Introduction
Cardiac hypertrophy is an adaptive response to
stress and occurs when the individual cellular volume of the heart
increases, an effect that is associated with neurohumoral
reactivation (1). In almost all
types of cardiac disease, cardiac hypertrophy is a critical
adaptive response in cardiomyocytes that aims to preserve the
output and efficiency of the heart in addition to optimize cardiac
pump function (2). However,
sustained cardiac hypertrophy is associated with myocardial
remodeling and dilatation of the left ventricle, which can
ultimately lead to dilated cardiomyopathy, arrhythmia, fibrotic
disease and heart failure (3). At
cellular level, cardiac hypertrophy is characterized by enlarged
cell size and increased synthesis of various cardiac hypertrophy
markers, including atrial natriuretic peptide (ANP) and brain
natriuretic peptide (BNP) (4).
The ubiquitin-proteasome system (UPS) protects
cardiomyocytes by preventing the build-up of misfolded proteins and
by removing pro-apoptotic signaling molecules (5). It has been previously demonstrated
that protein degradation via the UPS is activated in animal models
of cardiac hypertrophy and in cases of human cardiomyopathy)
(6). In addition, UPS dysfunction
can result in cardiac hypertrophy and defective cardiac responses
(7). Muscle really interesting new
gene (RING)-finger protein-1 (MuRF1) and muscle atrophy F-box
(MAFbx) are atrogenes that serve as early markers of skeletal
muscle atrophy and aid in the diagnosis of muscle disease (8). Previous studies have determined an
association between Atrogin-1/MAFbx with cardiac hypertrophy, such
that MuRF1 and MAFbx have been reported to be negative regulators
of cardiac hypertrophy (9,10). However, the precise mechanism
underlying the involvement of MuRF1 and MAFbx in cardiac
hypertrophy pathogenesis remains to be fully elucidated.
Metformin is currently the first-line blood
glucose-lowering agent for the treatment of type 2 diabetes
mellitus, with potential for future application in cardiovascular
diseases (11). It inhibits hepatic
gluconeogenesis, enhances glucose uptake in peripheral tissues and
reduces glucose absorption from the intestine (12). Furthermore, experimental studies
have previously revealed that metformin treatment can effectively
reverse infarct size, reduce vascular inflammation and myocardial
injury after ischemia, inhibit cardiomyocyte apoptosis and protect
against ischemia/reperfusion-induced cardiomyocyte death (13,14).
Therefore, considering the potential role of
metformin in the modulation of MuRF1 and MAFbx during angiotensin
II (Ang II)-induced cardiomyocyte hypertrophy, the present study
aimed to determine the effects of metformin on cardiomyocyte
hypertrophy, with the research focus on the MuRF1 and MAFbx
pathway, in an in vitro model of Ang II-induced
cardiomyocyte hypertrophy.
Materials and methods
Cell culture and treatment
Rat H9c2 myoblasts, a model for cardiomyocytes, were
purchased from the Cell Bank of Type Culture Collection of the
Chinese Academy of Sciences. These cells were routinely
authenticated and tested negative for Mycoplasma
contamination and were cultured in DMEM (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 1% streptomycin-penicillin and
10% FBS (Gibco; Thermo Fisher Scientific, Inc.) and 4 mM
L-glutamine (Gibco; Thermo Fisher Scientific, Inc.) in in an
atmosphere of 5% CO2 at 37˚C. Cells were collected and
centrifuged at 168 x g for 5 min at 18˚C, after which the
supernatant was discarded. The complete medium was changed every 3
days and cells were then sub-cultured when the density reached 80%
confluence. After incubation, cells were washed three times with
PBS and harvested by digestion with trypsin. The cells were then
placed into a single cell suspension, after which 1x105
cells/ml were seeded into six-well plates. The H9c2 cardiomyocytes
in all the experiments were passaged according to the ratio of 1:3
at 37˚C and 5% CO2 in saturated humidity conditions to
expand cultivation. Cells were subsequently divided into different
groups according to the experimental requirements.
To evaluate the effects of metformin on
cardiomyocyte hypertrophy, cultured cells were randomly divided
into the following four experimental groups: i) Control, untreated
H9c2 cells incubated in 10% FBS DMEM for 24 h at 37˚C; ii)
metformin, where cells were treated with 2 mM metformin
(Sigma-Aldrich; Merck KGaA) for 24 h at 37˚C (15); iii) Ang II, where cells were treated
with 100 nM Ang II (Beijing Solarbio Science & Technology Co.,
Ltd.) for 48 h at 37˚C; and iv) Ang II + metformin, where cells
were treated with 100 nM Ang II for 48 h, followed by metformin for
24 h at 37˚C.
To determine the role of MuRF1 and MAFbx after
metformin treatment in this angiotensin II-induced cardiomyocyte
hypertrophy model, cells were randomly divided into the following
seven experimental groups: i) Control; ii) metformin, treated with
2 mM metformin for 24 h at 37˚C; iii) Ang II, treated with 100 nM
Ang II for 48 h at 37˚C; iv) Ang II + metformin, treated with 100
nM Ang II for 48 h, followed by metformin for 24 h at 37˚C; v) Ang
II + Met + NC-siRNA groups; vi) Ang II + metformin + MAFbx-siRNA;
and vii) Ang II + metformin + MuRF1-siRNA. Groups v, vi and vii
were first treated with 100 nM Ang II for 48 h, followed by
metformin for 24 h. After siRNA targeting MuRF1 and MAFbx was used
to knock down MuRF1 and MAFbx expression, changes in the surface
area and the protein degradation rate in cells were examined.
