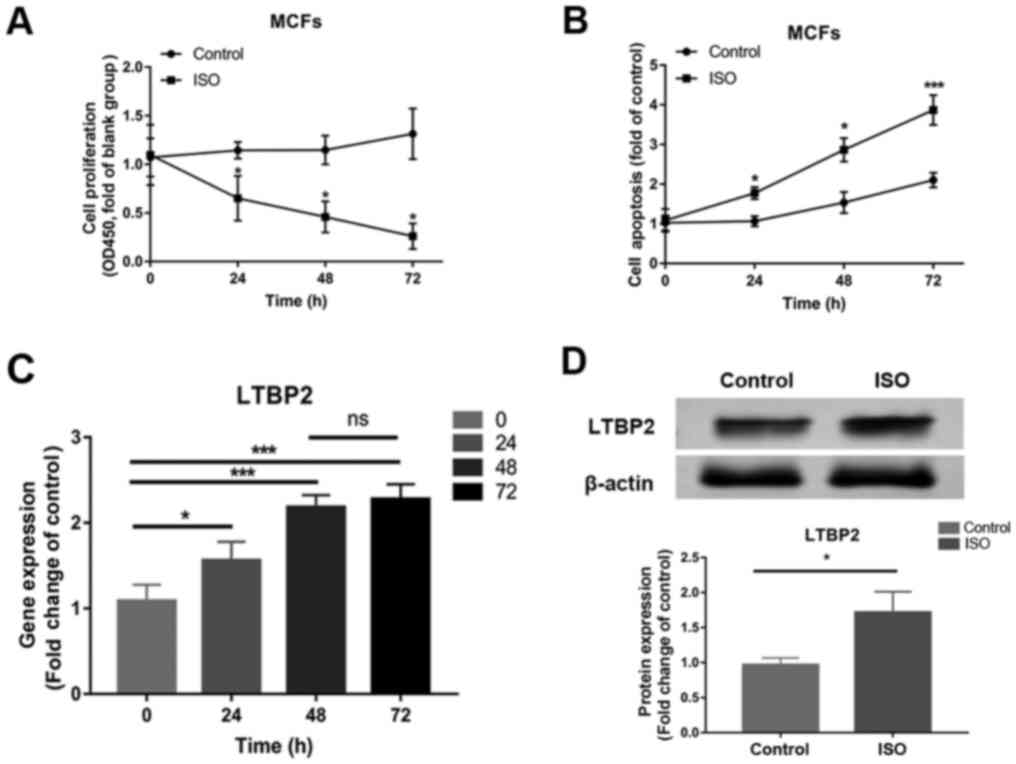
Compared with control, MCFs treated with ISO at 100
µM/l for 24 h had decreased proliferation at 0, 24, 48 and 72 h
(Fig. 1A), while cell apoptosis was
enhanced in a time-dependent manner in ISO-treated cells at 24, 48
and 72 h (Fig. 1B). Similar to the
changes observed in cell apoptosis, LTBP2 mRNA expression
significantly increased in cells treated with ISO and demonstrated
a time-dependent increase (Fig.
1C). Since there are no significant changes in LTBP2 mRNA
expression at 72 h compared with 48 h, the latter timepoint was
chosen for protein detection, which demonstrated increased LTBP2
protein expression following ISO treatment compared with the
control (Fig. 1D). The results
revealed that cell apoptosis and LTBP2 expression increased in a
time-dependent manner in MCFs treated with ISO.
