Introduction
The inflammasome is a multiprotein complex that
promotes interleukin (IL)-1β secretion and pyroptosis, and is
associated with the pathogenesis of inflammatory diseases,
including sepsis, atherosclerosis and type-2 diabetes (1). The NLR family pyrin domain containing
3 (NLRP3) inflammasome is the most widely studied inflammasome, and
has been reported to participate in numerous diseases (2). The NLRP3 inflammasome consists of the
NOD-like receptor, the adaptor protein, apoptosis-associated
speck-like protein containing CARD (ASC) and caspase 1, which can
be activated during the immune response to pathogens (2). The degradation of NLRP3 acts as the
central process of the suppression of inflammasome. A previous
study proposed two crucial mechanisms of NLRP3 degradation via
autophagy and the proteasome (1).
Both mechanisms were reported to regulate NLRP3 activation
(2). Although some crucial
mechanisms and regulators have been discovered in the regulation of
NLRP3 inflammasome activation, how the inflammasome is naturally
suppressed during inflammation and which degradation is crucial in
the inflammasome remain unknown.
Resolvins, which are metabolites of essential
ω3-polyunsaturated fatty acids, are a class of endogenous lipid
regulators discovered in recent years as suppressors of
inflammation (3). According to the
different precursors and synthesis pathways, resolvin E (RvE)
series derive from eicosapentaenoic acid and resolvin D (RvD)
series derive from docosahexaenoic acid (3). Previous studies reported that these
endogenous substances have a regulatory effect on inflammation and
inhibit the infiltration of inflammatory cells and cytokine
secretion, promote the suppression of inflammation and enhance the
removal of pathogenic microorganisms in the body (4,5).
Resolvin D2 (RvD2) is one type of RvD, and previous studies have
demonstrated that RvD2 has a vital role in promoting the regression
of inflammation (5,6). The results of a previous study
demonstrated that RvD2 can stimulate the production of phagocytic
reactive oxygen species in human multinucleated neutrophils, reduce
neutrophils recruitment during peritonitis and improve the survival
rate of patients with sepsis (6).
In animal models, it has been reported that RvD2 can alleviate the
development of inflammatory bowel disease colitis (7) and inflammation in fibromyalgia
(8). In addition, it was
demonstrated that RvD2 improves the survival rate of mice model of
burn injury, reduce liver and kidney injury (9), reduce the inflammatory response to
periodontitis (10) and reduce the
nerve injury in Parkinson's disease (11). However, the possible effects of
RvD2 on the NLRP3 inflammasome remain unclear.
The present evaluated the effects of RvD2 on the
activation of inflammasome in macrophages both in vitro and
in vivo. The role of RvD2 in NLRP3 inflammasome and its
underlying mechanism were also evaluated. The present study
highlighted the crucial role of RvD2 in NLRP3 inflammasome and
indicated the possible application of RvD2 in the management of
NLRP3 inflammasome-related inflammatory diseases.
Materials and methods
Animal model and reagents
A total of 200 mice (6-8-week-old; weight, 20-25 g)
were purchased from the Shanghai Laboratory Animal Center. Mice
were raised in a specific pathogen-free environment at a
temperature of 18-22˚C, 50-60% humidity and 12 h light/dark cycle.
Food and water were freely accessible to mice. The Ethics Committee
of No. 906 Hospital of the Chinese People's Liberation Army Joint
Logistic Support Force approved this study (approval no.
201904050024). All procedures were performed under anesthesia with
3-4% sevoflurane. Animals that died (n=10) after sevoflurane
induction were excluded from the experiments. In the mice
peritonitis models, mice were injected with monosodium (MSU; 2
mg/mouse; Sigma-Aldrich; Merck KGaA) or alum (2 mg/mouse; Thermo
Fisher Scientific, Inc.) or lipopolysaccharide (LPS; 10 mg/kg,
Escherichia coli O111:B4; Sigma-Aldrich; Merck KGaA) diluted
in 200 µl PBS. A single injection of RvD2 (1 µg/mouse;
Sigma-Aldrich; Merck KGaA) was administered intraperitoneally
immediately after the MSU, LPS or alum injection. Following 6 h
after the injection, mouse were sacrificed. A total of 5 ml normal
saline was injected into the peritoneum and peritoneal lavage
fluids (PLF) were collected at room temperature. The PLF was
subsequently centrifuged at 1000 x g for 5 min at room temperature
and the peritoneal macrophages were obtained (1). For blood collection, mice were
anesthetized with sevoflurane 5% for induction and 2% sevoflurane
for maintenance. While anesthetized, a total of 300 µl blood was
collected through heart puncture following 6-h treatment of LPS,
MSU or alum injection with or without RvD2. Mice were sacrificed by
cervical dislocation. Blood was centrifuged at 3,000 x g for 15 min
at room temperature, and the serum was collected and stored at
-80˚C for subsequent analysis.
Macrophage preparation and
stimulation
Peritoneal macrophages were generated through single
peritoneal injection of thioglycolate in mice, (0.5 ml per mice; BD
Biosciences) which induces the aseptic inflammation and recruits
macrophages in the peritoneum. Three days after the injection, PLF
was collected, macrophages were centrifuged at 1,000 x g for 5 min
at room temperature, resuspended at 2-4x106 cells/ml,
cultured in RPMI 1640 culture medium supplemented with 10% FBS
(Gibco; Thermo Fisher Scientific, Inc.) and placed at 37˚C in a
humidified incubator containing 5% CO2.