Cell surface area measurement
To determine changes in cell size, cells (n=100)
were randomly selected from each experimental group. Images of 3
fields per dish were captured with an average of 30-40 cells per
field. and imaged using an Olympus BX51 microscope (Olympus
Corporation) at a magnification of x200. The cell surface area was
measured using the Image-J, 1.37v Image software (National
Institutes of Health).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted using TRIzol®
reagent according to manufacturer's protocol (Thermo Fisher
Scientific, Inc.). The RNA sample (5 µg) was treated with DNase and
subsequently used for cDNA synthesis. The mRNA was reverse
transcribed into cDNA using HiScript® II 1st Strand cDNA
Synthesis Kit (Vazyme Biotech Co., Ltd; cat. no. R101-01/02)
according to the manufacturer's protocol. qPCR was performed in
Applied Biosystems QuantStudio 6 Flex Real-Time PCR System (Thermo
Fisher Scientific, Inc.) using SYBR® Green Master Mix
(Vazyme Biotech Co., Ltd.; cat. no. Q111-02) under the following
thermocycling conditions: Initial denaturation at 95˚C for 5 min,
followed by 40 cycles at 94˚C for 30 sec, 60˚C for 30 sec and 72˚C
for 1 min. To assess the concentration and purity of total RNA, the
optical density (OD) 260: OD 280 (1.8:2.0) ratio of the extracted
RNA sample was measured. The concentration of RNA was subsequently
calculated according to the OD value, with use of the following
formula: Total RNA concentration (µg/µl) = OD260 x
40x10-3. The following primers were used: Rat β-actin
forward, 5'-CACGATGGAGG GGCCGGACTCATC-3' and reverse,
5'-TAAAGACCTCT ATGCCAACACAGT-3'; rat MAFbx forward, 5'-TCCCTGAG
TGGCATCGCCCAAAAGA-3' and reverse, 5'-ACACAGGCA GGT CGGTGATCGT-3';
rat MuRF1 forward, 5'-GGTGCCTA CTTGCTCCTTGTGC-3' and reverse,
5'-AGTCTGAACTCGGTCG TTCCCT-3'; rat ANP forward, 5'-GGAAGTCAACCCG TC
TCA-3' and reverse, 5'-GGGCTCCAATCCTGTCAAT-3' and rat BNP forward,
5'-AGATGATTCTGCTCCTGCTTT-3' and reverse,
5'-TGAGCCATTTCCTCTGACTTT-3'. The PCR reactions yielded 240, 286,
145, 167 and 145 bp fragments for β-actin, MAFbx, MuRF1, MuRF1, ANP
and BNP, respectively. The relative gene expression was normalized
to that of the house keeping gene β-actin using the
2-ΔΔCq method (16).
Western blotting
H9C2 cells were lysed in RIPA buffer (Shanghai
Biyuntian Biotechnology Co., Ltd.) containing a mixture of protease
inhibitors (Puregene AG) and the total protein content of each
sample and maker was determined using the bicinchoninic acid
protein assay kit (Puregene AG). Each protein sample was diluted 20
times, after which standard protein dilutions were prepared using
bovine serum albumin (2 mg/ml) in duplicate at concentrations of 1,
0.8, 0.6, 0.4 and 0.2 mg/ml. An equal amount of protein was added
per lane (40 µg), separated by 10% SDS-PAGE and transferred onto
PVDF membranes.
The membranes were blocked with 5% skimmed milk
powder in Tris buffered saline with 0.5% Tween-20 (TBS-T) at room
temperature for 2 h. The primary antibodies used for western
blotting were as follows: ANP (1:1,000; cat. no. DF6497; Affinity
Biosciences, Ltd.), MuRF1 (E3 Ub-ligases TRIM63; 1:1,000; cat. no.
DF7178; Affinity Biosciences, Ltd.), MAFbx (1:800; cat. no. DF7075;
Affinity Biosciences, Ltd.), β-actin (1:500; cat. no. BM0627; Wuhan
Boster Biological Technology, Ltd.) and BNP (1:500; cat. no.
ab19645; Abcam). Primary antibody incubation was performed at 4˚C
overnight. After washing in TBS-T five times, the membrane was
incubated with horseradish-peroxidase-conjugated secondary
antibodies (1:50,000; Wuhan Boster Biological Technology, Ltd.;
cat. no. BA1051 and BA1051) at room temperature for 2 h. Protein
bands were detected by an enhanced-chemiluminescent reagent (Vazyme
Biotech Co., Ltd.) and exposed to X-ray film. Band density was
analyzed and quantified using the Image-J imaging software (1.37v;
National Institutes of Health).
