Introduction
Cerebral infarction, also known as ischemic stroke,
refers to the local ischemic necrosis or encephalomalacia of brain
tissue caused by disruption to the blood supply, leading to
ischemia and hypoxia (1-3).
Cerebral infarction is the most common type of cerebrovascular
disease, accounting for ~70% of all cerebrovascular diseases
(4). The most common clinical
causes of cerebral infarction include the formation of a cerebral
thrombus, lacunar infarction and cerebral embolism (5,6).
Although the different types may share common etiological
characteristics, there also exhibit specific differences. Diseases
and conditions commonly associated with cerebral infarction include
diabetes, obesity, hypertension, rheumatic heart disease,
arrhythmia, dehydration due to various reasons, several types of
arteritis, shock and a rapid or excessive drop in blood pressure
(7-9).
Cerebral infarction is considered as a medical emergency and is
associated with high disability and mortality rates. Therefore, it
is crucial to devise more effective treatments for cerebral
infarction. Previous studies have reported that microglia
activation may promote the excessive release of numerous
inflammatory cytokines, thereby exacerbating neuronal damage after
an ischemic stroke (10,11). Therefore, the present study
conducted a series of experiments using microglia to further study
their role in cerebral infarction and elucidate the underlying
mechanism.
Long non-coding RNAs (lncRNAs) are a class of
non-coding RNAs of >200 nucleotides in length, which are
transcribed in most eukaryotic genomes (12,13).
To date, most lncRNAs have been classified; however, their precise
functions require further investigations. An increasing number of
studies have shown that lncRNAs play key roles in regulating growth
and development, cell differentiation, subcellular structure
distribution and evolutionary selection, in addition to their roles
in numerous types of human disease, including vascular,
neurological and inflammatory diseases, as well as cancer (14-16).
In fact, numerous lncRNAs, such as metastasis-associated lung
adenocarcinoma transcript 1, maternally expressed 3, CDKN2B
antisense RNA 1 and small nucleolar RNA host gene 1 (SNHG1), have
been reported to be involved in regulating the pathogenesis of
cerebral infarction (17-20).
SNHG1 is a lncRNA, comprising 3,927 nucleotides, which is
transcribed from the SNHG1 gene on chromosome 11(21). Chen et al (20) found that SNHG1 exerted a protective
effect against cerebral infarction by regulating the PI3K/AKT
signaling pathway. Other studies have revealed that regulating the
endogenous activation of microglia may represent a new target for
ischemic stroke treatment (22,23).
In addition, an oxygen-glucose deprivation (OGD) model of BV-2
cells has been widely used to study cerebral infarction in
vitro (24-26).
However, to the best of our knowledge, whether SNHG1 participates
in cerebral infarction by affecting the activation of microglia has
not been investigated to date.
The present study was undertaken to investigate the
effects of SNHG1 on the activation of microglia and further
determine its underlying molecular mechanism. The results may
provide novel insight into potential clinical strategies for the
treatment of cerebral infarction.
Materials and methods
Cell culture and OGD model
establishment
The BV-2 microglial cell line was purchased from the
American Type Culture Collection. Cells were cultured in DMEM
(HyClone; Cytiva) supplemented with 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.), and maintained at 37˚C with 5%
CO2.
The establishment of the OGD model of BV-2 cells was
performed according to a previous study (25). Briefly, BV-2 cells were cultured in
serum/glucose-free DMEM with 95%N2 and 5% CO2
at 37˚C for 12, 24 or 48 h. Following incubation, the cells were
collected to determine the levels of SNHG1 and miR-329-3p.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). Total RNA was reverse transcribed into cDNA using a
SuperScript® VILO™ cDNA Synthesis kit (Invitrogen;
Thermo Fisher Scientific, Inc.). qPCR was subsequently performed on
a Prism 7000 Real-Time PCR Detection system (Applied Biosystems;
Thermo Fisher Scientific, Inc.) using SYBR qPCR Master mix (Thermo
Fisher Scientific, Inc.), according to the manufacturer's protocol.
The primers used for the qPCR were purchased from Genscript and the
primer sequences are listed as follows:
GAPDH, forward 5'-CTTTGGTATCGTGGAAGGACTC-3' and
reverse, 5'-GTAGAGGCAGGGATGATGTTCT-3'; U6, forward,
5'-GCTTCGGCAGCACATATACTAAAAT-3' and reverse,
5'-CGCTTCACGAATTTGCGTGTCAT-3'; lncRNA SNHG1, forward,
5'-CCAAACTCAGGCACTGTATAGAT-3' and reverse,
5'-ACAGACACGAAGTGGAGTTATG-3'; and miR-329-3p, forward,
5'-GTGGAACAGACCTGGTAAAC-3' and reverse, 5'-CAAGTGCGAGTCGTGCAGT-3'.
The following thermocycling conditions were used for the qPCR:
Initial denaturation for 5 min at 95˚C, followed by 40 cycles of
95˚C for 10 sec and 60˚C for 30 sec. The relative mRNA expression
levels of SNHG1 and microRNA (miRNA/miR)-329-3p were calculated
using the 2-ΔΔCq method (27), and GAPDH or U6 were used as the
internal controls for normalization of SNHG1 and miR-329-3p
expression, respectively.
Cell transfection
The SNHG1 sequence was synthesized based on the
SNHG1 sequence and then sub-cloned into the pcDNA3.1 vector
(SNHG1-plasmid; Shanghai GeneChem Co., Ltd.). The empty pcDNA3.1
vector was used as a control (control-plasmid). BV-2 cells were
plated into 6-well plates for 24 h, then transfected with 1 µg
control-plasmid, 1 µg SNHG1-plasmid, 50 nM mimic control
(5'-UUCUCCGAACGUGUCACGUTT-3'; GeneCopoeia, Inc.), 50 nM miR-329-3p
mimic (3'UUUUUCCAAUCGACCCACACAA5'; GeneCopoeia, Inc.), 1 µg
SNHG1-plasmid + 50 nM mimic control, 1 µg SNHG1-plasmid + 50 nM
miR-329-3p mimic, 100 nM inhibitor control
(5'CAGUACUUUUGUGUAGUACAA3'; GeneCopoeia, Inc.) or 100 nM miR-329-3p
inhibitor (5'AAAAAGGUUAGCUGGGUGUGUU3'; GeneCopoeia, Inc.) using
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). Following transfection for 24 h, the cells were
collected to determine the transfection efficiency using
RT-qPCR.