To induce the inflammasome activation, cells were
first treated with LPS (100 ng/ml) for 3 h, and nigericin (Nig; 20
µM), muramyl dipeptide (MDP; 200 ng/ml), flagellin (10 µM) or
poly(dA:dT; 1 µg/ml) were added for 30 min. A concentration of 10
nM RvD2 was added simultaneously with LPS in cell experiments. The
supernatant was collected 30 min after the nigericin stimulation
and subjected to further analysis. The inhibitors bafilomycin A1,
an autophagy inhibitor, (1 nM; incubated for 30 min; Cayman
Chemical Company), MG-132, a proteasome inhibitor, (20 µM;
incubated for 30 min; Selleck Company) and 3-methyladenine, an
autophagy inhibitor, (3-MA; 5 mM; incubated for 30 min; Merck KGaA)
were added individually to the culture medium 30 min before the
second induction [Nig, MDP, flagellin or poly(dA:dT)]. The p65
inhibitor (10 µM; Helenalin, MCE) was added 30 min before LPS and
Nigercin stimulation and incubated for 3.5 h.
Cytokine analysis
ELISA assays were used for cytokine analysis. IL-1β
(cat. no. 88-7013-88), IL-6 (cat. no. BMS603-2) and tumor necrosis
factor (TNF)-α (cat. no. BMS607-3TEN) levels were evaluated using
ELISA kits (Invitrogen; Thermo Fisher Scientific, Inc.) according
to the manufacturer's protocol. Absorbance was read on a microplate
reader (Tecan Group, Ltd.) and the concentration of the cytokines
was calculated using standard curves.
Western blotting
Western blotting was performed as previously
described (1). Cells were lysed in
RIPA buffer at 4˚C (Beyotime Institute of Biotechnology) and
protein concentration was determined using the BCA method (Thermo
Fisher Scientific, Inc.). Proteins (~20 µg) were separated by 10%
SDS-PAGE and transferred onto PVDF membranes (Merck KGaA). For
supernatant protein analysis, 200 µl supernatant was collected,
directly degenerated with 5X loading buffer (Beyotime Institute of
Biotechnology) at 80˚C for 5 min and subjected to SDS-PAGE.
Membranes were blocked with 5% non-fat dry milk in PBS with 10%
Tween (PBST) at pH 7.5 at room temperature for 30 min. Membranes
were then incubated with primary antibodies against ASC (1:5,000;
cat. no. 67824; Cell Signaling Technology, Inc.), NLRP3 (1:5,000;
cat. no. 15101; Cell Signaling Technology, Inc.),
microtubule-associated proteins 1A/1B light chain 3B (LC3B;
1:5,000; cat. no. 43566; Cell Signaling Technology, Inc.), β-actin
(1:5,000; cat. no. 4970; Cell Signaling Technology, Inc.),
pro-IL-1β (1:5,000; cat. no. 12242; Cell Signaling Technology,
Inc.), caspase 1 (1:5,000; cat. no. 24232; Cell Signaling
Technology, Inc.) and pro-caspase 1 (1:5,000; cat. no. AG-46B-0003;
Adipogen) for 4 h or overnight at 4˚C. Membranes were then
incubated with horseradish peroxidase (HRP)-conjugated secondary
antibodies for 3 h at room temperature (1:10,000; cat. no. 13999;
Cell Signaling Technology, Inc.). Enhanced chemiluminescence
reagent (Pierce; Thermo Fisher Scientific, Inc.). The data were
quantified by ImageJ (version 1.53; National Institutes of
Health)
Flow cytometry
Cells were diluted in PBS and stained with the
corresponding antibody at 4˚C for 20 min. The antibody used was as
follows: Macrophage (FITC-conjugated anti-mouse-F4/80; 1:2,000;
cat. no. 11-4801-85; Invitrogen; Thermo Fisher Scientific, Inc.).
Cells were washed three times with PBS and centrifuged at 1,000 x g
for 10 min at room temperature. Cells were then diluted in Dilution
Buffer (cat. no. 345035; BD Biosciences). Fluorescence was
evaluated for 105 events per sample using a FACS LSR II (BD
Bioscience) and data were analyzed using FlowJo software (version
10; FlowJo LLC).
Statistical analysis
Data were presented as the means ± standard error of
the mean. One-way ANOVA followed by Tukey's post hoc test was used
for multiple comparisons. P<0.05 was considered to indicate a
statistically significant difference.
Results
RvD2 suppresses inflammasome-mediated
peritoneal inflammation in vivo
To investigate the possible effects of RvD2 on
inflammasome, inflammasome-mediated peritonitis was generated in
mice through intraperitoneal injection of MSU, alum and LPS. The
results from in Fig. 1
demonstrated that MSU injection significantly increased the
macrophage cell count and peritoneal exudate cells (PECs) isolated
from PLF (Fig. 1A and B). Furthermore, IL-1β level in PLF was
also significantly increased (Fig.
1C). However, in mice injected with MSU and RvD2, these three
parameters were all significantly decreased, indicating the
inhibition of peritonitis (Fig.
1A-C). Similar results were observed in the alum-induced and
LPS-induced peritonitis model (Fig.
1D-I). Furthermore, IL-6, TNF-α and IL-1β levels were evaluated
in mice treated with LPS, and the results demonstrated that RvD2
could significantly decrease the levels of these three cytokines in
mice serum (Fig. 1J-L). Taken
together, these results indicated that injection of RvD2 could
suppress the inflammasome-mediated peritoneal inflammation in
vivo.