LTBP2 is involved in ISO-induced MCF
apoptosis
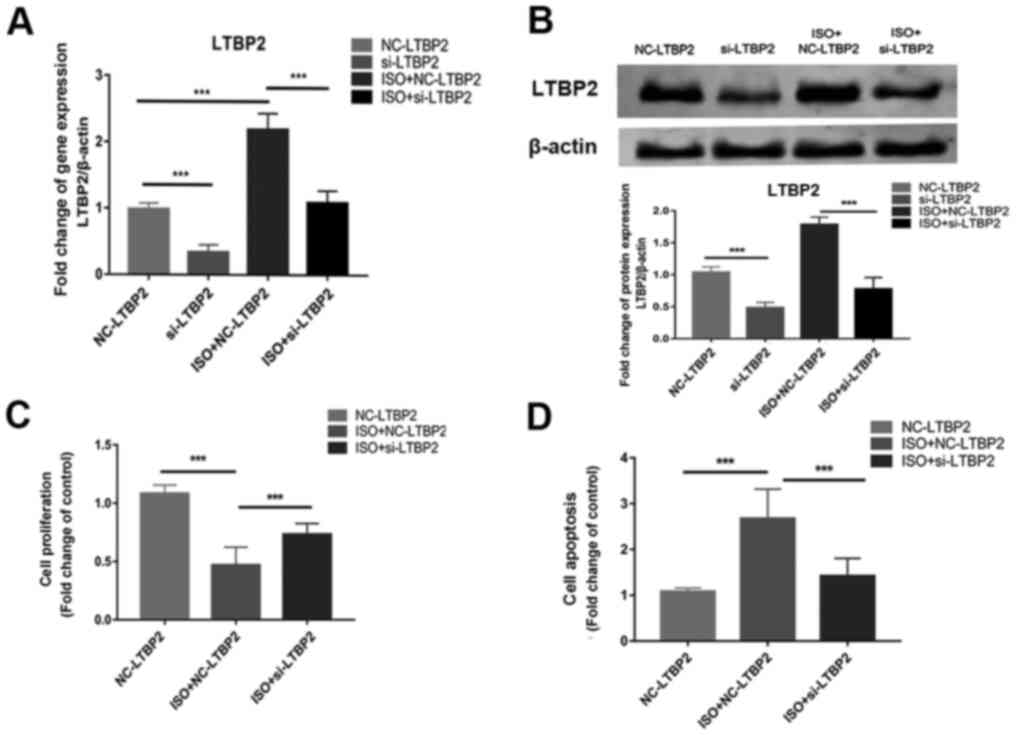
siRNA transfection was used to knockdown the
expression of LTBP2 and the role of LTBP2 in ISO-induced MCFs
apoptosis was investigated. RT-qPCR and western blotting
demonstrated successful LTBP2 knockdown (Fig. 2A and B), and results of the CCK-8 assay
demonstrated that cell proliferation was inhibited by ISO (Fig. 2C). Knocking down LTBP2 partially
reversed the inhibition induced by ISO compared with scrambled
siRNA (Fig. 2C), as well as abated
the enhanced apoptosis caused by ISO treatment compared with
scrambled siRNA (Fig. 2D). The