MuRF1 and MAFbx knockdown
The MuRF1 and MAFbx small interfering (si) RNA
oligonucleotides (20 nM) were purchased from Thermo Fisher
Scientific, Inc. H9c2 cells were first seeded at a density of
5x105 cells/well into 6-well plates and were transfected
using Opti-MEM (Gibco; Thermo Fisher Scientific, Inc.) containing
Lipofectamine® RNAiMAX (Invitrogen; Thermo Fisher
Scientific, Inc.). Cells were updated with fresh DMEM medium
supplemented with 2% FBS after transfection 4 h before being
subjected to either treatments or assays for knockdown efficiency,
and then collected 24 h later. The following siRNA sequences were
used: MAFbx-851 siRNA sense, 5'-GGCAGCUGGAUUGGAAGAATT-3' and
antisense, 5'-UUCUUCCAAUCCAGCUGCCTT-3'; MuRF1-327 siRNA sense,
5'-GCCGCCAUGAAGUGAUCAUTT-3' and antisense,
5'-AUGAUCACUUCAUGGCGGCTT-3'; negative control (NC)-siRNA sense,
5'-UUCUCCGAACGUGUCACGUTT -3' and antisense,
5'-ACGUGACACGUUCGGAGAATT-3'.
Statistical analysis
A total of three independent experimental repeats
were carried out for each experiment and data are presented as the
mean ± SEM and analyzed using one-way ANOVA followed by Bonferroni
post hoc test with the SPSS 22.0 statistical software (IBM Corp.).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Inhibitory effects of metformin on
cardiomyocyte hypertrophy
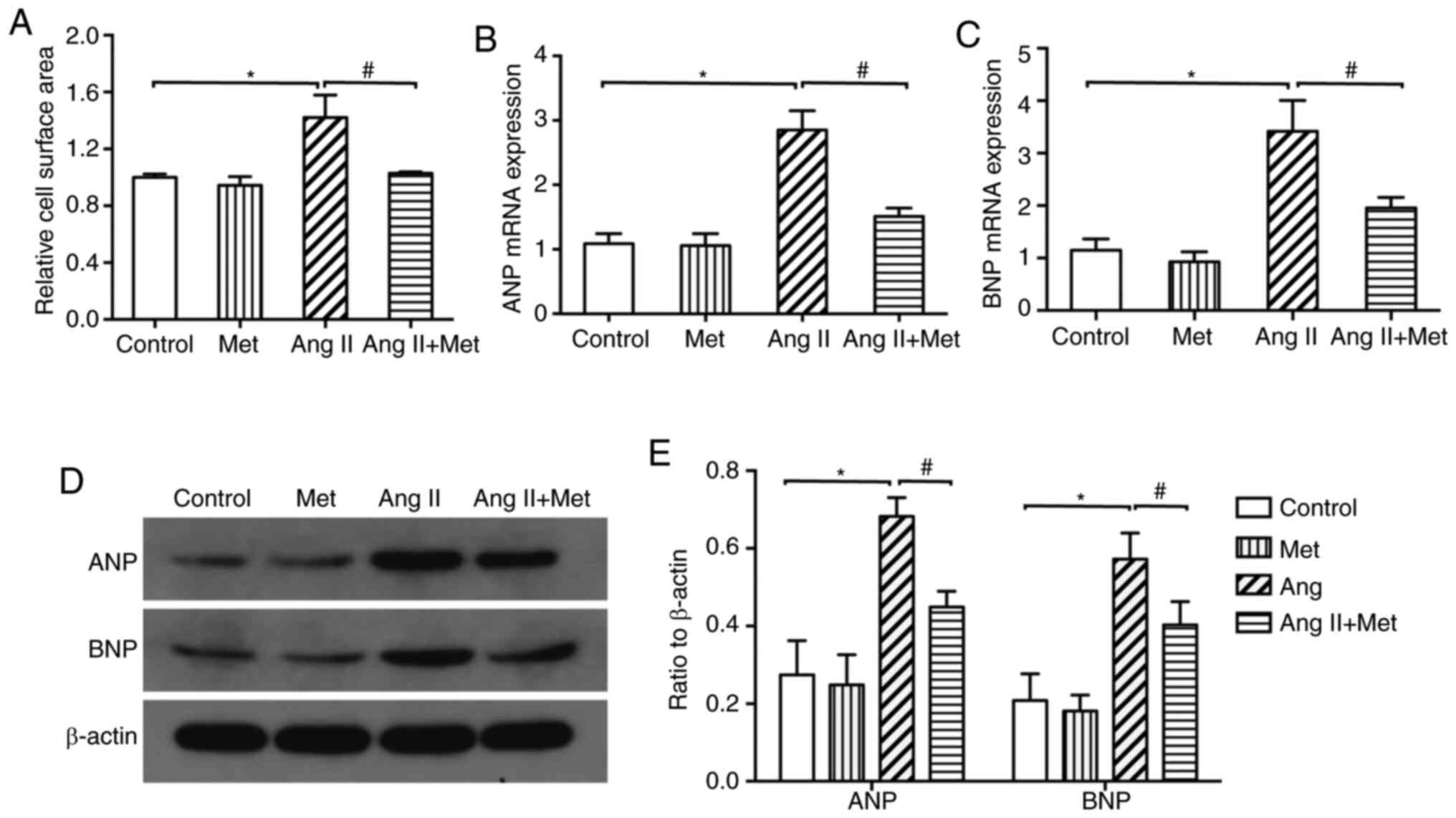
Treatment with Ang II was found to significantly
increase the surface area of cardiomyocytes compared with those in
control (Figs. 1 and 2). Additionally, compared with those in
control, protein and mRNA expression levels of ANP and BNP were
significantly increased by Ang II treatment (P<0.05; Fig. 1B and C). Subsequent experiments demonstrated
that the Ang II-induced increments in cardiomyocyte surface area,
mRNA and protein expression levels of ANP and BNP were all
significantly reversed by metformin treatment for 24 h (P<0.05;
Fig. 1). These results suggest that
metformin can attenuate Ang II-induced cardiomyocyte
hypertrophy.