To investigate the role of SNHG1 in OGD induced BV-2
cells, BV-2 cells were cultured in serum/glucose-free DMEM with 95%
N2 and 5% CO2 at 37˚C for 48 h, then the
cells were transfected with control-plasmid, SNHG1-plasmid,
SNHG1-plasmid + mimic control, or SNHG1-plasmid + miR-329-3p mimic
at 37˚C for another 24 h. Cells were divided into the following six
groups: i) Control group; ii) OGD group; iii) OGD + control-plasmid
group; iv) OGD + SNHG1-plasmid group; v) OGD + SNHG1-plasmid +
mimic control group; and vi) OGD + SNHG1-plasmid + miR-329-3p mimic
group.
To investigate the role of miR-329-3p downregulation
in OGD induced BV-2 cells, BV-2 cells were cultured in
serum/glucose-free DMEM with 95% N2 and 5%
CO2 at 37˚C for 48 h, then the cells were transfected
with inhibitor control or miR-329-3p inhibitor at 37˚C for 24 h.
Cells were divided into the following four groups: i) Control
group; ii) OGD group; iii) OGD + inhibitor group; and iv) OGD +
miR-329-3p inhibitor group.
miRNA target analysis and dual
luciferase reporter assay
The binding relationship between miR-329-3p and
SNHG1 was identified using starBase (http://starbase.sysu.edu.cn/). The 3'-untranslated
region (UTR) sequences of SNHG1 containing the target sequence of
miR-329-3p were obtained by RT-qPCR and cloned into a pmirGLO
vector (Promega Corporation) to construct the SNHG1-wild-type (WT)
reporter gene vector. A SNHG1-mutated type (MUT) reporter gene
vector was also constructed. The BV-2 cells were cultured for 24 h,
then co-transfected with the SNHG1-WT or SNHG1-MUT reporter gene
vector and miR-329-3p mimic (3'-UUUUUCCAAUCGACCCACACAA-5') or mimic
control (5'-UUCUCCGAACGUGUCACGUTT-3') using Lipofectamine 2000
reagent at 37˚C for 48 h. The relative luciferase activity was
measured using a Dual Luciferase Reporter assay system (Promega
Corporation) according to the manufacturer's protocol. Luciferase
activity was normalized to Renilla luciferase activity.
ELISA
Following 24 h of transfection, the cell culture
supernatants were collected through centrifugation at 500 x g at
4˚C for 5 min. Then, the levels of TNF-α in the supernatant of BV-2
cells were detected using an ELISA kit (cat. no. SMTA00B; R&D
Systems, Inc.) according to the manufacturer's protocol.
Measurement of nitric oxide (NO)
production
The production of NO was evaluated by detecting the
nitrate levels in the cell supernatant. Briefly, equal volumes of
BV-2 cell supernatant and Griess reagent were mixed and incubated
for 10 min at room temperature. The absorbance was measured at a
wavelength of 550 nm using a microplate reader (Bio-Rad
Laboratories, Inc.).
Flow cytometric analysis of
apoptosis
Flow cytometry was used to detect the levels of cell
apoptosis using Annexin V-FITC/PI apoptosis detection kit (Beyotime
Institute of Biotechnology). Briefly, the transfected cells were
collected by trypsinization after transfection and resuspended in
1X binding buffer. Then, 100 µl cell suspension was incubated with
5 µl Annexin V-FITC and 5 µl PI (Beyotime Institute of
Biotechnology) according to the manufacturer's protocol. Apoptotic
cells were analyzed using a FACSCalibur flow cytometer (BD
Biosciences) and FlowJo software (version 7.2.4; FlowJo LLC).
Detection of caspase-3 activity
Caspase-3 activity was measured using a colorimetric
assay kit (Beyotime Institute of Biotechnology) according to the
manufacturer's protocol.
Western blotting
Following incubation for 24 h, total protein was
extracted from BV-2 cells using RIPA lysis buffer (Beyotime
Institute of Biotechnology) and centrifugation at 10,000 x g for 15
min at 4˚C. Total protein was quantified using a BCA protein assay
kit (Bio-Rad Laboratories, Inc.) and separated via 10% SDS-PAGE.
The separated proteins were subsequently transferred onto a PVDF
membrane and blocked with PBS-0.1% Tween-20 (PBST) containing 5%
non-fat milk at room temperature for 1 h. The membranes were then
incubated with the following primary antibodies at 4˚C overnight:
Anti-cleaved caspase-3 (cat. no. 9664; dilution 1:1,000; Cell
Signaling Technology, Inc.), anti-caspase-3 (cat. no. 14220;
dilution 1:1,000; Cell Signaling Technology, Inc.) or anti-GAPDH
(cat. no. 5174; dilution 1:1,000; Cell Signaling Technology, Inc.).
Following primary antibody incubation, the membrane was washed with
PBST three times and incubated with the secondary antibody [goat
anti-rabbit IgG H&L (HRP) preadsorbed; cat. no. 97080; dilution
1:5,000; Abcam] for 1 h at room temperature. Protein bands were
visualized using an ECL substrate (Cytiva) on an Amersham
ImageQuant UV western blotting system (Cytiva) according to the
manufacturer's instructions.