results demonstrated the involvement of LTBP2 in the ISO-induced
apoptosis of MCF.
Caspase-3 expression and activity
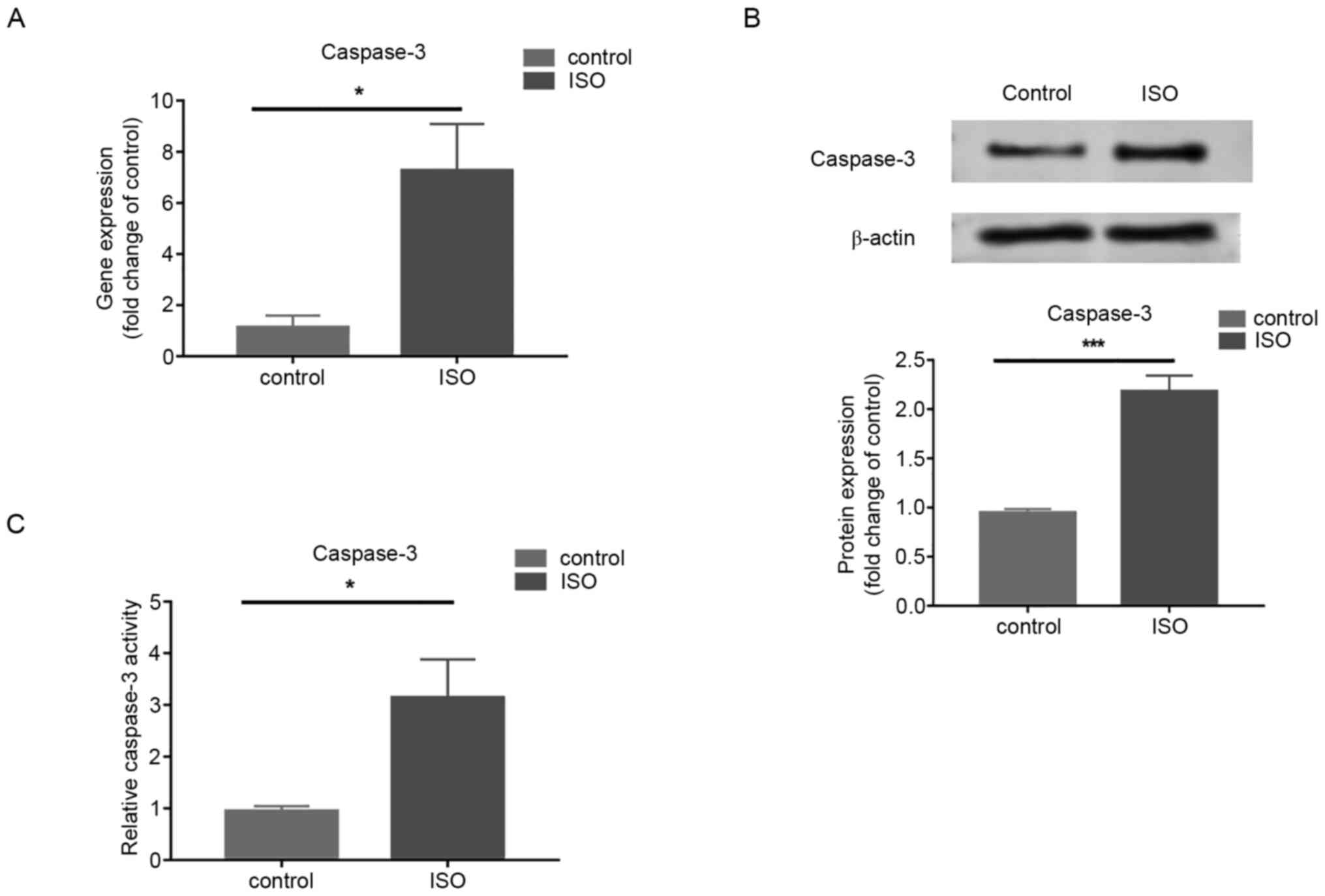
increases in MCFs treated with ISO
Gene and protein expression of caspase-3 measured by
RT-qPCR and western blotting respectively, increased in cells
treated with ISO at 100 µM/l for 72 h compared with that in control
cells (Fig. 3A and B). In addition, enhanced caspase-3
activity was demonstrated following ISO treatment compared with the
control group (Fig. 3C). The
results demonstrated that Caspase-3 was involved in ISO-induced MCF
apoptosis.