Effects of metformin on MuRF1 and
MAFbx expression
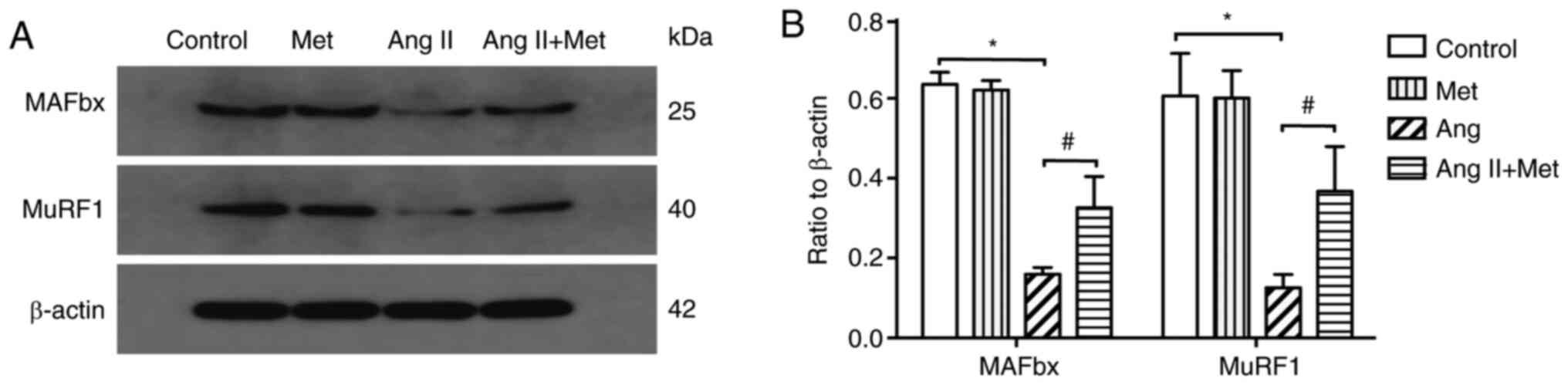
To investigate if MuRF1 and MAFbx are involved in
the attenuation of Ang II-induced cardiomyocyte hypertrophy by
metformin, the effects of Ang II and metformin on the protein
expression of MuRF1 and MAFbx were assessed. The results revealed
that compared with those in the control group, Ang II treatment
significantly downregulated MuRF1 and MAFbx protein expression
levels in cultured cardiomyocytes (P<0.05). However, metformin
treatment significantly reversed this reduction in MuRF1 and MAFbx
protein expression in Ang II stimulated cells (P<0.05; Fig. 3).
MuRF1-siRNA and MAFbx-siRNA blocks the
antihypertrophic effects of metformin
Transfection with MuRF1-siRNA and MAFbx-siRNA
resulted in an average, 82 and 73% knockdown efficiency compared
with control siRNAs, respectively (Figs. 4A, 4D and 4E).
The results revealed that compared with those transfected with
control siRNA, knocking down MuRF1 and MAFbx expression using siRNA
significantly increased the cell surface area of cardiomyocytes in
the presence of Ang II and metformin (P<0.05;Figs. 2 and 4C). Furthermore, the inhibitory effects of
metformin on the Ang II-induced elevations of ANP and BNP mRNA and
protein expression were significantly reversed by MuRF1-siRNA and
MAFbx-siRNA transfection (P<0.05; Fig. 4B, F
and G). Specifically, following Ang
II and metformin treatment, the expression of ANP and BNP was also
significantly higher in the MuRF1-siRNA and MAFbx-siRNA groups
compared with that in the NC-siRNA group (P<0.05;Fig. 4B, F
and G). These results suggested
that metformin upregulated the MuRF1 and MAFbx pathway to inhibit
Ang II-induced cardiomyocyte hypertrophy (Figs. 2 and 4).