Statistical analysis
Statistical analysis was performed using the SPSS
software (version 18.0; SPSS, Inc.). Data are presented as the mean
± SD of three independent experiments. Statistical differences
between groups were determined using an unpaired Student's t-test
or one-way ANOVA followed by Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
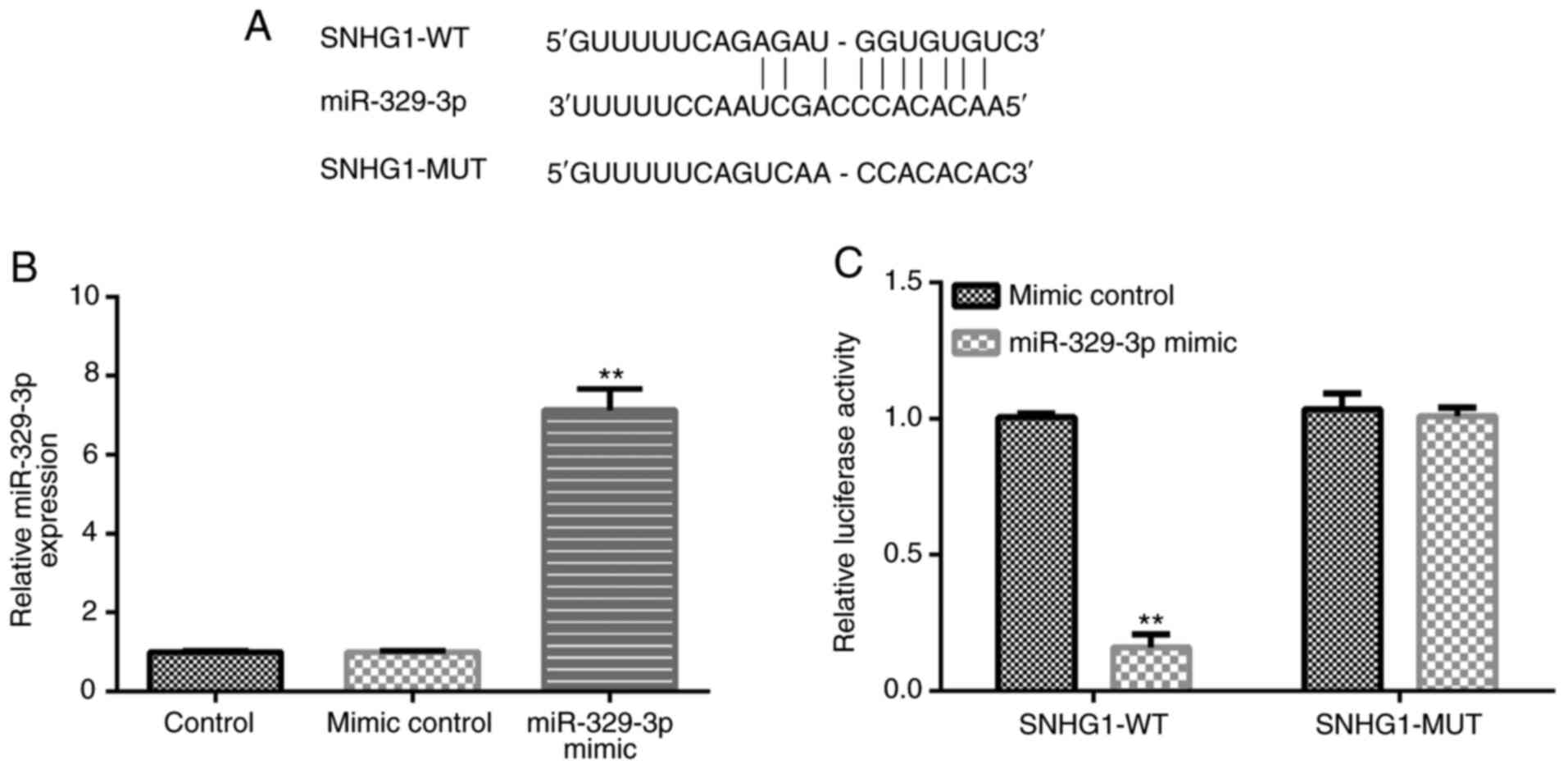
SNHG1 is a direct target gene of
miR-329-3p
Bioinformatics analysis using starBase identified a
binding site between SNHG1 and miR-329-3p (Fig. 1A). Compared with the mimic control
group, transfection with the miR-329-3p mimic significantly
upregulated miR-329-3p expression levels in BV-2 cells (Fig. 1B). Then, the binding site between
SNHG1 and miR-329-3p was confirmed using a dual luciferase reporter
assay (Fig. 1C).
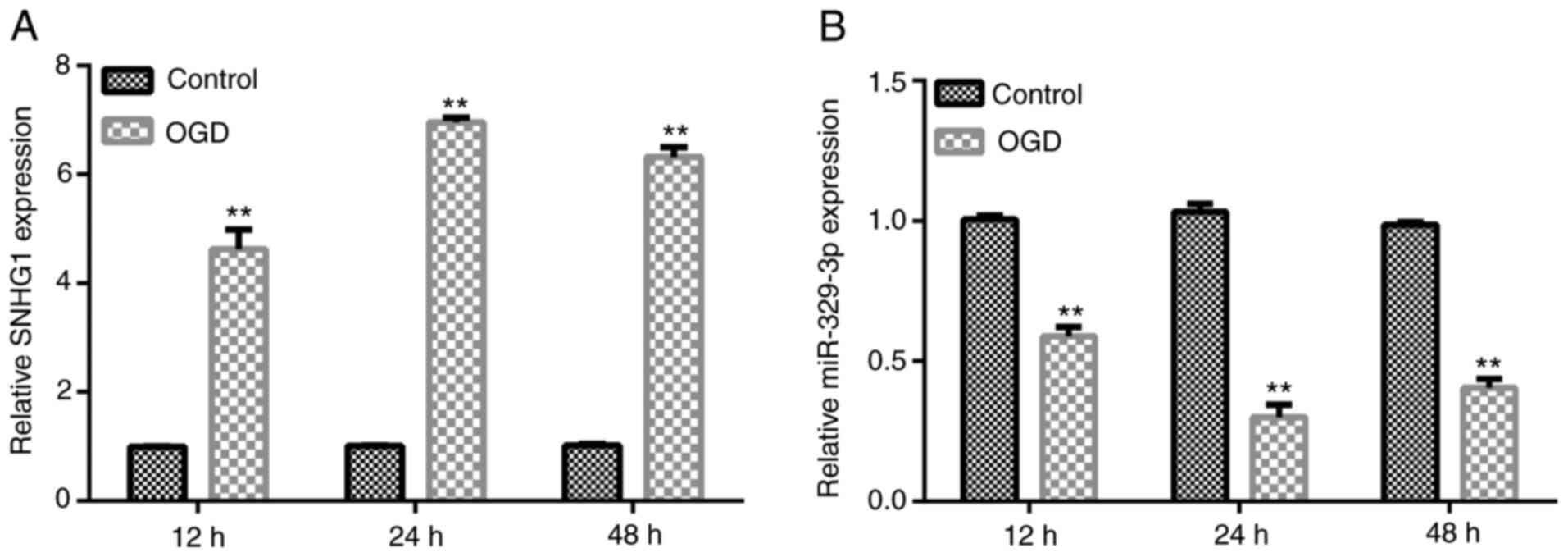
Expression levels of SNHG1 and
miR-329-3p in the OGD-induced BV-2 cell model
An OGD cell model was established via OGD induction
as previously described (25).
