Introduction
Cardiac arrhythmias leading to sudden cardiac death
are a common health issue worldwide (1). Arrhythmia includes abnormal cardiac
rhythm, such as bradyarrhythmia, atrial fibrillation and abnormal
electrical signal conduction in cardiac tissue (2). Cardiac rhythm abnormalities tend to
worsen with increasing age and are more prevalent among male
Caucasians. Other risk factors include cardiac comorbidities, such
as hypertension, diabetes and congestive heart failure (3). Atrial fibrillation is a common
complication of silent hypertension, the inefficient management of
which often leads to stroke (4).
Improved therapies and updated treatments are urgently required to
reduce the mortality associated with arrhythmias (5). Unfortunately, inadequate research into
the progression of arrhythmias and the underlying mechanisms that
link them with heart failure has hindered the efficiency of
treatment (6).
To improve the understanding of arrhythmias, it is
necessary to determine which genes are involved in the disturbance
of electrophysiology and in the progression of arrhythmogenesis
(7). Syndecan-1 is a transmembrane
heparan sulfate proteoglycan that plays a major role in
atherogenesis and cardiac fibrosis, mostly in association with
angiotensin II (8). It is involved
in the regulation of inflammation, response to injury, regeneration
and remodeling, and is protective following myocardial infarction
(9).
Generally, caspases are major influencers of
inflammation and apoptosis (10).
Caspase-6 acts as a substrate for numerous neuronal-associated
proteins, some of the most important of which are
microtubule-associated proteins (11). It has been reported that following
its induction by tumor necrosis factor (TNF)-α, caspase-6 cleaves
the cardiac intermediate filament desmin to initiate cardiac
failure (12). In the present
study, the aim was to determine the inductive role of syndecan-1 in
the regulation of caspase-6 at different stages of the development
of cardiac arrhythmia. In the present study, Leptin
receptor-deficient (C57BL/KS-leprdb/leprdb)
mice were used, which normally develop type 2 diabetes, in addition
to developing initial- and end-stage cardiac arrhythmia after 5 and
8 months, respectively.
Materials and methods
Experimental animals and study
design
Leptin receptor-deficient male mice (13) (n=36; average 23 g; 2 months old)
with an altered genetic background
(C57BL/KS-leprdb/leprdb) were purchased from
Jackson Laboratory. The mice were housed in a laboratory
environment and carefully monitored until they were acclimated to
the new environment (one week). The mice were maintained at 24˚C
with relative humidity of 55-60%, with free access to water and
regular chow and with a 12-h light/dark cycle. The genetically
modified C57BL/KS-leprdb/leprdb mice (n=24)
were maintained for 5 and 8 months (n=12 per time point) to develop
initial and advanced stages of cardiac arrhythmia, respectively.
The control C57BL/KS mice (n=12) were maintained under the same
conditions, with the exception that they followed a regular diet
throughout the experiments. At the end of 5 and 8 months, the
animals (n=6) were sacrificed in each group by cervical dislocation
and for control mice 8 month samples were taken for experimental
analysis. Prior to sacrifice, the body weight and blood glucose
level in a blood sample taken from the tail tip were measured. The
cardiac weight was determined after sacrifice. The experimental
procedure, animal handling, maintenance and protocol were
pre-approved by and carried out according to the guidelines of the
Ethics Committee of Affiliated Wujiang Hospital of Nantong
University (ref. no. AWHNU06/04/18).
Electrocardiogram (ECG) analyses
To analyze their electrophysiological condition, the
control and leptin receptor-deficient diabetic mice were subjected
to ECG analysis, as previously described (14). In brief, the mice were anesthetized
with 1% isoflurane vapor, and an ECG electrode needle was inserted
subcutaneously in the upper right limb and lower left abdomen. The
ECG was recorded until the signal returned to baseline and the
heart rate was stable (maximum of 8 min). The average times between
successive P waves (PP interval), from the onset of the P wave to
the start of the QRS complex (PR interval) and between QRS
complexes (RR interval) were determined from the ECGs.
Histology and
immunohistochemistry
After sacrifice, the mouse heart was carefully
excised by thoracotomy. The cardiac tissue was washed with 1X
phosphate-buffered saline (PBS) and quickly transferred to 4%
formalin solution for fixation at 37˚C for 48 h. Following
fixation, the tissues were placed in increasing concentrations of
isopropanol for dehydration. After clearing with xylene, the
tissues were embedded with paraffin. The tissues were then sliced
transversely using a microtome set to 6 µm. The sections were
dewaxed with xylene, dehydrated with isopropanol and stained with
hematoxylin (7 min) and eosin (20 sec) at room temperature for
subsequent imaging using a Nikon light microscope (Eclipse Ei;
Nikon Corporation). The cardiac tissue hardening and loosening were
analyzed based on the cellular variations observed within the
particular area.
For immunohistochemical analyses, the antigens were
unmasked by boiling the sections for 10 min in 10 mM sodium citrate
buffer (pH 6.0). Cellular endogenous peroxidase was blocked using
methanol in 2% H2O2 for 20 min at 24˚C. After
washing with 1X PBS, the non-specific sites in the sections were
blocked using 4% bovine serum albumin (Sigma-Aldrich; Merck KGaA)
in 1X Tris-buffered saline containing 0.1% Tween-20 (TBST) for 1 h
at room temperature. The primary antibodies anti-syndecan-1
(ab128936; Abcam) and anti-caspase-6 (ab231349; Abcam) were added
at 1:300 dilutions over the sections, which were kept at 4˚C for 5
h. The unbound antibody was washed away thrice with 1X
Tris-buffered saline containing TBST and further incubated with a
horseradish peroxidase (HRP)-conjugated secondary antibody (cat.
no. ab205718; Abcam, 1:3,000 dilution) for 1 h at room temperature.