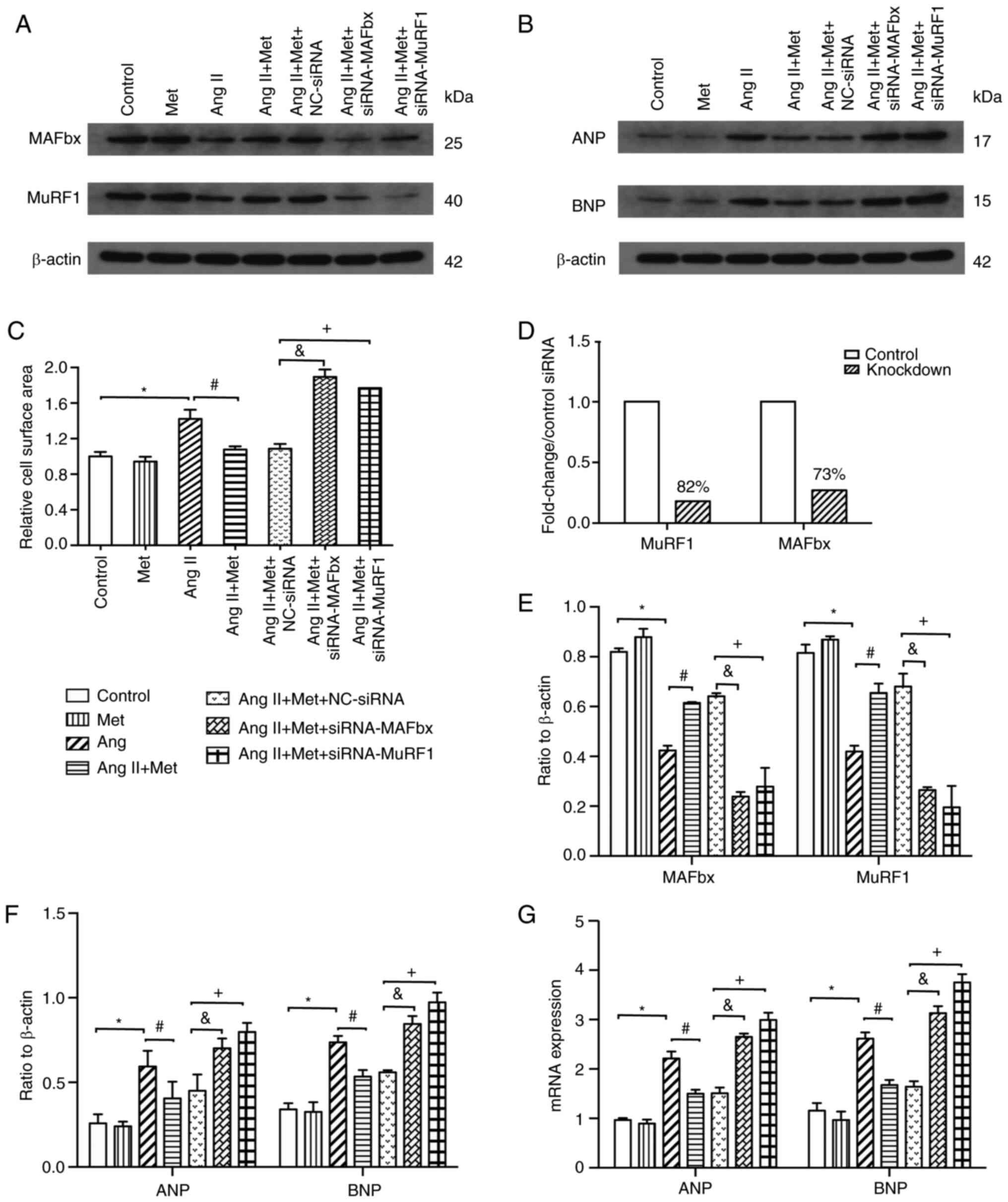 | Figure 4Effects of the MuRF1 and MAFbx
signaling pathway on the effects of Metformin on Ang II-induced
cardiomyocyte hypertrophy. (A-G) After treatment with Ang II and
metformin, H9c2 cells were transfected with either NC-siRNA,
MuRF1-siRNA or MAFbx-siRNA. In the presence of Ang II and
metformin, MuRF1-siRNA and MAFbx-siRNA transfection (A) inhibit the
expression of MuRF1 and MAFbx protein, (B) increase the expression
of ANP and BNP protein and (C) increase cardiomyocyte size compared
with NC-siRNA. (D) Measurement of MuRF1 and MAFbx expression
following transfection with MuRF1-siRNA and MAFbx-siRNA by western
blot presented in (A), which resulted in 82 and 73% knockdown
efficiency compared with control siRNAs, respectively. (E)
Densitometric analysis of MuRF1 and MAFbx expression presented in
(A). (F) Densitometric analysis of ANP and BNP expression presented
in (B). (G) MAFbx-siRNA and MuRF1-siRNA increases the mRNA
expression of ANP and BNP. n=3 for each group.
*P<0.05. #P<0.05.
&P<0.05. +P<0.05. Ang, angiotensin;
MuRF1, muscle RING-finger protein-1; MAFbx, muscle atrophy F-box;
siRNA, small interfering RNA; ANP, atrial natriuretic peptide; BNP,
brain natriuretic peptide; Met, metformin. |
Discussion
The present study investigated the potential effects
of metformin on a model of cardiomyocyte hypertrophy and the
underlying mechanism by which it functions. The results revealed
that metformin attenuated cell hypertrophy by activating the MuRF1
and MAFbx pathway. It was also revealed that Ang II treatment
significantly increased the surface area of cells and increased the
mRNA and protein expression of ANP and BNP whilst reducing the mRNA
and protein expression levels of MuRF1 and MAFbx. These data
suggest that the MuRF1 and MAFbx signaling pathways may serve an
important role in Ang II-induced cardiomyocyte hypertrophy.