After OGD induction for 12, 24 or 48 h, RT-qPCR was performed to
analyze the expression levels of SNHG1 and miR-329-3p in BV-2 cells
compared with the control group. The results revealed that the
expression levels of SNHG1 were significantly upregulated, while
the expression levels of miR-329-3p were significantly
downregulated in the OGD group (Fig.
2A and B).
SNHG1-plasmid downregulates the
expression levels of miR-329-3p in BV-2 cells
BV-2 cells were transfected with control-plasmid,
SNHG1-plasmid, mimic control, miR-329-3p mimic, SNHG1-plasmid +
mimic control or SNHG1-plasmid + miR-329-3p mimic, and the
transfection efficiencies were determined using RT-qPCR. The
results demonstrated that transfection with the SNHG1-plasmid or
miR-329-3p mimic markedly upregulated the expression levels of
SNHG1 and miR-329-3p, respectively, in BV-2 cells (Fig. 3A and B). Compared with the control-plasmid
group, transfection with the SNHG1-plasmid significantly
downregulated the expression levels of miR-329-3p in BV-2 cells,
while this downregulation was reversed following transfection with
the miR-329-3p mimic (Fig. 3C).
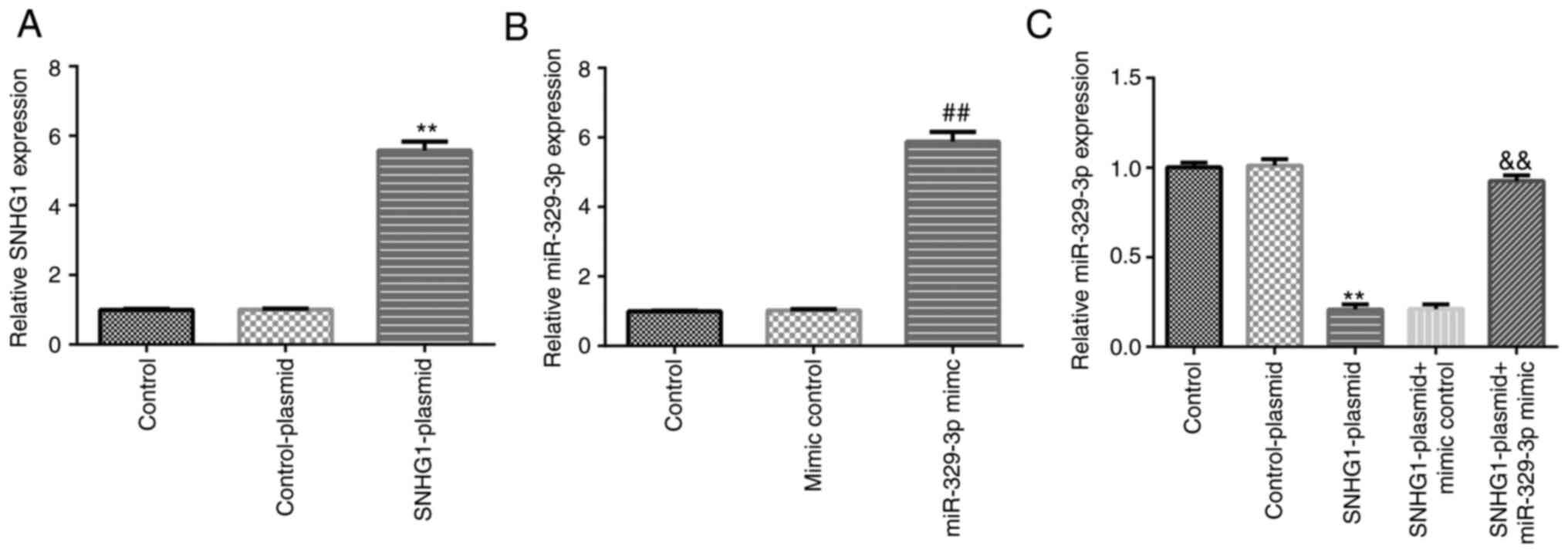 | Figure 3Transfection efficiency of
SNHG1-plasmid and miR-329-3p mimic in BV-2 cells. BV-2 cells were
transfected with control-plasmid, SNHG1-plasmid, mimic control,
miR-329-3p mimic, SNHG1-plasmid + mimic control, or SNHG1-plasmid +
miR-329-3p mimic for 24 h. (A) Reverse transcription-quantitative
PCR was performed to analyze the mRNA expression levels of SNHG1 in
BV-2 cells following transfection with control-plasmid or
SNHG1-plasmid. (B) Reverse transcription-quantitative PCR was
performed to analyze the expression levels of miR-329-3p in BV-2
cells following transfection with mimic control or miR-329-3p
mimic. (C) Reverse transcription-quantitative PCR was performed to
analyze the expression levels of miR-329-3p in BV-2 cells
transfected with control-plasmid, SNHG1-plasmid, SNHG1-plasmid +
mimic control, or SNHG1-plasmid + miR-329-3p mimic.