LTBP2 increases MCF apoptosis by
regulating caspase-3 expression and activity
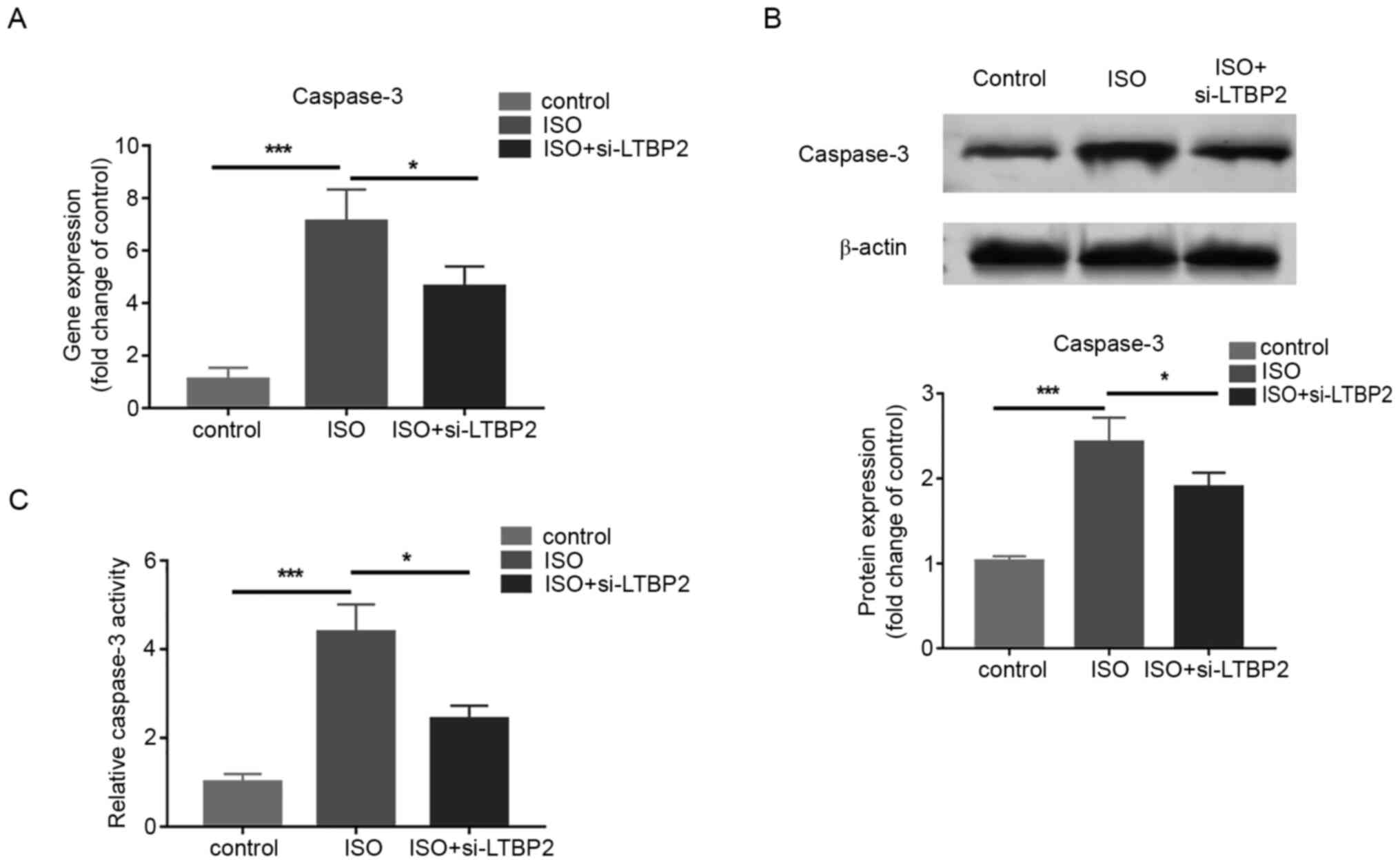
To examine whether the effect of LTBP2 on MCF
apoptosis is associated with caspase-3, LTBP2 was knocked down and
the gene and protein expressions of caspase-3 was quantified in
cells treated with ISO. The RT-qPCR and western blotting results
demonstrated that LTBP2 deficiency partially reversed the increase
in caspase-3 expression induced by ISO treatment (Fig. 4A and B). In addition, the results indicated that
knocking down LTBP2 inhibited the increase in caspase-3 activity
caused by ISO compared with the control group (Fig. 4C). The results revealed that LTBP2
affects MCF apoptosis induced by ISO through caspase-3.
Discussion
LTBP2 is a protein that associates with the
extracellular matrix and binds to it in a fibrillin-dependent
manner (22,23). In humans, it has been confirmed that
LTBP2 serves a pivotal role in primary congenital glaucoma with
defects in the trabecular network (16,24),
and loss of LTBP2 also results in an autosomal recessive ocular
syndrome (25). In addition, a
series of clinical studies demonstrated that LTBP2 is related to
coronary artery disease and heart failure (26,27),
and a recent study revealed increased expression and localization
of LTBP2 in the fibrotic regions of the myocardium after injury in
mice and in patients with heart failure (11). Together, the findings of the
aforementioned studies suggested that LTBP2 may play a role in the
development of heart failure. However, its function as well as the
underlying mechanism by which LTBP2 may be involved in heart
failure remains unknown. Cardiac fibroblasts play a key role in
heart failure (5,6); hence, there is great value in
investigating the function of LTBP2 in this process.