The peroxidase signals, which indirectly reflect the binding of
primary antibody, were visualized using a 3,3'-diaminobenzidine
(DAB) kit (Sigma-Aldrich; Merck KGaA). The sections were
counterstained with hematoxylin for 7 mins at room temperature,
which gave a blue-purple hue to the background to contrast with the
brown antibody signal under Nikon light microscope (Eclipse Ei;
Nikon Corporation).
Silencing syndecan-1 using small
interfering RNA (siRNA)
Syndecan-1 expression was silenced using siRNA
against syndecan-1 (siSyn; cat no. AM16708; Thermo Fisher
Scientific, Inc.). The negative control used was a non-targeted
siRNA with limited sequence similarity to the targeted gene (siCtr;
cat no. AM4611; Thermo Fisher Scientific, Inc.). siRNA against
GAPDH was also used (siGAPDH; cat no. AM4631; Thermo Fisher
Scientific, Inc.) as a control, The siRNAs were suspended in water
to a final concentration of 0.5 µg/µl. To facilitate the
transfection, 0.5 µl 50 nM Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) was mixed with the
siRNA 20 min prior to injection. The control, initial- and
late-stage cardiac arrhythmia groups were each divided into four
groups as follows: Untreated, siCtr, siSyn and siGAPDH. Each group
had six mice. The siRNA was injected into the tail veins of mice at
the ages of 4 months (mice developing with initial-stage cardiac
arrhythmia) or 7 months (mice developing with late-stage cardiac
arrhythmia). The mice were sacrificed 1 month later (at the end of
5 and 8 months, respectively) and cardiac tissue samples were
collected for analysis by western blotting.
Western blotting
The dissected heart was immediately placed in
ice-cold 1X PBS and the right auricle tissue was used for protein
sample preparation. The tissues were mechanically homogenized with
ice-cold 2X Laemmli buffer (Bio-Rad Laboratories, Inc.). The
homogenized tissue was then immediately heated in a boiling water
bath for 5 min to release proteins from the cell lysate. The
protein concentration in each sample was estimated using the Lowry
method. The proteins were separated by SDS-PAGE (12% gel, 70 µg
protein/well) for 4 h at 50 V. The separated proteins were then
transferred to a polyvinyl difluoride membrane using the semi-dry
transfer method. The membrane was blocked with 4% skimmed milk
powder for 2 h at room temperature to prevent non-specific binding,
and the target proteins were detected by incubation for 4 h at 4˚C
with the primary antibodies anti-syndecan-1 (cat. no. ab128936;
Abcam; 1:1,000 dilution), anti-caspase-6 (cat. no. ab231349; Abcam;
1:500 dilution) and anti-β actin (cat. no. ab8227; Abcam; 1:1,000
dilution). Following gentle rocking for 5 min, the membrane was
then incubated for 1 h at room temperature with goat anti-rabbit
HRP-conjugated secondary antibody (ab6721; dilution, 1:4,000;
Abcam). The membrane was washed thoroughly with 1X TBST buffer for
10 min and then counter reacted with DAB to obtain the protein
signals. The intensity of the signal band was documented using Gel
documentation system (InGenius3; Syngene) and analyzed using ImageJ
software (v1.52h, National Institutes of Health).
Statistical analysis
The experiments or observations were performed at
least three times, and the obtained data are presented as the mean
± standard deviation. The differences between two groups were
compared using two-tailed Student's t-test and among multiple
groups using two-way ANOVA followed by Tukey's post hoc tests for
multiple comparisons. P<0.05 was considered to indicate a
statistically significant result.
Results
Mouse model develops progressive
cardiac arrhythmia
In comparison with the control mice, the leptin
receptor-deficient mice exhibited abnormal weight gain with
advancing age. The control C57BL/KS mice (n=12) exhibited an
average mean weight of 30±2 g after 5 months and 34±2 g after 8
months. By comparison, the leptin receptor-deficient diabetic mice
(n=12) exhibited mean weights of 41±3 g (37% higher than that of
the control mice) after 5 months and 48±3 g after 8 months (41%
higher than that of the control mice).
The leptin receptor-deficient mice were also
hyperglycemic at 5 and 8 months. At 5 months, the control mice had
fasting blood glucose levels of 80±4 mg/dl, while the leptin
receptor-deficient diabetic mice had fasting blood glucose levels
of 452±14 mg/dl. Similarly, at 8 months, the fasting blood glucose
level of the leptin receptor-deficient mice was 517±16 mg/dl, while
that of the control mice was 88±7 mg/dl.