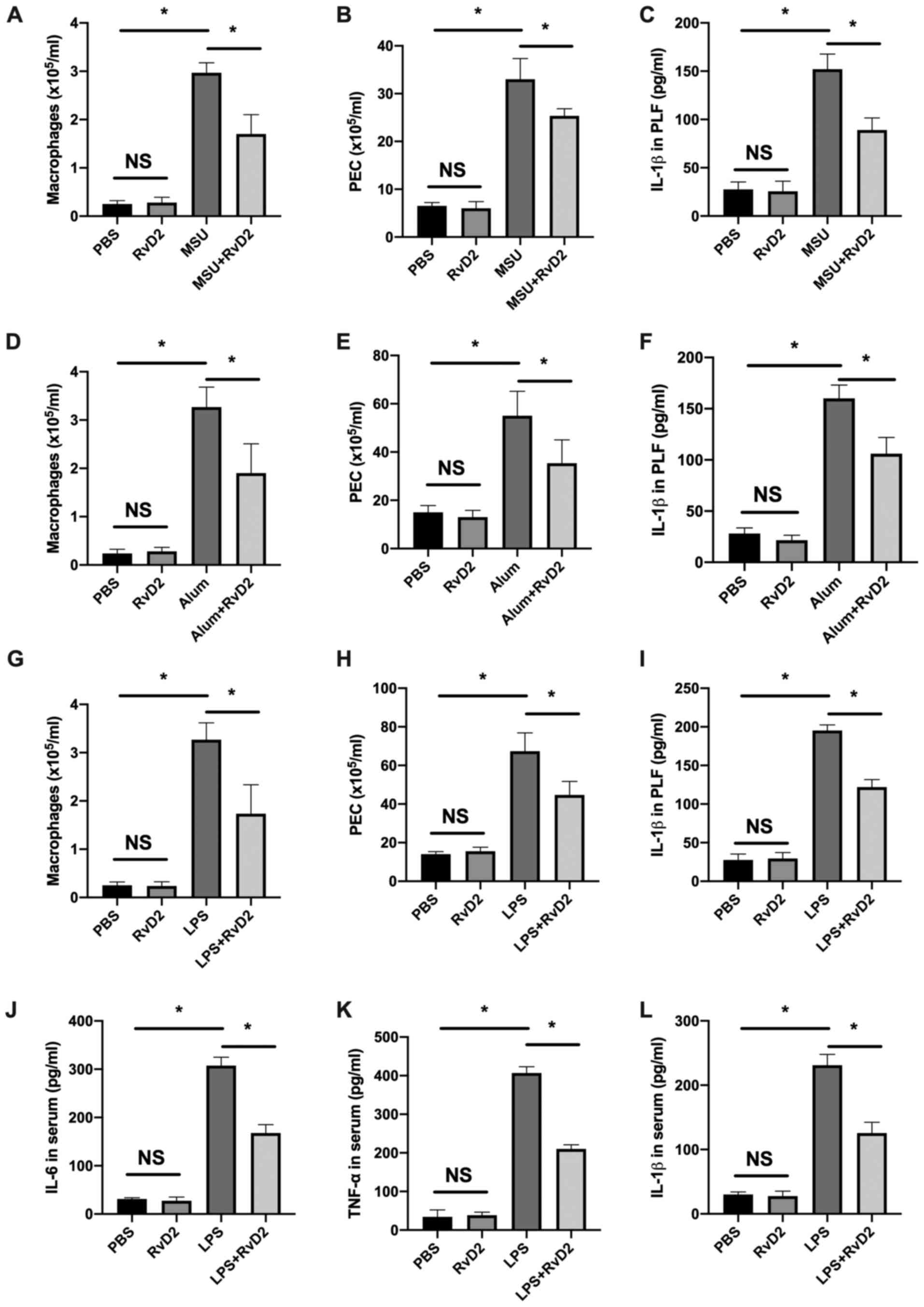 | Figure 1RvD2 suppresses inflammasome-mediated
peritoneal inflammation in vivo. (A-I) RvD2 (1 µg/mouse)
inhibited (A-C) MSU-, (D-F) alum- and (G-I) LPS-induced peritoneal
inflammation (n=3). Flow cytometry revealed that (A, D and G)
macrophages (F4/80+) number, (B, E and H) PEC number and
(C, F and I) IL-1β level in the peritoneal lavage fluid were
decreased in the RvD2 pretreatment group. (J-L) Serum levels of (J)
IL-6, (K) TNF-α and (L) IL-1β were decreased in the RvD2+LPS group
(n=3). *P<0.05. IL, interleukin; LPS,
lipopolysaccharide; MSU, monosodium; NS, non-significant; PEC,
peritoneal exudate cell; RvD2, resolvin D2; TNF-α, tumor necrosis
factor-α. |
RvD2 suppresses NLRP3 inflammasome
activation in macrophage
Different inflammasomes participate in different
inflammatory responses (1). To
investigate which inflammasome RvD2 specifically regulates, we
evaluated the effects of RvD2 on AIM2 inflammasome [induced by
poly(dA:dT)], NLRC4 inflammasome (induced by flagellin), NALP3
(induced by MDP), and NLRP3 inflammasome (induced by nigericin).
RvD2 suppressed IL-1β in the flagellin and nigericin group only,
while no effects were demonstrated in the Poly(dA/dT) and MDP group
(Fig. 2A and B). The results demonstrated that RvD2 did
not suppress the activation of AIM2 and NLRC4 inflammasome
(Fig. 2A and B). Furthermore, although RvD2 inhibited
the activation of NLRP3 and NALP3 inflammasome (Fig. 2C), RvD2 showed the most significant
suppression in NLRP3 inflammasome activation in a dose-dependent
manner (Fig. 2D). In addition,
RvD2 suppressed the NLRP3 inflammasome activation both before the
LPS and Nig induction, and following the Nig induction, indicated
by the decreased IL-1β levels in all three groups (Fig. 2D), suggesting that RvD2 suppressed
the priming and secondary signal induction process during the
inflammasome activation. However, NF-κB inhibition using p65
inhibitor could not abolish the suppressive effects of RvD2 on
NLRP3 activation (Fig. S1A and
B), suggesting that RvD2
regulation on NLRP3 was dependent on a secondary signal.