The findings of the present study demonstrated that
LTBP2 expression increased in a time-dependent manner in
ISO-treated MCFs, which is consistent with a previous study
(11) and implied that LTBP2 is
involved in the development of cardiac fibrosis. In addition, the
present study demonstrated that compared with control, ISO
inhibited the proliferation of MCFs, which was alleviated by
knocking down LTBP2 expression. This implies a role for LTBP2 in
ISO-induced MCF apoptosis. LTBP2 has been reportedly involved in
other types of cell apoptosis in recent studies, but its function
in these processes varies. Liang et al (14) demonstrated that depletion of LTBP2
partly abolished the apoptosis of osteosarcoma cells induced by a
microRNA-421 inhibitor. Suri et al (15) demonstrated that knockdown of LTBP2
induced apoptosis in trabecular meshwork cells compared with the
control group. The present study demonstrated that LTBP2 deficiency
partially reversed ISO-induced apoptosis, which is consistent with
the results of Liang et al (14), but is contrary to that of Suri et
al (15). The reason behind
these contradictory results remains unknown; a possible explanation
for this may be that LTBP2 is regulated by different pathways in
different cells or when treated with different factors. Further
studies are required to confirm this.
In addition, the findings of the present study
revealed that LTBP2 enhances MCF apoptosis by regulating caspase-3
expression and activity which has not been previously reported.
Caspase-3 plays a key role in cell apoptosis (28) and activation of caspase-3 is related
to heart failure caused by cardiac fibrosis (19,29).
The results of the present study demonstrated that knocking down
LTBP2 led to a decrease in caspase-3 expression and inhibition of
caspase-3 activity in MCFs treated with ISO. The mechanism through
which LTBP2 regulates caspase-3 expression and activity is unclear.
As mentioned previously, TGF-β may serve as a mediator between
LTBP2 and caspase-3 and is a potential link that needs further
studies to be substantiated.
Although studies have shown that serum levels of
LTBP2 are increased in patients with heart failure, the effects of
LTBP2 on heart failure are currently not being investigated
(27). Appropriate proliferation of
cardiac fibroblasts is a means through which the heart repairs
itself in response to damage and restores function (7). However, excessive fibroblast
proliferation can also trigger cardiac fibrosis and lead to the
progression of heart failure (7).
Whether the apoptosis of cardiomyocytes triggered by upregulation
of LTBP2 promotes cardiac fibrosis and the progression of heart
failure or aids in its recovery needs to be further verified in
animal studies. In addition, since the TGFβ pathway is closely
associated with cardiac fibrosis (30), the effect of LTBP2 on cardiac
fibrosis may also be related to changes in the expression of
TGFβ.
There are several limitations of the present study.
Firstly, apoptosis was detected using the ELISA instead of flow
cytometry. CCK-8 assay can be used to determine cell viability,
proliferation and cytotoxicity, but is not a good measure of
apoptosis (31). Secondly, the
in vitro results of the present study were not verified in
animal models. Further research should be undertaken to investigate
whether LTBP2 deficiency in animal models can lead to cardiac
fibrosis as well as heart failure, and these results may provide
new targets for treatment and prevention of heart failure.
In conclusion, the results revealed that LTBP2 and
caspase-3 expression was increased in cells during ISO-induced MCF
apoptosis. Inhibition of LTBP2 expression alleviated ISO-induced
MCF apoptosis by suppressing caspase-3 expression. The results also
indicated that LTBP2 may influence the development of cardiac
fibrosis by regulating caspase-3-induced cardiac fibroblast
apoptosis. The results of the current study may provide novel ideas
for mitigating the progression of heart failure.