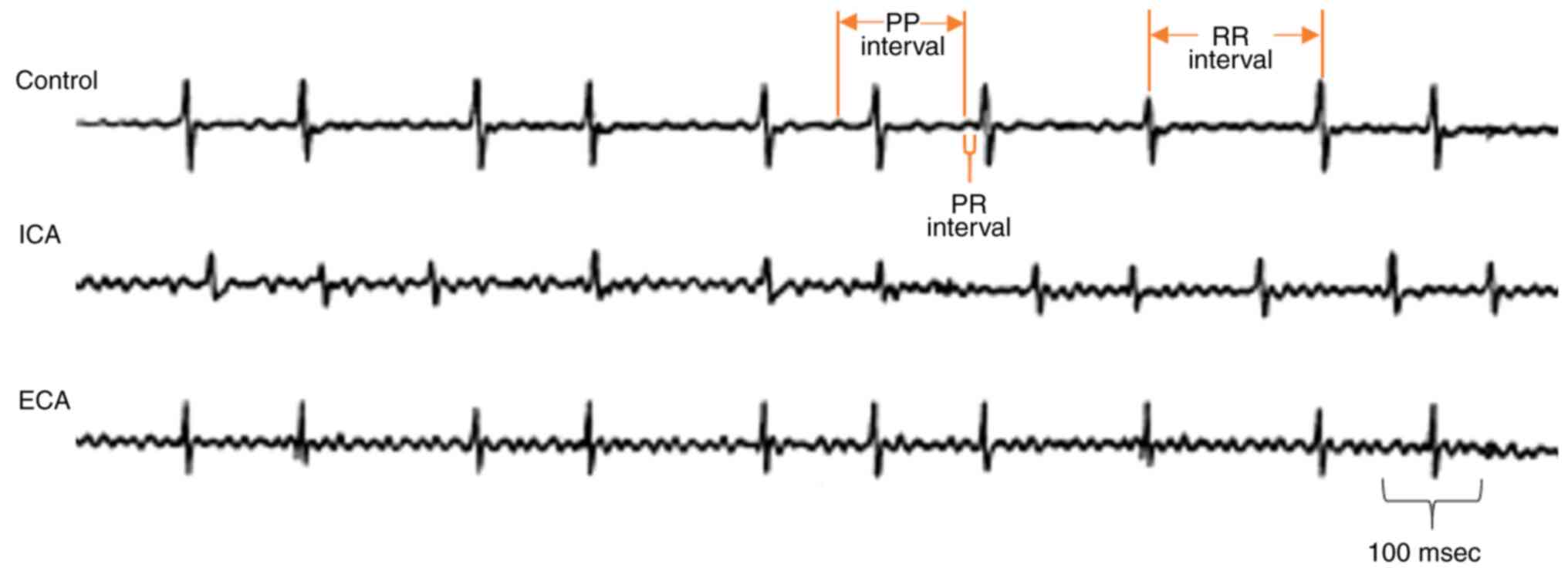
The development of arrhythmia was monitored by the
analysis of ECG recordings. The 5-month-old mice with leptin
receptor deficiency exhibited longer PP intervals (120 msec)
compared with the control mice (Fig.
1). Furthermore, the 8-month-old leptin receptor-deficient mice
exhibited shorter and longer PP intervals more frequently, which
directly reflect the development of adverse cardiac arrhythmia in
the form of atrial fibrillation (Fig.
1). Furthermore, the PR and RR intervals exhibited progressive
variations as the cardiac arrhythmia became severe (Fig. 1).
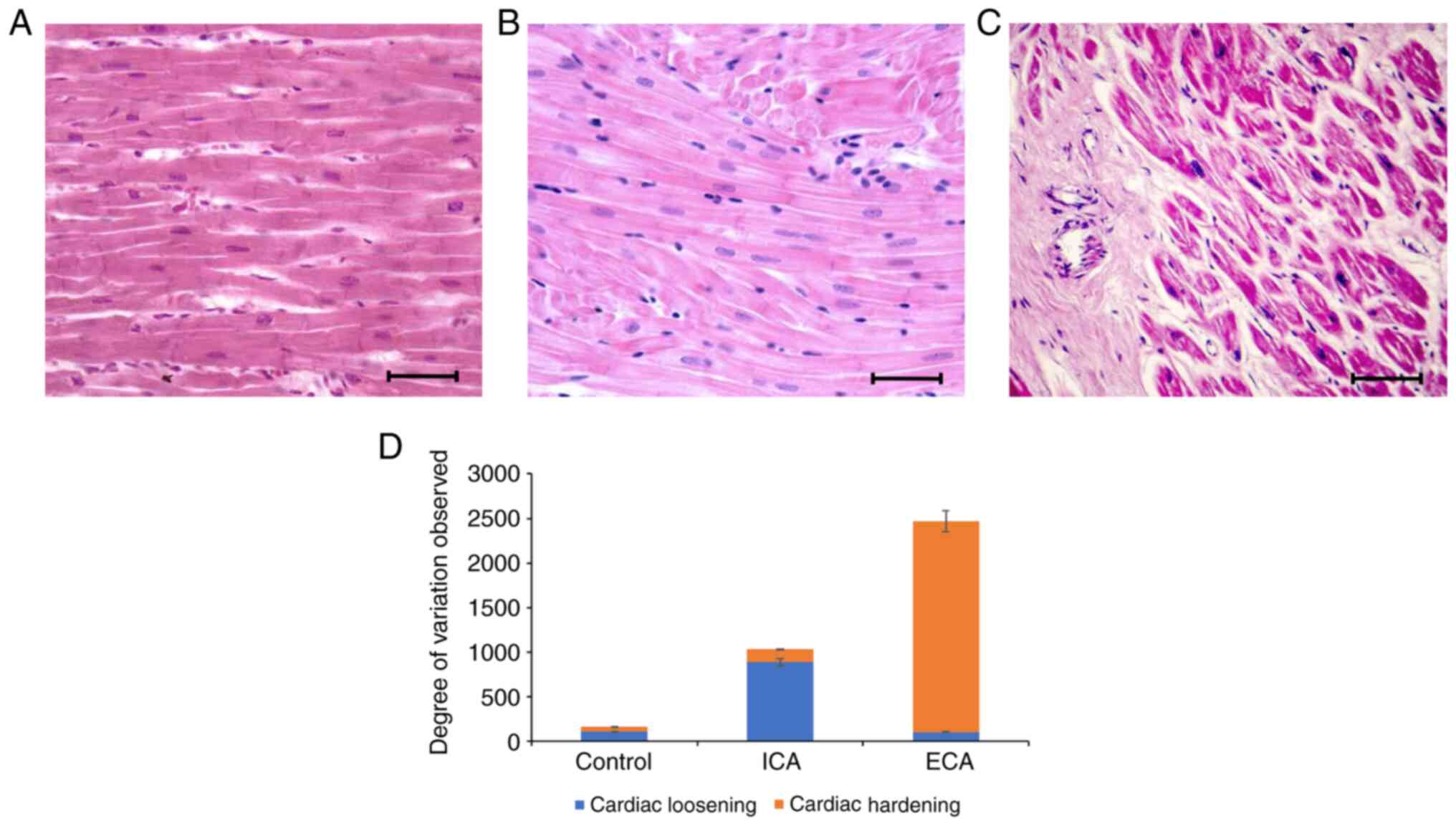
Histological analysis of initial and
end stage cardiac arrhythmia
Cardiac specimens from the control, initial- and
end-stage cardiac arrhythmic mice were sectioned and subjected to
histopathological analysis (Fig.