**P<0.01 vs. control-plasmid; ##P<0.01
vs. mimic control; &&P<0.01 vs. SNHG1-plasmid
+ mimic control. SNHG1, small nucleolar RNA host gene 1; miR,
microRNA. |
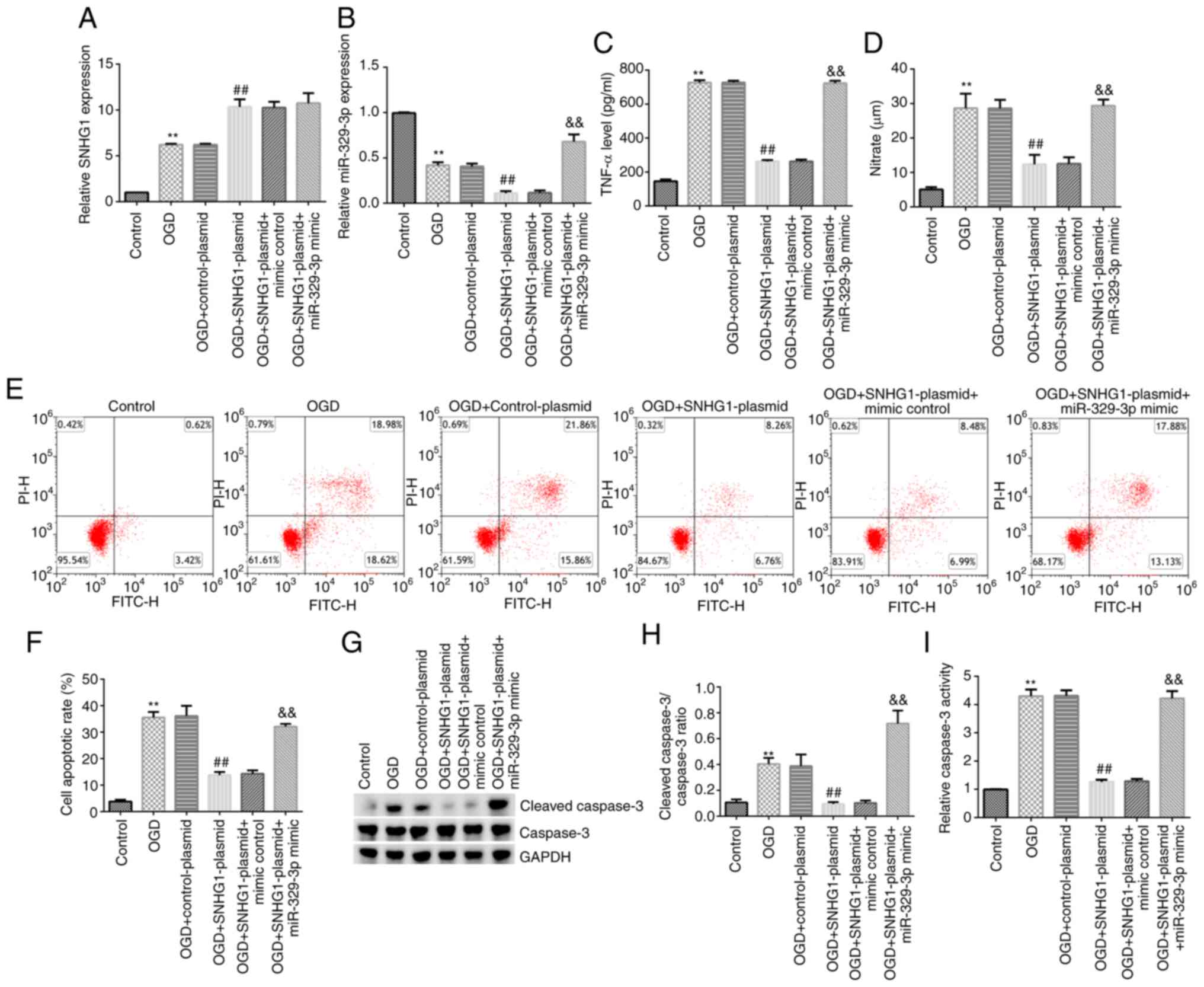
SNHG1 inhibits OGD-induced BV-2 cell
activation by downregulating the expression of miR-329-3p
BV-2 cells were transfected for 24 h and then
divided into the following six groups: i) Control group; ii) OGD
group; iii) OGD + control-plasmid group; iv) OGD + SNHG1-plasmid
group; v) OGD + SNHG1-plasmid + mimic control group; and vi) OGD +
SNHG1-plasmid + miR-329-3p mimic group. Compared with the control
group, the expression levels of SNHG1 were significantly
upregulated (Fig. 4A), the
expression levels of miR-329-3p were significantly downregulated
(Fig. 4B), and the release of TNF-α
and NO was significantly increased (Fig. 4C and D) in the OGD group. In addition, the
results of the flow cytometry, caspase-3 activity and western blot
analyses demonstrated that the levels of cell apoptosis, caspase-3
activity, cleaved caspase-3 protein expression and the cleaved
caspase-3/caspase-3 ratio were all markedly increased in the OGD
group compared with the control group (Fig. 4E-I).
Similarly, compared with the OGD + control-plasmid
group, SNHG1 expression levels were upregulated in the OGD +
SNHG1-plasmid group, while miR-329-3p expression levels, TNF-α and
NO release, cell apoptosis, cleaved caspase-3 protein expression,
the cleaved caspase-3/caspase-3 ratio and caspase-3 activity were
all significantly reduced in the OGD + SNHG1-plasmid group. All
these effects were significantly reversed following transfection
with the miR-392-3p mimic.