Acknowledgements
Not applicable.
Funding
Funding: The work was funded by the Master Plan of Natural Fund
in Liaoning (grant no. 2019-ZD-0424).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CS designed the study, conducted most of the
experiments and drafted the manuscript. XL, FH, XW and TJ conducted
experiments. BS and SL collected and analyzed the data. All authors
read and approved the final manuscript. All authors confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ponikowski P, Voors AA, Anker SD, Bueno H,
Cleland JG, Coats AJ, Falk V, González-Juanatey JR, Harjola VP,
Jankowska EA, et al: 2016 ESC Guidelines for the diagnosis and
treatment of acute and chronic heart failure: The Task Force for
the diagnosis and treatment of acute and chronic heart failure of
the European Society of Cardiology (ESC). Developed with the
special contribution of the Heart Failure Association (HFA) of the
ESC. Eur J Heart Fail. 18:891–975. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
González A, Schelbert EB, Díez J and
Butler J: Myocardial interstitial fibrosis in heart failure:
Biological and translational perspectives. J Am Coll Cardiol.
71:1696–1706. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Talman V and Ruskoaho H: Cardiac fibrosis
in myocardial infarction-from repair and remodeling to
regeneration. Cell Tissue Res. 365:563–581. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kong P, Christia P and Frangogiannis NG:
The pathogenesis of cardiac fibrosis. Cell Mol Life Sci.
71:549–574. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Porter KE and Turner NA: Cardiac
fibroblasts: At the heart of myocardial remodeling. Pharmacol Ther.
123:255–278. 2009.PubMed/NCBI View Article : Google Scholar
|
|
6
|
van den Borne SW, Diez J, Blankesteijn WM,
Verjans J, Hofstra L and Narula J: Myocardial remodeling after
infarction: The role of myofibroblasts. Nat Rev Cardiol. 7:30–37.
2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Travers JG, Kamal FA, Robbins J, Yutzey KE
and Blaxall BC: Cardiac fibrosis: The fibroblast awakens. Circ Res.
118:1021–1040. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kawaguchi M, Takahashi M, Hata T, Kashima
Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J,
et al: Inflammasome activation of cardiac fibroblasts is essential
for myocardial ischemia/reperfusion injury. Circulation.
123:594–604. 2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Frangogiannis NG: The inflammatory
response in myocardial injury, repair, and remodelling. Nature
Reviews Cardiology. 11:255–265. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Brown RD, Ambler SK, Mitchell MD and Long
CS: The cardiac fibroblast: Therapeutic target in myocardial
remodeling and failure. Annu Rev Pharmacol Toxicol. 45:657–687.
2005.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Park S, Ranjbarvaziri S, Lay FD, Zhao P,
Miller MJ, Dhaliwal JS, Huertas-Vazquez A, Wu X, Qiao R, Soffer JM,
et al: Genetic regulation of fibroblast activation and
proliferation in cardiac fibrosis. Circulation. 138:1224–1235.
2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Morén A, Olofsson A, Stenman G, Sahlin P,
Kanzaki T, Claesson-Welsh L, ten Dijke P, Miyazono K and Heldin CH:
Identification and characterization of LTBP-2, a novel latent
transforming growth factor-beta-binding protein. J Biol Chem.
269:32469–32478. 1994.PubMed/NCBI
|
|
13
|
Rifkin DB: Latent transforming growth
factor-beta (TGF-beta) binding proteins: Orchestrators of TGF-beta
availability. J Biol Chem. 280:7409–7412. 2005.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Liang X, Zhang L, Ji Q, Wang B, Wei D and
Cheng D: miR-421 promotes apoptosis and suppresses metastasis of
osteosarcoma cells via targeting LTBP2. J Cell Biochem: Mar 28,
2019 (Epub ahead of print). doi: 10.1002/jcb.28144.
|
|
15
|
Suri F, Yazdani S and Elahi E: LTBP2
knockdown and oxidative stress affect glaucoma features including
TGFβ pathways, ECM genes expression and apoptosis in trabecular
meshwork cells. Gene. 673:70–81. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ali M, McKibbin M, Booth A, Parry DA, Jain
P, Riazuddin SA, Hejtmancik JF, Khan SN, Firasat S, Shires M, et
al: Null mutations in LTBP2 cause primary congenital glaucoma. Am J
Hum Genet. 84:664–671. 2009.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Marin-Garcia J, Goldenthal MJ and Moe GW:
Mitochondrial pathology in cardiac failure. Cardiovasc Res.