2). The histomorphological analysis of the cardiac specimens
from the control mice revealed elongated, branched cells with
prominent nuclei (Fig. 2A). The
5-month-old leptin receptor-deficient mice exhibited
histopathological changes that included scattered and loosened
cardiac cells with enlarged, dark nuclei (Fig. 2B); after 8 months, when these mice
had developed a severe form of cardiac arrhythmia, a complete
change in cardiomyocyte structure was observed, with excessive
secretion of extracellular matrix tissue leading to hardening of
the tissue (Fig. 2C). These
histological variations observed in the mice with initial- and
end-stage cardiac arrhythmia were quantified and are graphically
presented in Fig. 2D. The data
indicate that cardiac tissue integrity progressed from a loosened
cellular structure to tissue hardening as the cardiac arrhythmia
progressed from initial- to end-stage (Fig. 2D).
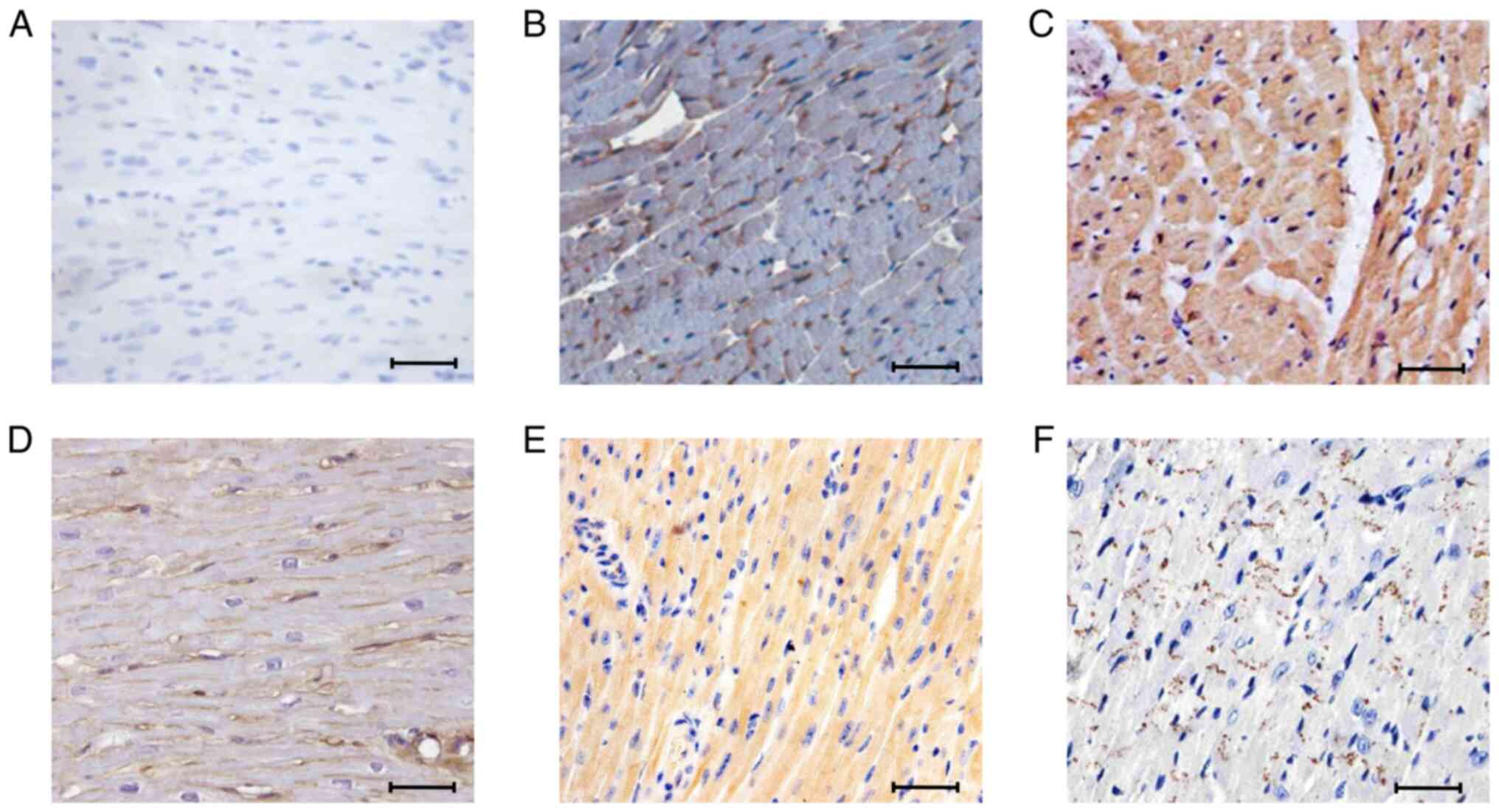
Caspase-6 exhibits elevated expression
in progressive stages of cardiac arrhythmia
Immunohistochemistry was used to analyze caspase-6
expression in the cardiac tissue of mice exhibiting initial- and
end-stage arrhythmia. In the control cardiac tissue, caspase-6 was
expressed at a low level (Fig. 3A).