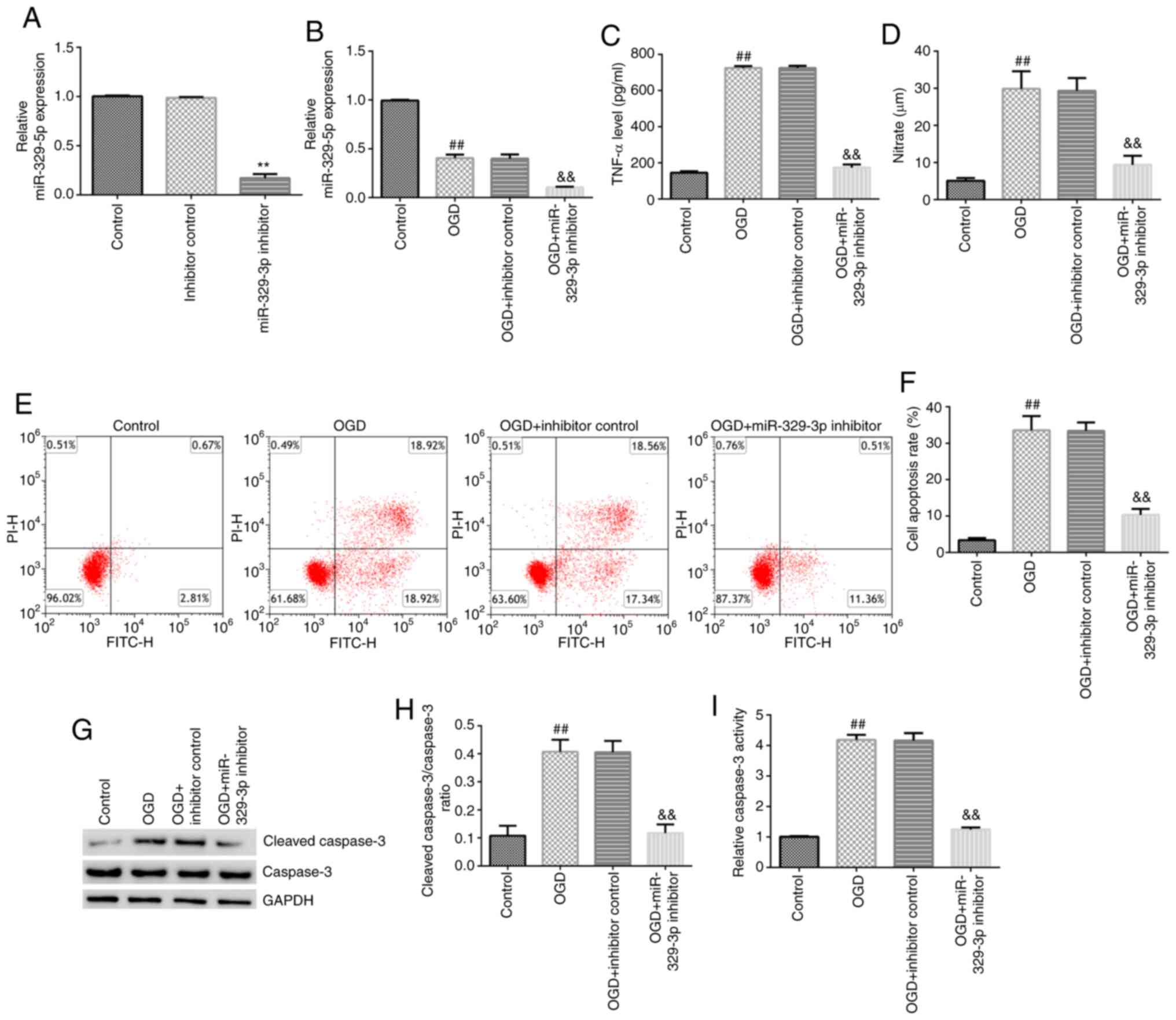
Knockdown of miR-329-3p expression
inhibits OGD-induced BV-2 cell activation
The transfection efficiency of the miR-329-3p
inhibitor into BV-2 cells was detected 24 h post-transfection, and
the results revealed that the miR-329-3p inhibitor significantly
downregulated the expression levels of miR-329-3p in BV-2 cells
(Fig. 5A). The cells were then
divided into the following four groups: i) Control group; ii) OGD
group; iii) OGD + inhibitor control; and iv) OGD + miR-329-3p
inhibitor group. Experiments were subsequently performed to
determine the molecular mechanisms underlying the effects of
miR-329-3p on BV-2 cells. As shown in Fig. 5B-I, compared with the control group,
miR-329-3p expression levels were downregulated in the OGD group,
while TNF-α and NO release, cell apoptosis, cleaved caspase-3
protein expression, the cleaved caspase-3/caspase-3 ratio and
caspase-3 activity were all significantly increased in the OGD
group. Similarly, compared with the OGD + inhibitor control group,
miR-329-3p expression, TNF-α and NO release, cell apoptosis,
cleaved caspase-3 protein expression, the cleaved
caspase-3/caspase-3 ratio and caspase-3 activity were all markedly
decreased in the OGD + miR-329-3p inhibitor group.
Discussion
Cerebral infarction is caused by blood circulation
disorders, which lead to ischemia, hypoxia and necrosis of the
brain areas affected (1-3).
The clinical symptoms of cerebral infarction are complex, and are
associated with the location of the brain damage, the size of the
cerebral ischemic blood vessels, the severity of the ischemia, the
influence of other diseases prior to the onset of ischemia and the
effects of the comorbidity of cerebral infarction and other types
of vital organ disease. Mild cases of cerebral infarction can be
completely asymptomatic, but they can also manifest as recurrent
limb paralysis or dizziness, which is known as a transient ischemic
attack. Individuals with severe cases of cerebral infarction may
not only experience limb paralysis, but acute coma and death may
also occur in some cases (28,29).
The expression levels of SNHG1 were previously found
to be upregulated in middle cerebral artery occlusion model mice
(30), indicating the potentially
important role of SNHG1 in cerebral infarction. Furthermore, the
level of SNHG1 is elevated in OGD and cerebral ischemic rodents and
exerts neuroprotective effects involving the PI3K/AKT pathway
(20). To investigate the
mechanisms underlying the role of lncRNA SNHG1 in cerebral
infarction, the present study predicted and verified the binding
site between miR-329-3p and SNHG1. Microglia are small cells found
in the nervous system, which are mostly localized in the gray
matter near the cell bodies of neurons and around small blood
vessels, but can also be found in the white matter of the brain
(31). Microglia can promote the
development of the nervous system and regulate the number of
neurons in the central nervous system (32). After an ischemic stroke, activated
microglia were discovered to promote the excessive release of
inflammatory cytokines, thereby exacerbating neuronal damage
(33,34). Therefore, regulating the activation
of endogenous microglia may represent a new treatment target in
ischemic stroke (22,23). Furthermore, OGD induced BV-2 cells
have been widely used to study cerebral infarction in vitro
(24-26).