MAFbx and MuRF1 are important factors in the UPS and
have been studied widely in the context of cardiac hypertrophy
(9,10). MAFbx and MuRF1 are also considered
to serve a role in protein degradation (17). Overexpression of MAFbx and MuRF1 may
therefore lead to excessive protein degradation and muscle atrophy
(18). Previous studies have
suggested that MuRF1 and MAFbx were involved in the regulation of
the hypertrophic response of cardiac tissue (19,20).
Reduced expression of MuRF1 and MAFbx in the myocardium have been
documented to result in hypertrophy (21). Furthermore, previous studies have
identified the pivotal role of certain E3 ubiquitin ligases in
cardiac hypertrophy, including MuRF1 and MAFbx (22,23).
In particular, a number of studies have demonstrated that MuRF1 and
MAFbx were activated during cardiac hypertrophy, where the precise
control of myofiber synthesis and turnover was essential for
maintaining structural integrity (23,24).
Metformin is a first-line pharmacological agent used
for the treatment of type 2 diabetes, which act to reduce the risk
of cardiovascular events and even mortality (11). The beneficial effects of metformin
on cardiac function have been attributed to direct actions on cell
metabolism, endothelial function, platelet reactivity and calcium
homeostasis (25). Since
hypertrophic stimuli, including angiotensin II and pressure
overload, may increase the expression of ANP and BNP (26), the mRNA levels of ANP and BNP
following metformin and/or Ang II treatment were investigated as
hypertrophic markers in the present study. The results revealed
that metformin significantly suppressed the development of Ang
II-induced cardiomyocyte hypertrophy as evidenced by reductions in
cardiomyocyte surface area, reduced expression of
hypertrophy-associated genes ANP and BNP in Ang II-stimulated
cardiomyocytes. Metformin has also been demonstrated to serve
cardioprotective effects by reducing left ventricular hypertrophy
in patients with coronary artery disease, insulin resistance and
pre-diabetes (27,28) Additionally, previous studies have
demonstrated that metformin can inhibit protein synthesis by
activating MAPK, functioning in the anti-hypertrophic pathway in
cardiomyocytes (29,30).
The present study revealed that metformin increased
the expression of MuRF1 and MAFbx. Furthermore, MuRF1 and MAFbx
knockdown resulted in the reversal of metformin-mediated
suppression of Ang II-induced cardiomyocyte hypertrophy. These
results indicated that metformin inhibited cardiomyocyte
hypertrophy by activating MuRF1and MAFbx signaling pathway.
UPS-dependent protein degradation is an energy-consuming process,
where 5'AMP-activated protein kinase (AMPK) serves a central role
by regulating ATP production (31).
Metformin has been previously demonstrated to increase AMPK
activation to decrease Ang II-induced cardiac hypertrophy, whilst
exerting cardioprotective effects (32). Consistent with this notion, the
present results indicated that metformin conferred cardioprotective
effects by regulating MuRF1and MAFbx expression. In addition, the
present study demonstrated that MAFbx silencing was accompanied by
markedly reduced MuRF1 expression, whereas MuRF1 silencing was also
accompanied by markedly reduced MuRF1 expression. The reason for
that remain unclear. Since both MAFbx and MuRF1 are crucial factors
of E3 proteins that serve roles in protein degradation (22), the possible direct relationship
between these two proteins require further study.
The expression of either MuRF1 or MAFbx is
sufficient to inhibit cardiac hypertrophy (33). Previous studies have revealed that
treatment with metformin increased MuRF1 and MAFbx transcription,
in addition to increasing AMPK phosphorylation (34,35).
This suggest that AMPK activation results in the transcriptional
activation of MuRF1 and MAFbx (36). In addition, previous studies have
also demonstrated that AMPK activation by AMPK activator
5-aminoimidazole-4-carboxamide ribonucleoside (AICAR )significantly
suppressed cardiomyocyte hypertrophy by increasing the activity of
forkhead box protein O1 (FOXO1) and upregulating the expression of
downstream atrogenes MuRF1 and MAFbx (37,38).
The effects of AICAR on cardiomyocyte hypertrophy were also
inhibited after silencing MuRF1 and MAFbx (37,38).
In conclusion, the present study revealed that the
treatment of hypertrophied cardiomyocytes caused by Ang II with
metformin attenuated cardiomyocyte hypertrophy through the MuRF1
and MAFbx pathway. The antihypertrophic effects of metformin may
result from the upregulation of MuRF1 and MAFbx expression in
cardiomyocytes. Although previous studies demonstrated that the
AMPK/FOXO1 and MuRF1/MAFbx pathways may serve an important role in
this process (37,38), further studies are required for
confirmation, especially in an in vivo setting. The
AMPK/FOXO1 pathway was not measured in the present study, meaning
that the mechanism by which AMPK/FOXO1 affects the
metformin-induced attenuation of cardiomyocyte hypertrophy and
MuRF1 and MAFbx transcriptional activity remain to be fully
elucidated. Further studies are required to determine how the
AMPK/FOXO1 and MuRF1/MAFbx pathways are regulated during cardiac
hypertrophy.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Guizhou
Provincial Science and Technology Department Fund of China [grant
no. Qian (2017)1103], The Science and Technology Fund of Guizhou
Provincial Health Commission (grant nos. gzwjkj2018-1-005 and
gzwjkj2019-1-097) and The Clinical Research Center Project of
Department of Science & Technology of Guizhou Province [grant
no. (2017) 5405].