Subsequently, NLRP3 inflammasome activation was evaluated via
western blotting, and the results demonstrated that RvD2 could
decrease caspase 1-p20 expression in the supernatant, as well as
pro-IL-1β, NLRP3 and caspase 1-p20 expression in the cell lysate,
suggesting that RvD2 could inhibit NLRP3 inflammasome
activation.
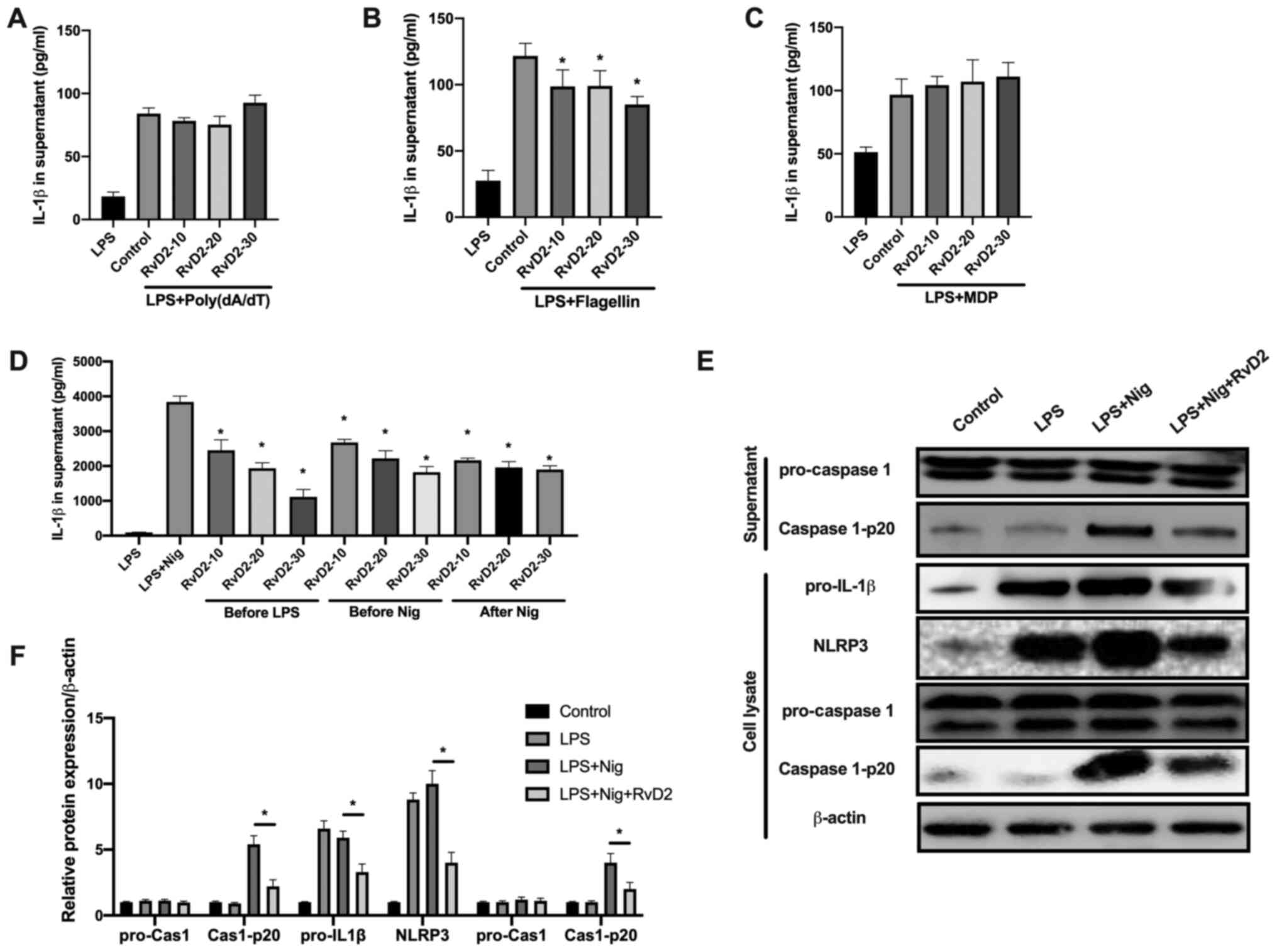 | Figure 2RvD2 inhibits NLRP3 inflammasome
activation in macrophages. (A-C) IL-1β level in the supernatant of
macrophages following treatment with (A) poly(dA/dT), (B) MDP and
(C) flagellin (n=3). (D) IL-1β level in the supernatant of
macrophages treated with RvD2 (10 nM) 30 min before LPS
stimulation, 30 min before Nig treatment or 15 min after Nig
treatment (n=3). (E and F) Western blot analysis and quantification
of pro-caspase 1, Cas-1-p20 in supernatant and of pro-IL-1β, NLRP3,
pro-caspase 1, Cas-1-p20 and β-actin in cell lysate after indicated
treatments (n=3). *P<0.05. Cas, caspase; LPS,
lipopolysaccharide; MDP, muramyl dipeptide; Nig, nigericin; NLRP3,
NLR family pyrin domain containing 3; RvD2, resolvin D2. |
RvD2 promotes NLRP3 degradation
through autophagy
Previous studies reported the inhibiting role of
RvD2 on NF-κB signaling (4,6). The
present study investigated therefore how RvD2 could inhibit NLRP3
activation. The degradation of NLRP3 has been reported to be
associated with autophagy and the proteasome pathway (1). In the present study, we used
inhibitors of autophagy (3-MA and Bafilomycin A1) and proteasome
(MG-132) to determine which process is crucial in RvD2-mediated
NLRP3 degradation. The results from macrophages in vitro
experiments demonstrated that the inhibition of autophagy through
3-MA and bafilomycin A reversed the RvD2-mediated suppression of
IL-1β (Fig. 3A and B), whereas the inhibition of proteasome
via MG-132 had no effects on RvD2-mediated suppression of IL-1β
(Fig. 3C). The results from
western blotting demonstrated that RvD2 induced the increase in LC3
expression, and that 3-MA abolished the decrease in NLRP3
expression (Fig. 3D). However,
MG-132 had no effects on LC3 and NLRP3 expression (Fig. 3E). These findings suggested that
RvD2 could inhibit NLRP3 activation by promoting NLRP3 degradation
via autophagy.