In the present study, BV-2 cells were used to establish an in
vitro OGD model, and the results revealed that the expression
levels of SNHG1 were upregulated in OGD-induced cells, while
miR-329-3p expression levels were downregulated.
To determine whether SNHG1 affected the activation
of BV-2 cells by regulating miR-329-3p expression, experiments with
BV-2 cells were performed following overexpression of SNHG1 and
downregulation of miR-329-3p. The results demonstrated that
transfection with the SNHG1-plasmid downregulated miR-329-3p
expression, while this effect was reversed following transfection
with the miR-329-3p mimic. TNF-α and NO are important
proinflammatory factors that have been found to play important
roles in the occurrence and development of the systemic
inflammatory response (35,36). Thus, the present study measured the
release of TNF-α and NO in the supernatant of OGD-induced BV-2
cells. The results revealed that the overexpression of SNHG1 or
knockdown of miR-329-3p expression inhibited the release of TNF-α
and NO in the supernatant of OGD-induced BV-2 cells. Contrary to
our findings, an in vitro study on Parkinson's disease
demonstrated that LPS increased SNHG1 expression in BV-2 cells,
leading to NLRP3 activation and cytokine production (37). Moreover, the findings of present
study indicated that the overexpression of SNHG1 or knockdown of
miR-329-3p expression reduced OGD-induced BV-2 cell apoptosis. Of
note, all the effects of SNHG1 overexpression on OGD-induced BV-2
cells were significantly reversed by miR-329-3p overexpression.
Other previous studies have also reported that SNHG1 may act as a
sponge RNA to interfere with the actions of target miRNAs (38,39).
The findings of the present study confirmed that the effects of
SNHG1 on OGD-induced BV-2 cells are mediated through downregulating
miR-329-3p levels.
The present study was only a preliminary in
vitro study on the role of lncRNA SNHG1 in cerebral infarction.
In order to further elucidate the role of lncRNA SNHG1 in cerebral
infarction, extensive in-depth research is required. For example,
the role of lncRNA SNHG1 silencing or miR-329-3p overexpression
alone in cerebral infarction should be investigated. Furthermore,
the downstream targets of SNHG1 and miR-329-3p should be further
analyzed. In addition, the role of lncRNA SNHG1 and miR-329-3p in
cerebral infarction should be studied in vivo. These issues
will be addressed in future studies.
In conclusion, the results of the present study
indicated that lncRNA SNHG1 may play an important role in microglia
activation by modulating the expression of miR-329-3p. Therefore,
targeting SNHG1 and miR-329-3p may provide novel strategies for the
treatment of cerebral infarction.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Science and
Technology Development Foundation of Nanjing Medical University
(grant no. 2017NJMU083), the Zhejiang Chinese Medical University
Field of Scientific Research Foundation (grant no. 2018ZY23) and
the Hangzhou Agriculture and Social Development Research Project
Foundation (grant no. 20191203B84).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JH designed the study, in addition to performing all
experiments, analyzing the data and preparing the manuscript. XX,
MJ, JL, and NL contributed to performing the experiments and data
collection. TN analyzed and interpreted the data, and revised the
manuscript. JH and TN confirm the authenticity of all the raw data.
All the authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sun W, Li G, Zeng X, Lai Z, Wang M, Ouyang
Y, Zeng G, Peng J, Zhong J, Xiao D, et al: Clinical and imaging
characteristics of cerebral infarction in patients with nonvalvular
atrial fibrillation combined with cerebral artery stenosis. J
Atheroscler Thromb. 25:720–732. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Choi JW, Chong S, Phi JH, Lee JY, Kim HS,
Chae JH, Lee J and Kim SK: Postoperative symptomatic cerebral
infarction in pediatric moyamoya disease: Risk factors and clinical
outcome. World Neurosurg. 136:e158–e164. 2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wang YW and Zhang GM: New silent cerebral
infarction in patients with acute non-cerebral amyloid angiopathy
intracerebral hemorrhage as a predictor of recurrent
cerebrovascular events. Med Sci Monit. 25:418–426. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Sveinsson OA, Kjartansson O and
Valdimarsson EM: Cerebral ischemia/infarction-epidemiology, causes
and symptoms. Laeknabladid. 100:271–279. 2014.PubMed/NCBI View Article : Google Scholar : (In Icelandic).
|
|
5
|
Rojsanga W, Sawanyawisuth K, Chotmongkol
V, Tiamkao S, Kongbonkiat K and Kasemsap N: Clinical risk factors
predictive of thrombotic stroke with large cerebral infarction.
Neurol Int. 11(7941)2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Jin L, Zhou J, Shi W, Xu L, Sheng J, Fan
J, Yuan Y and Yuan H: Effects of six types of aspirin combination
medications for treatment of acute cerebral infarction in China: A
network meta-analysis. J Clin Pharm Ther. 44:91–101.
2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Orlický M, Hrbáč T, Sameš M, Vachata P,
Hejčl A, Otáhal D, Havelka J, Netuka D, Herzig R, Langová K and
Školoudík D: Anesthesia type determines risk of cerebral infarction
after carotid endarterectomy. J Vasc Surg. 70:138–147.
2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Tu J, Wang LX, Wen HF, Xu YC and Wang PF:
The association of different types of cerebral infarction with
post-stroke depression and cognitive impairment. Medicine
(Baltimore). 97(e10919)2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Xu W, Xie N, Zhang C and Huang Q: Imaging
characteristics and pathogenesis of intracranial artery stenosis in
patients with acute cerebral infarction. Exp Ther Med.
15:4564–4570. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Rodríguez-Gómez JA, Kavanagh E,
Engskog-Vlachos P, Engskog MKR, Herrera AJ, Espinosa-Oliva AM,
Joseph B, Hajji N, Venero JL and Burguillos MA: Microglia: Agents
of the CNS pro-inflammatory response. Cells. 9(1717)2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kinuthia UM, Wolf A and Langmann T:
Microglia and inflammatory responses in diabetic retinopathy. Front
Immunol. 11(564077)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Szcześniak MW, Wanowska E, Mukherjee N,
Ohler U and Makałowska I: Towards a deeper annotation of human
lncRNAs. Biochim Biophys Acta Gene Regul Mech.