49:17–26. 2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chen QM and Tu VC: Apoptosis and heart
failure: Mechanisms and therapeutic implications. Am J Cardiovasc
Drugs. 2:43–57. 2002.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Narula J, Pandey P, Arbustini E, Haider N,
Narula N, Kolodgie FD, Dal Bello B, Semigran MJ, Bielsa-Masdeu A,
Dec GW, et al: Apoptosis in heart failure: Release of cytochrome c
from mitochondria and activation of caspase-3 in human
cardiomyopathy. Proc Natl Acad Sci USA. 96:8144–8149.
1999.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Robertson IB, Horiguchi M, Zilberberg L,
Dabovic B, Hadjiolova K and Rifkin DB: Latent TGF-β-binding
proteins. Matrix Biol. 47:44–53. 2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Hyytiäinen M, Taipale J, Heldin CH and
Keski-Oja J: Recombinant latent transforming growth factor
beta-binding protein 2 assembles to fibroblast extracellular matrix
and is susceptible to proteolytic processing and release. J Biol
Chem. 273:20669–20676. 1998.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Vehviläinen P, Hyytiäinen M and Keski-Oja
J: Matrix association of latent TGF-beta binding protein-2 (LTBP-2)
is dependent on fibrillin-1. J Cell Physiol. 221:586–593.
2009.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Narooie-Nejad M, Paylakhi SH, Shojaee S,
Fazlali Z, Rezaei Kanavi M, Nilforushan N, Yazdani S, Babrzadeh F,
Suri F, Ronaghi M, et al: Loss of function mutations in the gene
encoding latent transforming growth factor beta binding protein 2,
LTBP2, cause primary congenital glaucoma. Hum Mol Genet.
18:3969–3977. 2009.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Désir J, Sznajer Y, Depasse F, Roulez F,
Schrooyen M, Meire F and Abramowicz M: LTBP2 null mutations in an
autosomal recessive ocular syndrome with megalocornea,
spherophakia, and secondary glaucoma. Eur J Hum Genet. 18:761–767.
2010.PubMed/NCBI View Article : Google Scholar
|
|
26
|
De Sutter J, Pardaens S, Van Hercke D, Van
De Veire N, De Buyzere M, Philippe J, Vanpoucke G and Thomas G:
Latent Transforming growth factor Binding Protein 2 (LTBP2) is
related to phenotypic changes suggestive for HFPEF in stable
outpatients with coronary artery disease and preserved ejection
fraction. Eur Heart J. 34 (Suppl 1)(P5726)2013.
|
|
27
|
Bai Y, Zhang P, Zhang X, Huang J, Hu S and
Wei Y: LTBP-2 acts as a novel marker in human heart failure-a
preliminary study. Biomarkers. 17:407–415. 2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Jänicke RU, Sprengart ML, Wati MR and
Porter AG: Caspase-3 is required for DNA fragmentation and
morphological changes associated with apoptosis. J Biol Chem.
273:9357–9360. 1998.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Yang B, Ye D and Wang Y: Caspase-3 as a
therapeutic target for heart failure. Expert Opin Ther Targets.
17:255–263. 2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Goumans MJ, van Zonneveld AJ and ten Dijke
P: Transforming growth factor beta-induced
endothelial-to-mesenchymal transition: A switch to cardiac
fibrosis? Trends Cardiovasc Med. 18:293–298. 2008.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Wang YJ, Zhou SM, Xu G and Gao YQ:
Interference of phenylethanoid glycosides from cistanche tubulosa
with the MTT Assay. Molecules. 20:8060–8071. 2015.PubMed/NCBI View Article : Google Scholar
|