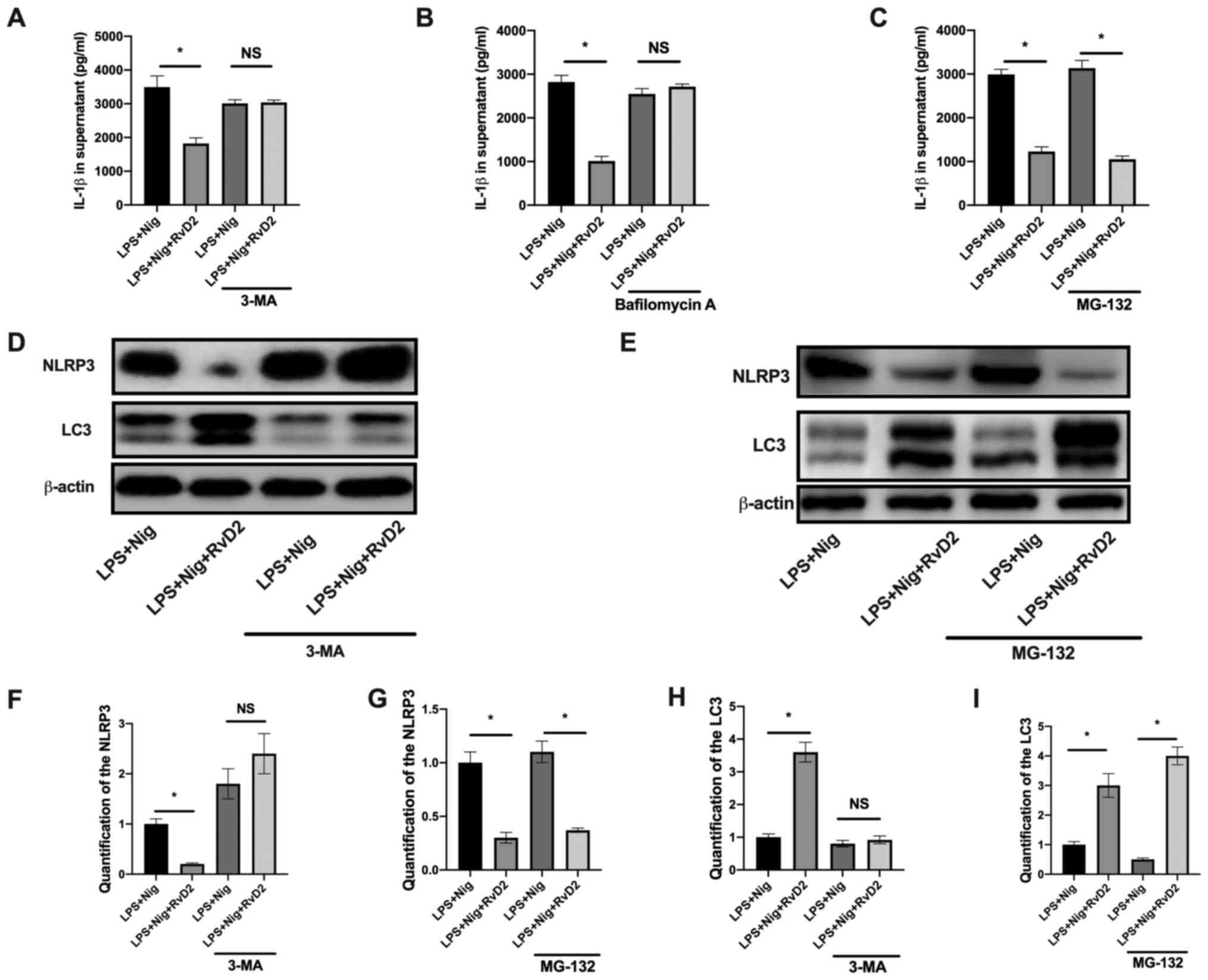 | Figure 3RvD2 promotes NLRP3 degradation
through autophagy. (A-C) IL-1β level in the supernatant of
macrophage pretreated for 1 h with the autophagy inhibitors 3-MA
and bafilomycin A, the proteasome inhibitor MG-132, and stimulated
with LPS and Nig. RvD2 (10 nM) was added for 30 min before LPS
stimulation (n=3). (D and E) Western blot analysis and (F-I)
quantification of (F and G) NLRP3, (H and I) LC3 and β-actin in
cell lysate in the presence of (D,. F,. G) 3-MA or (E, H, I) MG-132
(n=3). *P<0.05. 3-MA, 3-methyladenine; IL,
interleukin; LC3, microtubule-associated proteins 1A/1B light chain
3; LPS, lipopolysaccharide; Nig, nigericin; NLRP3, NLR family pyrin
domain containing 3; NS, non-significant; RvD2, resolvin D2. |
Inhibition of autophagy abolishes the
RvD2-mediated IL-1β downregulation in vivo
The in vitro experiments suggested that RvD2
promoted the NLRP3 degradation through autophagy. Subsequently, we
investigated whether inhibition of autophagy could abolish the
effects of RvD2 in vivo. MSU- and LPS-induced peritonitis
model were generated, and we evaluated whether 3-MA could abolish
the protective effects of RvD2 in mouse peritonitis. The results
demonstrated that in the MSU-induced peritonitis model, 3-MA
abolished the decrease in macrophage cell count (Fig. 4A) and partially reversed the IL-1β
and PEC downregulation in PLF (Fig.