The expression of caspase-6 was markedly elevated at the initial
stage of cardiac arrhythmia (Fig.
3B), and was elevated even further at the end stage of cardiac
arrhythmia (Fig. 3C).
Syndecan-1 is induced in the initial
stage of cardiac arrhythmia and repressed in the later stage
To understand the role of syndecan-1 in cardiac
pathogenesis, immunohistochemical analyses of syndecan-1 expression
were performed at progressive stages of cardiac arrhythmia
(Fig. 3D-F). In the control cardiac
tissue, syndecan-1 expression was observed primarily on cell
surfaces (Fig. 3D). In cardiac
tissues from the mice with initial-stage cardiac arrhythmia,
syndecan-1 was expressed throughout the cardiac tissue with
prominent expression on the cell surface and in the cytoplasm,
which implies expression at the transcriptional level (Fig. 3E). However, syndecan-1 appeared to
be downregulated in cardiac tissues from end-stage arrhythmic mice
(Fig. 3F), relative to those from
control mice (Fig. 3D).
Western blot analysis of caspase-6 and
syndecan-1 in tissues from cardiac arrhythmic mice
Western blotting was also carried out to evaluate
the expression of caspase-6 and syndecan-1 in the cardiac tissues
of the mice (Fig. 4). In agreement
with the immunohistochemistry data (Fig. 3), the expression of caspase-6 was
observed to increase over the course of development of cardiac
arrhythmia. In the cardiac tissues from the mice with initial-stage
cardiac arrhythmia, the level of caspase-6 was 1.5-fold that of the
control cardiac samples. Furthermore, in the cardiac tissues from
the mice with end-stage cardiac arrhythmia, caspase-6 was expressed
at 3.6-fold the level detected in control cardiac samples. Western
blotting analysis also showed that syndecan-1 is extremely
upregulated in the initial stage of cardiac arrhythmia, with
5.8-fold greater expression compared with that in the control
samples. Surprisingly, however, syndecan-1 expression was reduced
at the end stage of cardiac arrhythmia and was downregulated to
almost half the level in the control cardiac tissue. In
syndecan-1-silenced tissue at the initial stage of cardiac
arrhythmia, the caspase-6 level was elevated to 1.8-fold that in
the control siRNA treated group, while at the end stage, the
caspase-6 level in the syndecan-1-silenced tissue was 1.2-fold that
in the control siRNA treated group (Fig. 4). In this experiment, non-targeting
siRNA served as a negative control and siRNA against GADPH served
as an additional control.
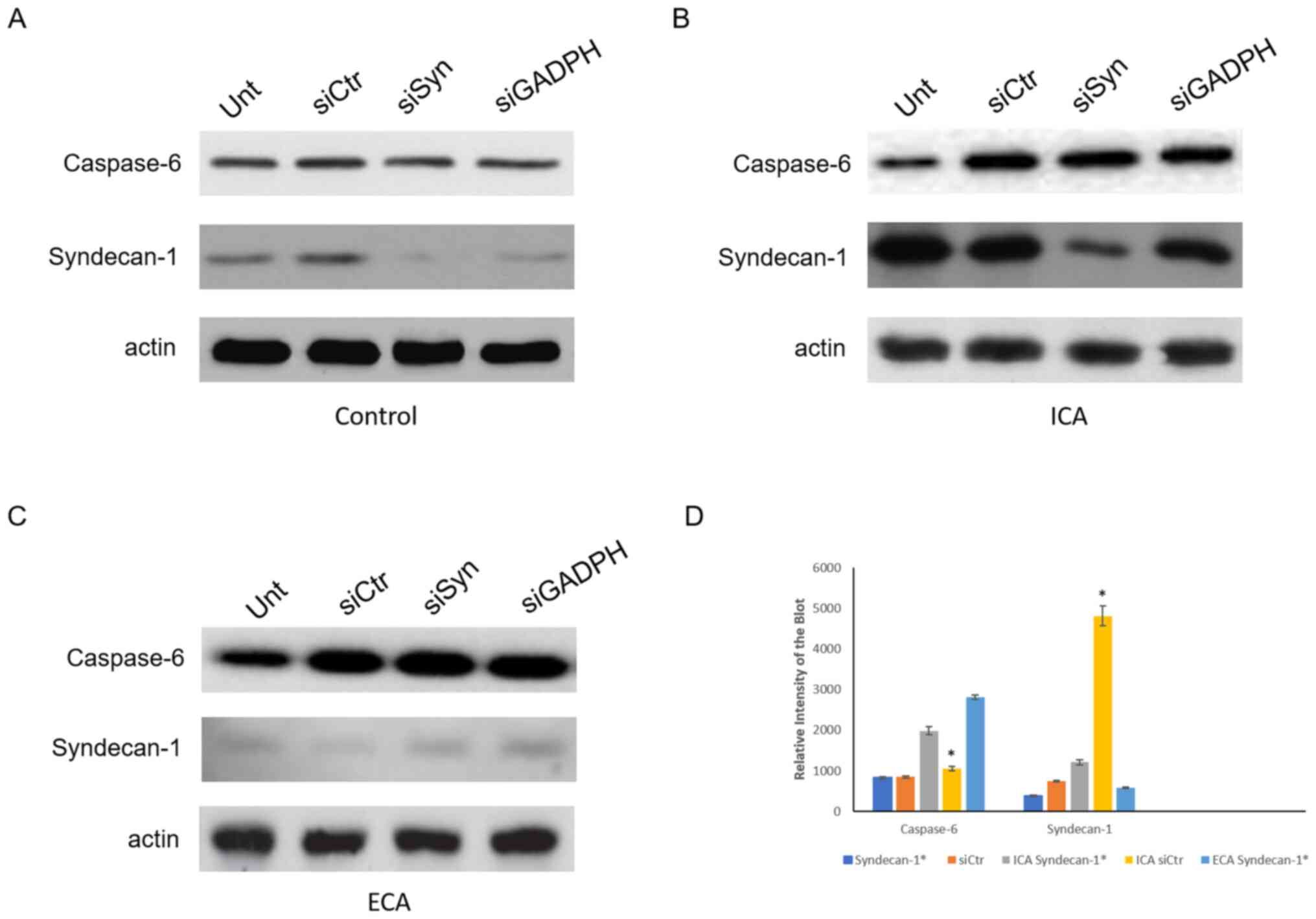 | Figure 4Western blots of the protein lysates
of cardiac tissue from mice at various stages of cardiac
arrhythmia. Expression of caspase-6 and syndecan-1 in cardiac
tissue from the Unt, siCtr, siSyn and siGADPH groups of (A) control
mice, (B) mice with ICA and (C) mice with ECA. (D) Quantification
of caspase-6 and syndecan-1. *P<0.01. The results are
normalized to the β-actin loading control. Unt, untreated; siCtr,
control siRNA; siSyn, siRNA against syndecan-1; siGADPH, siRNA
against GAPDH; siRNA, small interfering RNA; ICA, initial-stage
cardiac arrhythmia; ECA, end-stage cardiac arrhythmia.