1863(194385)2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Puvvula PK: LncRNAs regulatory networks in
cellular senescence. Int J Mol Sci. 20(2615)2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Simion V, Haemmig S and Feinberg MW:
LncRNAs in vascular biology and disease. Vascul Pharmacol.
114:145–156. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Lekka E and Hall J: Noncoding RNAs in
disease. FEBS Lett. 592:2884–2900. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Dinescu S, Ignat S, Lazar AD, Constantin
C, Neagu M and Costache M: Epitranscriptomic signatures in lncRNAs
and their possible roles in cancer. Genes (Basel).
10(52)2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Shi YL, Wang Q and Wei JC: Influence of
lncRNA-MALAT1 on neuronal apoptosis in rats with cerebral
infarction through regulating the ERK/MAPK signaling pathway. Eur
Rev Med Pharmacol Sci. 23:8039–8048. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Shen J, Zhao Z, Shang W, Liu C, Zhang B,
Xu Z and Cai H: Fabrication of a nano polymer wrapping Meg3 ShRNA
plasmid for the treatment of cerebral infarction. Artif Cells
Nanomed Biotechnol. 46 (Suppl 2):S894–S903. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhao JH, Wang B, Wang XH, Wang JR and Xu
CW: Influence of lncRNA ANRIL on neuronal apoptosis in rats with
cerebral infarction by regulating the NF-κB signaling pathway. Eur
Rev Med Pharmacol Sci. 23:10092–10100. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Chen J, Zhang W, Wu YQ, Chen H and Zhao
JF: LncRNA SNHG1 inhibits neuronal apoptosis in cerebral infarction
rats through PI3K/Akt signaling pathway. Eur Rev Med Pharmacol Sci.
23:5366–5373. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Huang L, Jiang X, Wang Z, Zhong X, Tai S
and Cui Y: Small nucleolar RNA host gene 1: A new biomarker and
therapeutic target for cancers. Pathol Res Pract. 214:1247–1252.
2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kaur C, Sivakumar V, Zou Z and Ling EA:
Microglia-derived proinflammatory cytokines tumor necrosis
factor-alpha and interleukin-1beta induce Purkinje neuronal
apoptosis via their receptors in hypoxic neonatal rat brain. Brain
Struct Funct. 219:151–170. 2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ma Y, Wang J, Wang Y and Yang GY: The
biphasic function of microglia in ischemic stroke. Prog Neurobiol.
157:247–272. 2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Mo Z, Tang C, Li H, Lei J, Zhu L, Kou L,
Li H, Luo S, Li C, Chen W and Zhang L: Eicosapentaenoic acid
prevents inflammation induced by acute cerebral infarction through
inhibition of NLRP3 inflammasome activation. Life Sci.
242(117133)2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Qi X, Shao M, Sun H, Shen Y, Meng D and
Huo W: Long non-coding RNA SNHG14 promotes microglia activation by
regulating miR-145-5p/PLA2G4A in cerebral infarction. Neuroscience.
348:98–106. 2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Lu D, Shen L, Mai H, Zang J, Liu Y, Tsang
CK, Li K and Xu A: HMG-CoA reductase inhibitors attenuate neuronal
damage by suppressing oxygen glucose deprivation-induced activated
microglial cells. Neural Plast. 2019(7675496)2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Chen XY, Wang Q, Wang X and Wong KS:
Clinical features of thalamic stroke. Curr Treat Options Neurol.
19(5)2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Liang Y, Wu J, Liu J, Liu H and Chen J:
The clinical implications of thrombelastography in the diagnosis of
acute cerebral infarction. Clin Lab. 64:147–152. 2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhang L, Luo X, Chen F, Yuan W, Xiao X,
Zhang X, Dong Y, Zhang Y and Liu Y: LncRNA SNHG1 regulates
cerebrovascular pathologies as a competing endogenous RNA through
HIF-1α/VEGF signaling in ischemic stroke. J Cell Biochem.
119:5460–5472. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Prinz M, Jung S and Priller J: Microglia
biology: One century of evolving concepts. Cell. 179:292–311.
2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Nayak D, Roth TL and McGavern DB:
Microglia development and function. Annu Rev Immunol. 32:367–402.
2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Wolf SA, Boddeke HW and Kettenmann H:
Microglia in physiology and disease. Annu Rev Physiol. 79:619–643.
2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Bartels T, De Schepper S and Hong S:
Microglia modulate neurodegeneration in Alzheimer's and Parkinson's
diseases. Science. 370:66–69. 2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Aggarwal R, Jain AK, Mittal P, Kohli M,
Jawanjal P and Rath G: Association of pro- and anti-inflammatory
cytokines in preeclampsia. J Clin Lab Anal.
33(e22834)2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Li Q, Hu X, Xuan Y, Ying J, Fei Y, Rong J,
Zhang Y, Zhang J, Liu C and Liu Z: Kaempferol protects
ethanol-induced gastric ulcers in mice via pro-inflammatory
cytokines and NO. Acta Biochim Biophys Sin (Shanghai). 50:246–253.
2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Cao B, Wang T, Qu Q, Kang T and Yang Q:
Long noncoding RNA SNHG1 promotes neuroinflammation in Parkinson's
disease via regulating miR-7/NLRP3 pathway. Neuroscience.
388:118–127. 2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Deng R, Zhang J and Chen J: lncRNA SNHG1
negatively regulates miRNA-101-3p to enhance the expression of
ROCK1 and promote cell proliferation, migration and invasion in
osteosarcoma. Int J Mol Med. 43:1157–1166. 2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Yan SM, Li H, Shu Q, Wu WJ, Luo XM and Lu
L: LncRNA SNHG1 exerts a protective role in cardiomyocytes
hypertrophy via targeting miR-15a-5p/HMGA1 axis. Cell Biol Int.
44:1009–1019. 2020.PubMed/NCBI View Article : Google Scholar
|