Introduction
Glioblastoma multiforme (GBM) accounts for 40-50% of
all gliomas (1,2). Patients with GBM have a
progression-free survival of only 6.2-7.5 months after diagnosis
and a median survival of 14.6-16.7 months (3-5),
while the 5-year survival rate is only 10% (6). As a highly malignant tumor, recurrence
of GBM following surgery is inevitable and adjuvant chemotherapy
following GBM resection is essential. Temozolomide (TMZ), a
second-generation alkylating agent, methylates the O6
guanine site, which causes a G-T mismatch during DNA replication
(7). DNA repair mechanisms, such as
mismatch repair systems are then activated, thereby triggering cell
cycle checkpoint protein production and stress response-related
proteins, finally resulting in cell cycle arrest or apoptosis
(8). Large-scale clinical studies
have demonstrated that adherence to a TMZ chemotherapy regime
following surgery prolongs the survival of patients with GBM by 2-4
months (3,9). However, due to the instability of the
tumor cell genome, the expression of
O6-methylguanine-DNA methyltransferase and the
involvement of various different mechanisms facilitating apoptosis
escape, chemotherapy resistance is the main factor affecting the
efficacy of TMZ (10). Of the
multiple studies aimed at enhancing TMZ efficacy, the present study
has focused on heat shock protein 90 (Hsp90)-related research.
According to the latest classification, the Hsp90
family includes five members, of which Hsp90AA1, Hsp90AA2 and
Hsp90AB1 are mainly located in the cytoplasm, Hsp90B1 in the
endoplasmic reticulum and TNF receptor-associated protein 1 (TRAP1)
in the mitochondrial matrix with a few molecules present in the
intermembrane space of mitochondria (11-13).
Although the members of the Hsp90 family have a similar structure,
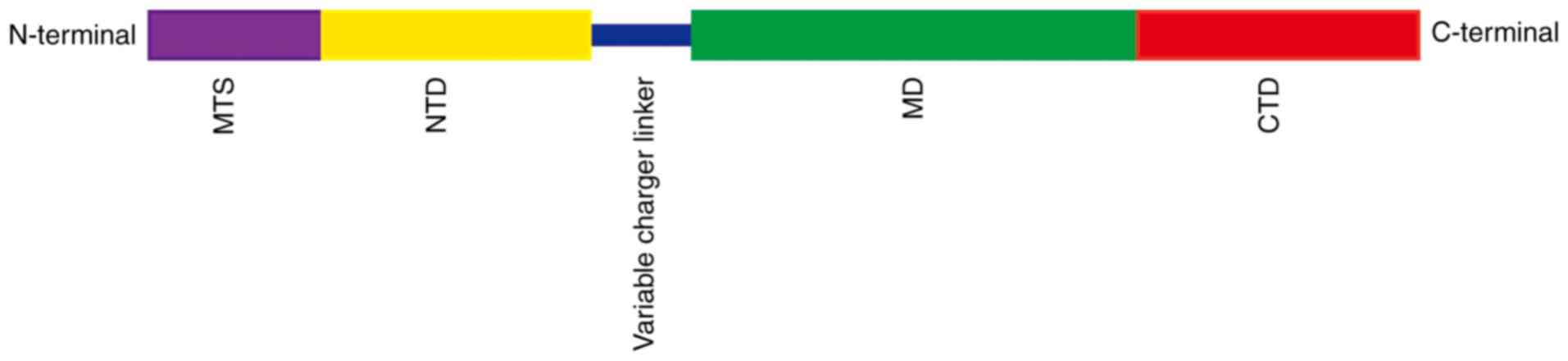
TRAP1 has its own specific characteristics. It consists of four
domains, mitochondria targeting domain (MTS), N-terminal domain
(NTD), middle domain (MD) and C-terminal domain (CTD; Fig. 1) (11) of which it is MTS that is the
mitochondrial targeting sequence. Thus, TRAP1 precursors are
targeted to mitochondria and, after passing through the inner
membrane, the MTS sequence is cleaved by mitochondrial processing
peptidase to form mature TRAP1. The other domains have different
functions as follows: NTD binds and hydrolyzes ATP (11,14),
MD acts as a substrate protein or chaperone-binding domain
(11,15) and CTD contains a homodimerization
binding domain that regulates N-terminal ATPase activity (11,16).
This is necessary for the ATP binding and ADP release cycle.
Changes to the dimer conformation of Hsp90 members help to shape
the structure of the substrate proteins (11).