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FD conducted the experiments, data collection and
interpretation. BC participated in study design, coordination of
the experiments and data collection. YR and YC analyzed and
interpreted the data, QW was a major contributor in the conception
and design of this study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Frey N and Olson EN: Cardiac hypertrophy:
The good, the bad, and the ugly. Annu Rev Physiol. 65:45–79.
2003.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Horton JS, Shiraishi T, Alfulaij N,
Small-Howard AL, Turner HC, Kurokawa T, Mori Y and Stokes AJ:
‘TRPV1 is a component of the atrial natriuretic signaling complex,
and using orally delivered antagonists, presents a valid
therapeutic target in the longitudinal reversal and treatment of
cardiac hypertrophy and heart failure’. Channels (Austin). 13:1–16.
2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Patten RD and Hall-Porter MR: Small animal
models of heart failure: Development of novel therapies, past and
present. Circ Heart Fail. 2:138–144. 2009.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Huang L, Xi Z, Wang C, Zhang Y, Yang Z,
Zhang S, Chen Y and Zuo Z: Phenanthrene exposure induces cardiac
hypertrophy via reducing miR-133a expression by DNA methylation.
Sci Rep. 6(20105)2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Spänig S, Kellermann K, Dieterlen MT,
Noack T, Lehmann S, Borger MA, Garbade J, Barac YD and Emrich F:
The ubiquitin proteasome system in ischemic and dilated
cardiomyopathy. Int J Mol Sci. 20(6354)2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lino CA, Demasi M and Barreto-Chaves ML:
Ubiquitin proteasome system (UPS) activation in the cardiac
hypertrophy of hyperthyroidism. Mol Cell Endocrinol.
493(110451)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Cacciapuoti F: Role of
ubiquitin-proteasome system (UPS) in left ventricular hypertrophy
(LVH). Am J Cardiovasc Dis. 4:1–5. 2014.PubMed/NCBI
|
|
8
|
Cui X, Zhang Y, Wang Z, Yu J, Kong Z and
Ružić L: High-intensity interval training changes the expression of
muscle RING-finger protein-1 and muscle atrophy F-box proteins and
proteins involved in the mechanistic target of rapamycin pathway
and autophagy in rat skeletal muscle. Exp Physiol. 104:1505–1517.
2019.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Usui S, Chikata A, Takatori O, Takashima
SI, Inoue O, Kato T, Murai H, Furusho H, Nomura A, Zablocki D, et
al: Endogenous muscle atrophy F-box is involved in the development
of cardiac rupture after myocardial infarction. J Mol Cell Cardiol.
126:1–12. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Gupta I, Varshney NK and Khan S: Emergence
of members of TRAF and DUB of ubiquitin proteasome system in the
regulation of hypertrophic cardiomyopathy. Front Genet.
9(336)2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Marshall SM: 60 years of metformin use: A
glance at the past and a look to the future. Diabetologia.
60:1561–1565. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Markowicz-Piasecka M, Huttunen KM,
Mateusiak L, Mikiciuk-Olasik E and Sikora J: Is Metformin a Perfect
Drug? Updates in Pharmacokinetics and Pharmacodynamics. Curr Pharm
Des. 23:2532–2550. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Driver C, Bamitale KDS, Kazi A, Olla M,
Nyane NA and Owira PMO: Cardioprotective Effects of Metformin. J
Cardiovasc Pharmacol. 72:121–127. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Tseng YT: Cardioprotective effect of
metformin against doxorubicin cardiotoxicity in rats. Anatol J
Cardiol. 16:242–243. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Polianskyte-Prause Z, Tolvanen TA,
Lindfors S, Dumont V, Van M, Wang H, Dash SN, Berg M, Naams JB,
Hautala LC, et al: Metformin increases glucose uptake and acts
renoprotectively by reducing SHIP2 activity. FASEB J. 33:2858–2869.
2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Tsai CH, Tsai HC, Huang HN, Hung CH, Hsu
CJ, Fong YC, Hsu HC, Huang YL and Tang CH: Resistin promotes tumor
metastasis by down-regulation of miR-519d through the AMPK/p38
signaling pathway in human chondrosarcoma cells. Oncotarget.
6:258–270. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Sun Y, Liang X, Chen J, Tang R, Li L and
Li D: Change in Ubiquitin Proteasome System of Grass Carp
Ctenopharyngodon idellus Reared in the Different Stocking
Densities. Front Physiol. 9(837)2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Menconi M, Gonnella P, Petkova V, Lecker S
and Hasselgren PO: Dexamethasone and corticosterone induce similar,
but not identical, muscle wasting responses in cultured L6 and
C2C12 myotubes. J Cell Biochem. 105:353–364. 2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Willis MS, Ike C, Li L, Wang DZ, Glass DJ
and Patterson C: Muscle ring finger 1, but not muscle ring finger
2, regulates cardiac hypertrophy in vivo. Circ Res. 100:456–459.