4B and C). In the LPS-induced
peritonitis model, 3-MA also partially reversed the decrease in
macrophage cell count and IL-1β synthesis in PLF, but had minor
effects in PEC number in PLF (Fig.
4F). Furthermore, 3-MA could abolish the inhibiting effect of
RvD2 on IL-6 and IL-1β serum levels and partially reversed the
decrease in TNF-α level in the serum of mice with LPS-induced
peritonitis. These findings suggested that autophagy may serve a
crucial role in RvD2-mediated inhibition of inflammasome activation
in vivo.
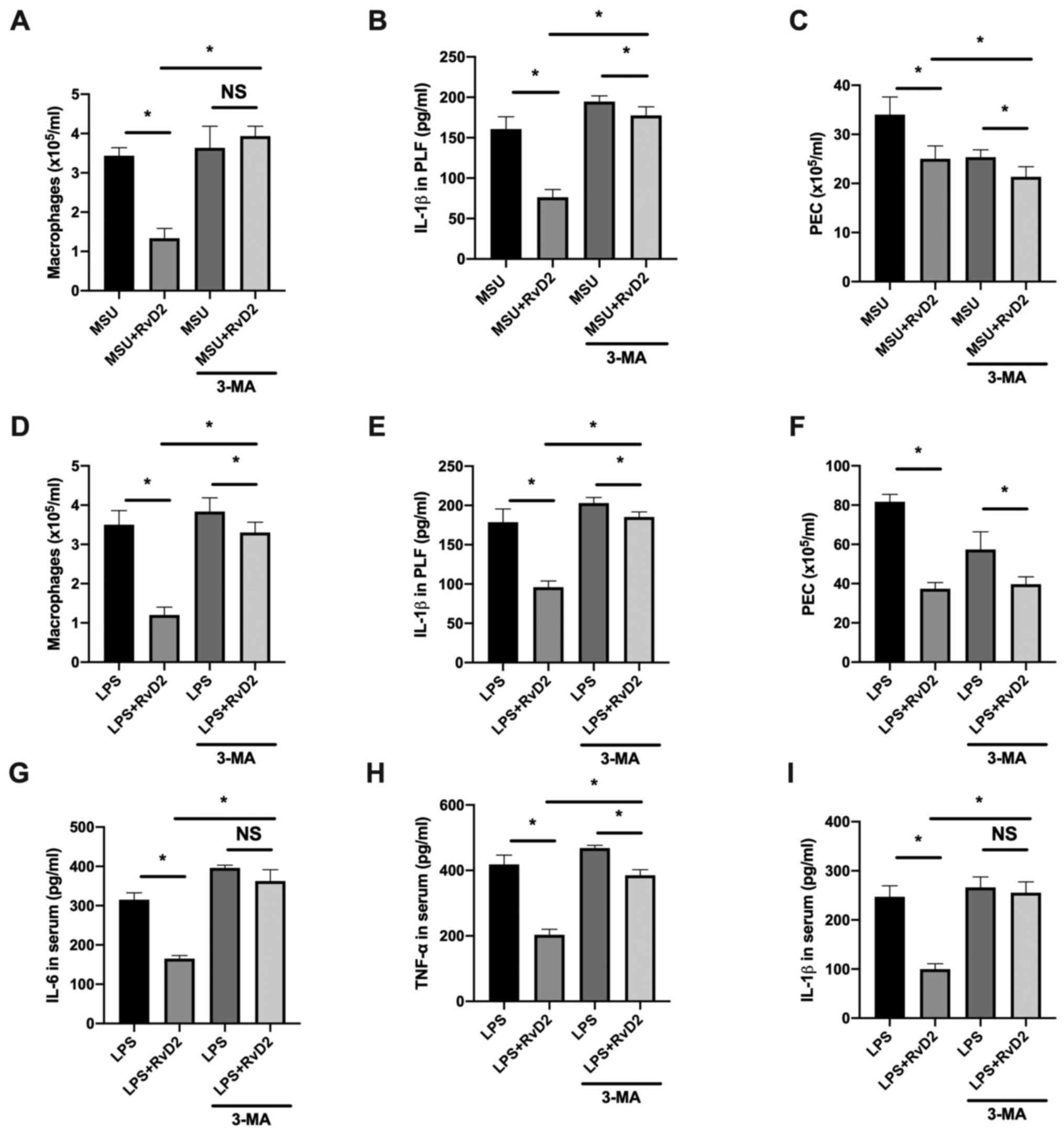 | Figure 4Inhibition of autophagy abolishes the
RvD2-mediated inhibition of IL-1β synthesis in vivo. (A-F)
IL-1β level in the PLF of mice treated with RvD2 (1 µg/mouse) and
(A-C) MSU or (G-I) LPS to induce peritoneal inflammation (n=3).