A previous study demonstrated that TRAP1 has
multiple functions in mitochondria, for example, playing a role in
the intra-mitochondria protein control system (17), regulating the opening of the
permeability transition pore by interacting with cyclophilin D
(13,18) and influencing the mitochondrial
respiratory process by interacting with mitochondrial respiratory
chain complexes (19). In the
present study, suppressing the function of TRAP1 was shown to
sensitize GBM cells to TMZ therapy. Furthermore, treatment with the
mitochondrial-targeting Hsp90 inhibitor Gamitrinib
triphenylphosphonium (G-TPP) together with TMZ exerted an additive
effect in reducing the viability of glioblastoma cells.
Materials and methods
Cell culture
SHG44, U251-MG and U87-MG (glioblastoma of unknown
origin) glioblastoma cell lines were obtained from the Cell Bank of
Type Culture Collection of the Chinese Academy of Sciences and were
grown in DMEM culture medium (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 10% fetal bovine serum (Thermo Fisher
Scientific, Inc.).
Reagents and antibodies
TMZ and G-TPP were purchased from MedChemExpress
company and the MTT assay was obtained from MilliporeSigma. The
MitoTracker™ (cat. no. 7512), Hoechst 33342 (cat. no.
H3570) and LysoTracker™ (cat. no. L7526) were all
obtained from Thermo Fisher Scientific, Inc. RIPA buffer (cat. no.
P0013B) and horse serum (cat. no. C0262) were purchased from
Beyotime Institute of Biotechnology. The antibodies used in the
present study were as follows: anti-Hsp10 (cat. no. ab210681;
Abcam), anti-phosphorylated-ubiquitin [serine 65 (Ser65) cat. no.
ABS1513-I] from Merck KGaA, anti-ubiquitin (cat. no. 13-1600) from
Thermo Fisher Scientific, Inc., anti-β actin (cat. no. 60008-1-Ig),
anti-Bax (cat. no. 50599-2-Ig), anti-PTEN-induced kinase 1 (PINK1;
cat. no. 23274-1-AP), anti-caspase-3 (cat. no. 19677-1-AP),
anti-caspase-9 (cat. no. 10380-1-AP), anti-Hsp60 (cat.
no.15282-1-AP) and anti-p53 (cat. no. 60283-2-Ig) from ProteinTech
Group, Inc., goat anti-mouse secondary fluorescent antibody (cat.
no. F2761) and goat anti-rabbit secondary fluorescent antibody
(cat. no. A-11012) from Thermo Fisher Scientific, Inc. and goat
anti-mouse, HRP-conjugated secondary antibody (cat. no. SA00001-1)
and goat anti-rabbit, HRP-conjugated secondary antibody (cat. no.
SA00001-2) from ProteinTech Group, Inc.
MTT assays
Cells were seeded into 96-well plates at a density
of 1x104 cells/well and cultured with TMZ or G-TPP,
after which 20 µl MTT solution (5 mg/ml) was introduced into each
well. After 4 h incubation in the CO2 incubator at 37˚C,
150 µl DMSO was added to each well. The plates were assessed using
a CLARIO star microplate reader (BMG Labtech GmbH) at an absorbance
of 570 nm.
Immunofluorescence (IF) assay
Cells grown on coverslips in 24-well plates were
treated with the different agents. After staining with MitoTracker
for 30 min in CO2 incubator at 37˚C, the coverslips were
rinsed with PBS and fixed with 4% (w/v) paraformaldehyde for 25 min
followed by a PBS rinse. Permeabilization with 0.1% (v/v) Triton
X-100 for 7 min was followed by blockade with 10% horse serum for
another 30 min at room temperature. Following incubation with
anti-p53 or anti-Hsp60 antibodies (1:100) at 4˚C for 12 h, the
coverslips were incubated with fluorescent secondary antibodies
(1:200) for 30 min at room temperature, then rinsed with cold PBS
and stained with Hoechst 33342 according to the manufacturer's
instructions for 7 min before rinsing with PBS again. A Revolve
Hybrid Microscope (Discover ECHO) was used for image capture.
Images were further processed by ImageJ Software version 1.52s
(National Institutes of Health) to assess the intensity of IF, as
well as the mitochondrial parameters. For the evaluation of
mitochondrial fusion, three images in each treatment group from
three independent experiments were evaluated using the ImageJ
Software and three parameters were used to assess the level of
mitochondrial fusion as follows: The mean size of mitochondrial
network (MS), the mean length of mitochondria (ML) and the number
of mitochondrial nets/individual mitochondria (N/I).