*Represents the expression of caspase-6 or syndecan-1 in
syndecan-1 silenced tissue. |
Discussion
Leptin deficiency is associated with
insulin-dependent metabolism, which affects cardiac rhythm, which
in turn has chronotropic effects (15-17).
Mice treated with streptozotocin also develop chronotropic
responses, as well as type 1 diabetes mellitus (18). Since they are genetically modified,
the leptin receptor-deficient
C57BL/KS-leprdb/leprdb mice provide more
accurate experimental results than do streptozotocin-induced mice
(13). In the present study, over
the course of 5-8 months, the
C57BL/KS-leprdb/leprdb mice exhibited
increasing weight gain, elevated blood glucose levels and ECG
abnormalities. A previous study showed that cardiac arrhythmia
develops due to initial metabolic disturbances, which in turn
affect cardioregulatory autonomic dysfunction (19). The present study is consistent with
this finding. The results demonstrated that the heterogeneous
pattern of ECG morphology became more complex over time; the PP
interval and its uniformity was disturbed, indicating severe
cardiac arrhythmia (20).
The histological data confirmed changes to the
cellular morphology of the cardiac tissue, with a loosened
structure appearing after 5 months. This loosened structure was
likely due to unstable cellular adhesion components, resulting in
the initiation of tissue remodeling and fibro-fatty replacement
(21,22). After 8 months, the
C57BL/KS-leprdb/leprdb mice exhibited cardiac
tissue thickening, which was likely due to the infiltration of fat
and/or fibrosis, affecting cardiac elasticity (23,24).
The cardiac tissue hardening may also have been due to the
excessive secretion of extracellular matrix to replace degenerated
tissue (25). The risk factors for
cardiac arrhythmia and heart dysfunction have been indicated to
involve the excessive stimulation of extracellular matrix protein
secretion, which leads to cardiac failure (26).
Caspase-6 has been associated with neurological
disorders; it cleaves neuronal microtubule-associated proteins,
causing neurodegeneration (27,28).
Caspase-6 also plays a role in apoptosis, and its principal
substrates are nuclear proteins and intermediate filaments
(29). However, little is known
about the role of caspase-6-mediated apoptosis in heart disease
(30,31). In the present study, the elevated
expression of caspase-6 in the cardiac tissue of the mice with
initial- and end-stage cardiac arrhythmia may indicate that cardiac
intermediate filaments and extracellular matrix were the targets of
caspase-6 activity, which would disrupt the normal cardiac
microarchitecture. In the initial stage of cardiac arrhythmia, the
expression of caspase-6 occurred primarily at the cell surface,
suggesting that disruption of the extracellular matrix was the
initiating event leading to cardiac arrhythmia. Later in the
progression of the arrhythmia, caspase-6 expression was observed
inside the cells, as well as at the surface, suggesting that at the
end stage, the intermediate filaments were also targeted. The
increased level of caspase-6 expression in the cytoplasm may also
be indicative of apoptosis (32).