Flow cytometry revealed that (A and D) macrophages
(F4/80+) number, (B and E) PEC number and (C and F)
IL-1β level in PLF were partially increased in the 3-MA
pretreatment groups (n=3). (G-I) Serum levels of (G) IL-6 and (I)
IL-1β were increased to a level similar to the LPS group and (H)
TNF-α levels were partially increased in the 3-MA treatment group
(n=3). *P<0.05. 3-MA, 3-methyladenine; IL,
interleukin; LPS, lipopolysaccharide; MSU, monosodium; NS,
non-significant; PEC, peritoneal exudate cell; PLF, peritoneal
lavage fluid; RvD2, resolvin D2; TNF-α, tumor necrosis
factor-α. |
Discussion
RvD2 has been identified as a potent immunoresolvent
and controller of inflammation and showed protective effects in
multiple diseases models, such as acute lung injury,
gastrointestinal inflammation and sepsis (5). Uncontrolled and excessive
inflammation are associated with diabetes, arthritis and systematic
inflammation (12). The innate
resolving effects of RvD2 demonstrated some limited but actively
orchestrated responses during inflammation (10). However, whether RvD2 could
influence inflammasome activation and through which mechanism
remain unknown. The present study demonstrated that RvD2 could
inhibit the activation of NLRP3 inflammasome but not of AIM2, NLRC4
or NALP3 inflammasomes. We also demonstrated the decreased IL-1β
secretion both in vivo and in vitro in the presence
of exogenous RvD2. These findings extended our knowledge on the
role of RvD2 in the regulation of inflammasome and suggested the
potential use of RvD2 in inflammasome-mediated diseases. In
addition, the results from the present study demonstrate that RvD2
could promote the degradation of NLRP3 inflammasome and may induce
the inhibition of NLRP3 inflammasome activation.
LPS intraperitoneal injection was previously
reported to stimulate inflammasome activation in peritoneal
macrophages (1). Therefore, we
used this model to analyze the levels of serum cytokines and
demonstrate the alteration of inflammasome activation. The effects
of RvD2 in MSU- and alum-induced peritonitis were also evaluated,
which are inflammasome-related models.
A previous study reported the suppressive effects of
RvD2 in TLR4 signaling (5).
However, as NLRP3 expression was decreased following p65 inhibitor
treatment, RvD2 continued to suppress the production of IL-1β. This
result suggested that NF-κB suppression by RvD2 is not a crucial
process in NLRP3 regulation.
NLRP3 degradation is a widely studied process in the
regulation of inflammasome activation (13). Previous studies reported that
proteasome- and autophagy-mediated degradations are two central
processes in NLRP3 regulation (1,14).
Once the degradation is activated, NLRP3 inflammasome cannot
recruit ASC to form the multiprotein platform and fail therefore to
activate the caspase 1 to cleave the pro-IL-1β into IL-1β, which
leads to the suppression of the inflammasome (13). The present study demonstrated that
treatment with RvD2 decreased the protein expression of NLRP3,
suggesting that NLRP3 might be degraded. Autophagy inhibitors
(bafilomycin A and 3-MA) and proteasome inhibitor (MG-132) were
used to evaluate the effects of RvD2 in inflammasome activation.
The results demonstrated that 3-MA and bafilomycin A could reverse
the inhibiting effect of RvD2 on IL-1β secretion, while MG-132 had
no effect, suggesting that autophagy may participate in NLRP3
degradation. Furthermore, RvD2 was shown to induce the increase in
LC-3II expression, and inhibition of autophagy could also partially
reverse the protective effects of RvD2 on inflammasome-mediated
peritonitis in vivo. These findings suggested that RvD2 may
promote NLRP3 degradation through autophagy.
The present study used LC-3 as the marker of
autophagy and performed experiments using inhibitors to demonstrate
its crucial role in NLRP3 degradation. However, how NLRP3
inflammasome is degraded should be further investigated with more
evidence about p62 and Atg family. Whether ubiquitin is involved in
this process should also be investigated, which may provide
additional information about the effects of RvD2 in NLRP3
regulation.
Although the present study demonstrated the effects
of RvD2 in NLRP3 degradation, the underlying mechanism should be
investigated in future study. A previous study reported that RvD2
could regulate multiple inflammatory signaling pathways, including
signal transducer and activator of transcription 3 (STAT3),
MAPK-Erk and Akt (5). The
regulation of RvD2 in STAT3 and MAPK signaling is mainly dependent
on the interaction between RvD2 and DRV2 (termed as G
protein-coupled receptor, GPR18), a G protein-coupled receptor for
RvD2(5). It has been reported that
the interaction between RvD2 and DRV2 could influence the level of
cyclic adenosine monophosphate (cAMP) and the phosphorylation of
STAT3(5). The cAMP further induces
protein kinase A (PKA) activation and influences therefore the
function of the macrophage (5).
The cAMP/PKA level has been demonstrated to regulate NLRP3
inflammasome through multiple mechanisms (15), in particular via promoting
autophagy, which leads to NLRP3 degradation (16,17).
These findings are in accordance with the present study
demonstrating that RvD2 could decrease the protein expression of
NLRP3 and increase LC-3II expression. Future studies may therefore
investigate the underlying mechanism of RvD2-mediated autophagy in
macrophages.