2007.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Arya R, Kedar V, Hwang JR, McDonough H, Li
HH, Taylor J and Patterson C: Muscle ring finger protein-1 inhibits
PKC{epsilon} activation and prevents cardiomyocyte hypertrophy. J
Cell Biol. 167:1147–1159. 2004.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Conraads VM, Vrints CJ, Rodrigus IE,
Hoymans VY, Van Craenenbroeck EM, Bosmans J, Claeys MJ, Van Herck
P, Linke A, Schuler G, et al: Depressed expression of MuRF1 and
MAFbx in areas remote of recent myocardial infarction: A mechanism
contributing to myocardial remodeling? Basic Res Cardiol.
105:219–226. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Bodine SC, Latres E, Baumhueter S, Lai VK,
Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K,
et al: Identification of ubiquitin ligases required for skeletal
muscle atrophy. Science. 294:1704–1708. 2001.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Fielitz J, Kim MS, Shelton JM, Latif S,
Spencer JA, Glass DJ, Richardson JA, Bassel-Duby R and Olson EN:
Myosin accumulation and striated muscle myopathy result from the
loss of muscle RING finger 1 and 3. J Clin Invest. 117:2486–2495.
2007.PubMed/NCBI View
Article : Google Scholar
|
|
24
|
Shaalan WM, El-Hameid NAA, El-Serafy SS
and Salem M: Expressions and characterization of MuRFs, Atrogin-1,
F-box25 genes in tilapia, Oreochromis niloticus, in response to
starvation. Fish Physiol Biochem. 45:1321–1330. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Nesti L and Natali A: Metformin effects on
the heart and the cardiovascular system: A review of experimental
and clinical data. Nutr Metab Cardiovasc Dis. 27:657–669.
2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ying H, Xu MC, Tan JH, Shen JH, Wang H and
Zhang DF: Pressure overload-induced cardiac hypertrophy response
requires janus kinase 2-histone deacetylase 2 signaling. Int J Mol
Sci. 15:20240–20253. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Li J, Minćzuk K, Massey JC, Howell NL, Roy
RJ, Paul S, Patrie JT, Kramer CM, Epstein FH, Carey RM, et al:
Metformin Improves Cardiac Metabolism and Function, and Prevents
Left Ventricular Hypertrophy in Spontaneously Hypertensive Rats. J
Am Heart Assoc. 9(e015154)2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Mohan M, Al-Talabany S, McKinnie A, Mordi
IR, Singh JSS, Gandy SJ, Baig F, Hussain MS, Bhalraam U, Khan F, et
al: A randomized controlled trial of metformin on left ventricular
hypertrophy in patients with coronary artery disease without
diabetes: The MET-REMODEL trial. Eur Heart J. 40:3409–3417.
2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Rena G, Hardie DG and Pearson ER: The
mechanisms of action of metformin. Diabetologia. 60:1577–1585.
2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Yang F, Qin Y, Wang Y, Meng S, Xian H, Che
H, Lv J, Li Y, Yu Y, Bai Y, et al: Metformin inhibits the NLRP3
inflammasome via AMPK/mTOR-dependent effects in diabetic
cardiomyopathy. Int J Biol Sci. 15:1010–1019. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Xiao B, Sanders MJ, Carmena D, Bright NJ,
Haire LF, Underwood E, Patel BR, Heath RB, Walker PA, Hallen S, et
al: Structural basis of AMPK regulation by small molecule
activators. Nat Commun. 4(3017)2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Benes J, Kazdova L, Drahota Z, Houstek J,
Medrikova D, Kopecky J, Kovarova N, Vrbacky M, Sedmera D, Strnad H,
et al: Effect of metformin therapy on cardiac function and survival
in a volume-overload model of heart failure in rats. Clin Sci
(Lond). 121:29–41. 2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Witt CC, Witt SH, Lerche S, Labeit D, Back
W and Labeit S: Cooperative control of striated muscle mass and
metabolism by MuRF1 and MuRF2. EMBO J. 27:350–360. 2008.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Krawiec BJ, Nystrom GJ, Frost RA,
Jefferson LS and Lang CH: AMP-activated protein kinase agonists
increase mRNA content of the muscle-specific ubiquitin ligases
MAFbx and MuRF1 in C2C12 cells. Am J Physiol Endocrinol Metab.
292:E1555–E1567. 2007.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Thomson DM: The Role of AMPK in the
Regulation of Skeletal Muscle Size, Hypertrophy, and Regeneration.
Int J Mol Sci. 19(3125)2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Vilchinskaya NA, Krivoi II and Shenkman
BS: AMP-Activated Protein Kinase as a Key Trigger for the
Disuse-Induced Skeletal Muscle Remodeling. Int J Mol Sci.
19(3558)2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Chen B, Wu Q, Xiong Z, Ma Y, Yu S, Chen D,
Huang S and Dong Y: Adenosine monophosphate-activated protein
kinase attenuates cardiomyocyte hypertrophy through regulation of
FOXO3a/MAFbx signaling pathway. Acta Biochim Biophys Sin
(Shanghai). 48:827–832. 2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Chen BL, Ma YD, Meng RS, Xiong ZJ, Wang
HN, Zeng JY, Liu C and Dong YG: Activation of AMPK inhibits
cardiomyocyte hypertrophy by modulating of the FOXO1/MuRF1
signaling pathway in vitro. Acta Pharmacol Sin. 31:798–804.
2010.PubMed/NCBI View Article : Google Scholar
|