Indeed, caspase-6 has been associated with myocardial infarction
and brain injury following cardiac arrest (33).
Syndecan-1 is reported to be a promising marker for
the degree of cardiac fibrosis (34). In general, syndecan-1 is involved in
the regulation of inflammation and regenerative responses, but its
mode of action is highly variable and influenced by numerous
independent factors such as liver fibrosis and growth stimulating
conditions (9). In the present
study, in the initial stage of cardiac arrhythmia, the expression
of syndecan-1 was elevated and appeared to be protective by
restricting caspase-6 expression. However, at the end-stage of
cardiac arrhythmia, syndecan-1 was downregulated, favoring the
overexpression of caspase-6 and the concomitant destruction of
cardiac tissue.
In conclusion, the model
C57BL/KS-leprdb/leprdb mice developed
initial- and end-stage cardiac arrhythmia after 5 and 8 months,
respectively. The overexpression of caspase-6 at the cell surface
observed during the initial stage of arrhythmia suggests that
caspase-6 initially targets the extracellular matrix. The later
expression of caspase-6 within the cells suggests that the
cytoskeleton is also targeted as the arrhythmia progresses. The
overexpression of syndecan-1 in the initial stage of cardiac
arrhythmia and its regenerative response suggests that syndecan-1
restricts the expression of caspase-6 in the initial stage, but
then the downregulation of syndecan-1 favors the destructive
overexpression of caspase-6 in the end-stage of cardiac
arrhythmia.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YW and CW contributed to the design of the study and
fund mobilization. YW performed histology, immunohistochemistry,
western blotting and silencing experiments and LC contributed to
the experiments, CW performed the statistical analysis, critically
reviewed the manuscript and provide support in the experiments. All
authors read and approved the final manuscript. YW and CW confirm
the authenticity of all the raw data.
Ethics approval and consent to
participate
The experimental procedure, animal handling,
maintenance and protocol were approved by the Ethics Committee of
Affiliated Wujiang Hospital of Nantong University (ref. no:
AWHNU06/04/18).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chugh SS, Roth GA, Gillum RF and Mensah
GA: Global burden of atrial fibrillation in developed and
developing nations. Glob Heart. 9:113–119. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Khurshid S, Choi SH, Weng LC, Wang EY,
Trinquart L, Benjamin EJ, Ellinor PT and Lubitz SA: Frequency of
cardiac rhythm abnormalities in a half million adults. Circ
Arrhythm Electrophysiol. 11(e006273)2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Rietbrock S, Heeley E, Plumb J and van
Staa T: Chronic atrial fibrillation: Incidence, prevalence, and
prediction of stroke using the Congestive heart failure,
Hypertension, Age> 75, Diabetes mellitus, and prior Stroke or
transient ischemic attack (CHADS2) risk stratification scheme. Am
Heart J. 156:57–64. 2008.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Abrich VA, Narichania AD, Love WT, Lanza
LA, Shen WK and Sorajja D: Left atrial appendage exclusion during
mitral valve surgery and stroke in atrial fibrillation. J Interv
Card Electrophysiol. 53:285–292. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wiseman T and Betihavas V: The association
between unexplained falls and cardiac arrhythmias: A scoping
literature review. Aust Crit Care. 32:434–441. 2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zhang Y, Du W and Yang B: Long non-coding
RNAs as new regulators of cardiac electrophysiology and
arrhythmias: Molecular mechanisms, therapeutic implications and
challenges. Pharmacol Ther. 203(107389)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Sinner MF, Tucker NR, Lunetta KL, Ozaki K,
Smith JG, Trompet S, Bis JC, Lin H, Chung MK, Nielsen JB, et al:
Integrating genetic, transcriptional, and functional analyses to
identify 5 novel genes for atrial fibrillation. Circulation.
130:1225–1235. 2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Schellings MW, Vanhoutte D, van Almen GC,
Swinnen M, Leenders JJ, Kubben N, van Leeuwen RE, Hofstra L,
Heymans S and Pinto YM: Syndecan-1 amplifies angiotensin II-induced
cardiac fibrosis. Hypertension. 55:249–256. 2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Miftode RS, Şerban IL, Timpau AS, Miftode
IL, Ion A, Buburuz AM, Costache AD and Costache II: Syndecan-1: A
review on its role in heart failure and chronic liver disease
patients' assessment. Cardiol Res Pract.
2019(4750580)2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Dagbay KB and Hardy JA: Multiple
proteolytic events in caspase-6 self-activation impact
conformations of discrete structural regions. Proc Natl Acad Sci
USA. 114:E7977–E7986. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Horowitz PM, Patterson KR,
Guillozet-Bongaarts AL, Reynolds MR, Carroll CA, Weintraub ST,
Bennett DA, Cryns VL, Berry RW and Binder LI: Early N-terminal
changes and caspase-6 cleavage of tau in Alzheimer's disease. J
Neurosci. 24:7895–7902. 2004.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Papathanasiou S, Rickelt S, Soriano ME,
Schips TG, Maier HJ, Davos CH, Varela A, Kaklamanis L, Mann DL and
Capetanaki Y: Tumor necrosis factor-α confers cardioprotection
through ectopic expression of keratins K8 and K18. Nat Med.