Taken together, the results from the present study
demonstrated the regulation of NLRP3 inflammasome activation in
macrophages by RvD2. In addition, RvD2 may promote NLRP3
degradation through autophagy. These findings may help
understanding the resolving-mediated regulation of inflammation and
help determining potential targets in the management of
inflammasome-mediated diseases.
Supplementary Material
Helenalin, a p65 inhibitor, inhibits
the NLRP3 activation but is not crucial for the NLRP3 regulation by
RvD2. (A and B) Western blot analysis and quantification of NLRP3
and β-actin of macrophage after indicated treatments (n=3).
*P<0.05. LPS, lipopolysaccharide; Nig, nigericin;
NLRP3, NLR family pyrin domain containing 3.
Acknowledgements
Not applicable.
Funding
Funding: This study was funded by the Health Commission of
Ningbo City (grant no. 2020Y26).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZL and LL designed the study and directed the
experiments. LC, YiyW and YinW performed the experiments. YiyW and
YinW prepared the manuscript with knowledgeable advice. FL assisted
with essential experimental techniques and performed part of the
experiments. YiyW and YinW confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
No. 906 Hospital of the Chinese People's Liberation Army Joint
Logistic Support Force (approval no. 201904050024).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Xu M, Jiang Z, Wang C, Li N, Bo L, Zha Y,
Bian J, Zhang Y and Deng X: Acetate attenuates inflammasome
activation through GPR43-mediated Ca2+-dependent NLRP3
ubiquitination. Exp Mol Med. 51:1–19. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Yang J, Liu Z and Xiao TS:
Post-translational regulation of inflammasomes. Cell Mol Immunol.
14:65–79. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Serhan CN, Clish CB, Brannon J, Colgan SP,
Chiang N and Gronert K: Novel functional sets of lipid-derived
mediators with antiinflammatory actions generated from omega-3
fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory
drugs and transcellular processing. J Exp Med. 192:1197–1204.
2000.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Bannenberg GL, Chiang N, Ariel A, Arita M,
Tjonahen E, Gotlinger KH, Hong S and Serhan CN: Molecular circuits
of resolution: Formation and actions of resolvins and protectins. J
Immunol. 174:4345–4355. 2005.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Chiang N, de la Rosa X, Libreros S and
Serhan CN: Novel Resolvin D2 Receptor Axis in Infectious
Inflammation. J Immunol. 198:842–851. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Spite M, Norling LV, Summers L, Yang R,
Cooper D, Petasis NA, Flower RJ, Perretti M and Serhan CN: Resolvin
D2 is a potent regulator of leukocytes and controls microbial
sepsis. Nature. 461:1287–1291. 2009.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Bento AF, Claudino RF, Dutra RC, Marcon R
and Calixto JB: Omega-3 fatty acid-derived mediators 17(R)-hydroxy
docosahexaenoic acid, aspirin-triggered resolvin D1 and resolvin D2
prevent experimental colitis in mice. J Immunol. 187:1957–1969.
2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Klein CP, Sperotto ND, Maciel IS, Leite
CE, Souza AH and Campos MM: Effects of D-series resolvins on
behavioral and neurochemical changes in a fibromyalgia-like model
in mice. Neuropharmacology. 86:57–66. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Bohr S, Patel SJ, Sarin D, Irimia D,
Yarmush ML and Berthiaume F: Resolvin D2 prevents secondary
thrombosis and necrosis in a mouse burn wound model. Wound Repair
Regen. 21:35–43. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Siddiqui YD, Omori K, Ito T, Yamashiro K,
Nakamura S, Okamoto K, Ono M, Yamamoto T, Van Dyke TE and Takashiba
S: Resolvin D2 Induces Resolution of Periapical Inflammation and
Promotes Healing of Periapical Lesions in Rat Periapical
Periodontitis. Front Immunol. 10(307)2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Tian Y, Zhang Y, Zhang R, Qiao S and Fan
J: Resolvin D2 recovers neural injury by suppressing inflammatory
mediators expression in lipopolysaccharide-induced Parkinson's
disease rat model. Biochem Biophys Res Commun. 460:799–805.
2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Maslowski KM, Vieira AT, Ng A, Kranich J,
Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, et al:
Regulation of inflammatory responses by gut microbiota and
chemoattractant receptor GPR43. Nature. 461:1282–1286.
2009.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yan Y, Jiang W, Liu L, Wang X, Ding C,
Tian Z and Zhou R: Dopamine controls systemic inflammation through
inhibition of NLRP3 inflammasome. Cell. 160:62–73. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wen H, Gris D, Lei Y, Jha S, Zhang L,
Huang MT, Brickey WJ and Ting JP: Fatty acid-induced NLRP3-ASC
inflammasome activation interferes with insulin signaling. Nat
Immunol. 12:408–415. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
15
|
Lee GS, Subramanian N, Kim AI,
Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner
DL and Chae JJ: The calcium-sensing receptor regulates the NLRP3
inflammasome through Ca2+ and cAMP. Nature. 492:123–127.
2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Inda C, Dos Santos Claro PA, Bonfiglio JJ,
Senin SA, Maccarrone G, Turck CW and Silberstein S: Different cAMP
sources are critically involved in G protein-coupled receptor CRHR1
signaling. J Cell Biol. 214:181–195. 2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Tresguerres M, Levin LR and Buck J:
Intracellular cAMP signaling by soluble adenylyl cyclase. Kidney
Int. 79:1277–1288. 2011.PubMed/NCBI View Article : Google Scholar
|