21:1076–1084. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Soltysinska E, Speerschneider T, Winther
SV and Thomsen MB: Sinoatrial node dysfunction induces cardiac
arrhythmias in diabetic mice. Cardiovasc Diabetol.
13(122)2014.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Sysa-Shah P, Sørensen LL, Abraham MR and
Gabrielson KL: Electrocardiographic characterization of cardiac
hypertrophy in mice that overexpress the ErbB2 receptor tyrosine
kinase. Comp Med. 65:295–307. 2015.PubMed/NCBI
|
|
15
|
Hasslacher C and Wahl P: Diabetes
prevalence in patients with bradycardiac arrhythmias. Acta Diabetol
Lat. 14:229–234. 1977.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Movahed MR, Hashemzadeh M and Jamal MM:
Increased prevalence of third-degree atrioventricular block in
patients with type II diabetes mellitus. Chest. 128:2611–2614.
2005.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Abenavoli T, Rubler S, Fisher VJ, Axelrod
HI and Zuckerman KP: Exercise testing with myocardial scintigraphy
in asymptomatic diabetic males. Circulation. 63:54–64.
1981.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Luo M, Guan X, Luczak ED, Lang D, Kutschke
W, Gao Z, Yang J, Glynn P, Sossalla S, Swaminathan PD, et al:
Diabetes increases mortality after myocardial infarction by
oxidizing CaMKII. J Clin Invest. 123:1262–1274. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
19
|
da Costa Goncalves AC, Tank J, Diedrich A,
Hilzendeger A, Plehm R, Bader M, Luft FC, Jordan J and Gross V:
Diabetic hypertensive leptin receptor-deficient db/db mice develop
cardioregulatory autonomic dysfunction. Hypertension. 53:387–392.
2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Rawles JM, Pai GR and Reid SR: A method of
quantifying sinus arrhythmia: Parallel effect of respiration on P-P
and P-R intervals. Clin Sci (Lond). 76:103–108. 1989.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Delmar M and McKenna WJ: The cardiac
desmosome and arrhythmogenic cardiomyopathies: From gene to
disease. Circ Res. 107:700–714. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Basso C, Czarnowska E, Della Barbera M,
Bauce B, Beffagna G, Wlodarska EK, Pilichou K, Ramondo A, Lorenzon
A, Wozniek O, et al: Ultrastructural evidence of intercalated disc
remodelling in arrhythmogenic right ventricular cardiomyopathy: An
electron microscopy investigation on endomyocardial biopsies. Eur
Heart J. 27:1847–1854. 2006.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Sen-Chowdhry S, Morgan RD, Chambers JC and
McKenna WJ: Arrhythmogenic cardiomyopathy: Etiology, diagnosis, and
treatment. Annu Rev Med. 61:233–253. 2010.PubMed/NCBI View Article : Google Scholar
|
|
24
|
MacKenna DA, Vaplon SM and McCulloch AD:
Microstructural model of perimysial collagen fibers for resting
myocardial mechanics during ventricular filling. Am J Physiol.
273:H1576–H1586. 1997.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Khan R and Sheppard R: Fibrosis in heart
disease: Understanding the role of transforming growth factor-beta
in cardiomyopathy, valvular disease and arrhythmia. Immunology.
118:10–24. 2006.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Travers JG, Kamal FA, Robbins J, Yutzey KE
and Blaxall BC: Cardiac fibrosis: The fibroblast awakens. Circ Res.
118:1021–1040. 2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Guo H, Albrecht S, Bourdeau M, Petzke T,
Bergeron C and LeBlanc AC: Active caspase-6 and caspase-6-cleaved
tau in neuropil threads, neuritic plaques, and neurofibrillary
tangles of Alzheimer's disease. Am J Pathol. 165:523–531.
2004.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Albrecht S, Bogdanovic N, Ghetti B,
Winblad B and LeBlanc AC: Caspase-6 activation in familial
Alzheimer disease brains carrying amyloid precursor protein or
presenilin i or presenilin II mutations. J Neuropathol Exp Neurol.
68:1282–1293. 2009.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Godefroy N, Foveau B, Albrecht S, Goodyer
CG and LeBlanc AC: Expression and activation of caspase-6 in human
fetal and adult tissues. PLoS One. 8(e79313)2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Nevière R, Fauvel H, Chopin C,
Formstecher P and Marchetti P: Caspase inhibition prevents cardiac
dysfunction and heart apoptosis in a rat model of sepsis. Am J
Respir Crit Care Med. 163:218–225. 2001.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Qiu L and Liu X: Identification of key
genes involved in myocardial infarction. Eur J Med Res.
24(22)2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Parrish AB, Freel CD and Kornbluth S:
Cellular mechanisms controlling caspase activation and function.
Cold Spring Harb Perspect Biol. 5(a008672)2013.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Krajewska M, Rosenthal RE, Mikolajczyk J,
Stennicke HR, Wiesenthal T, Mai J, Naito M, Salvesen GS, Reed JC,
Fiskum G and Krajewski S: Early processing of Bid and
caspase-6,-8,-10,-14 in the canine brain during cardiac arrest and
resuscitation. Exp Neurol. 189:261–279. 2004.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Lunde IG, Herum KM, Carlson CC and
Christensen G: Syndecans in heart fibrosis. Cell Tissue Res.
365:539–552. 2016.PubMed/NCBI View Article : Google Scholar
|