Western blot (WB) analysis
Cells exposed to the different agents were
harvested, washed at room temperature with cold PBS and lysed with
80 µl RIPA buffer. Cell lysates were centrifuged at 4,500 x g for
10 min at 4˚C and the supernatant proteins were harvested. A total
of 10 µg proteins were separated on 12 or 15% (w/v)
SDS-polyacrylamide gels and transferred to Immobilon™-P transfer
membranes (EMD Millipore). Next, 10% (w/v) skimmed milk was used to
block the membranes for 30 min at room temperature, before they
were incubated with primary antibodies (anti-Hsp10,
anti-phosphorylated-ubiquitin, anti-ubiquitin, anti-β-actin,
anti-Bax, anti-PINK1, anti-caspase-3, anti-caspase-9, anti-Hsp60 or
anti-p53; 1:1,000) overnight at 4˚C. The next day, after rinsing
with PBST, the membranes were incubated with goat anti-mouse or
goat anti-rabbit HRP-conjugated secondary antibodies (1:1,000) for
2 h at room temperature. After washing with PBST for three times,
10 min each, protein bands were visualized using the Syngene Bio
Imaging System (Synoptics). β-actin was used as loading control.
Quantification of protein bands was performed using the ImageJ
Software version 1.52s (National Institutes of Health).
Knocking down TRAP1
shRNA sequences targeting human TRAP1 and the empty
vector were purchased from Shanghai GenePharma Co., Ltd. The TRAP1
shRNA sequence was as follows:
5'-CCGGCAGAGCACTCACCCTACTATGC-TCGAGCATAGTAGGGTGAGTGCTCTGTTTTTG-3'.
Plasmids were transduced into cells using transfection reagent
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. Briefly, SHG44 cells grown in 6-well plates were
transfected with 4 µg TRAP1-shRNA or empty vector control and 6 µl
transfection reagent was added to each well. Cells were harvested
24 h later and samples were further analyzed by WB analysis.
Apoptosis analysis
In order to evaluate the efficacy of TMZ and G-TPP
treatment on apoptosis, SHG44 cells grown in 6-well plates were
exposed to 400 µM TMZ (20), 15 µM
G-TPP or 400 µM TMZ + 15 µM G-TPP for 24 h, then washed with cold
PBS for three times and trypsinized at room temperature for 5 min
by 0.25% trypsin solution without EDTA. Apoptosis assays were
performed using the Apoptosis Detection Kit (BD Biosciences)
according to the manufacturer's instructions. Cells were then
counted using a Guava® easyCyte flow cytometer (Merck
KGaA).
Effects of combination treatment
A combination index (CI) is used to evaluate the
synergistic effects of drugs according to the Chou-Talalay method
(21) as follows: Synergism,
CI<1; additive effect, CI=1; antagonism, CI >1. In the
present study, growth inhibition effects were evaluated by MTT
assays on SHG44 cells. To evaluate the effect of a combination
treatment of TMZ and G-TPP, independent assays were conducted with
different concentrations of TMZ (200, 400, 800, 1,000 and 1,600 µM)
and G-TPP (5, 10, 15, 20, 30 and 40 µM) for 24 h and the growth
inhibition effect of each treatment group was evaluated by MTT
assays. Moreover, 15 µM G-TPP was combined with difference
concentrations of TMZ (200, 400 and 800 µM) and in each independent
assay, the two agents were added together simultaneously. The
growth inhibition effect was evaluated by MTT following 24 h of
treatment. The CI was calculated using CalcuSyn version 1.0.1
(Biosoft).
Statistical analysis
Data from three independent experiments were
collected and analyzed using SPSS 22.0 (IBM Corp.) and Student's
t-test. One-way ANOVA followed by Dunnett's post hoc test was also
used to conduct multiple comparisons using Graphpad Prism 7.00
(GraphPad Software, Inc.). Data are presented as the means ±
standard deviation and P<0.05 was considered to indicate a
statistically significant difference.
Results
Inhibition of TRAP1 sensitizes GBM
cells to TMZ
G-TPP combines the ATPase inhibition module of the
Hsp90 inhibitor, 17-allylamino-17-demethoxy-geldanamycin and the
mitochondrial targeting portion of TPP. This enables the drug to
target and concentrate in the mitochondria and also exert the Hsp90
inhibitory function (22). In the
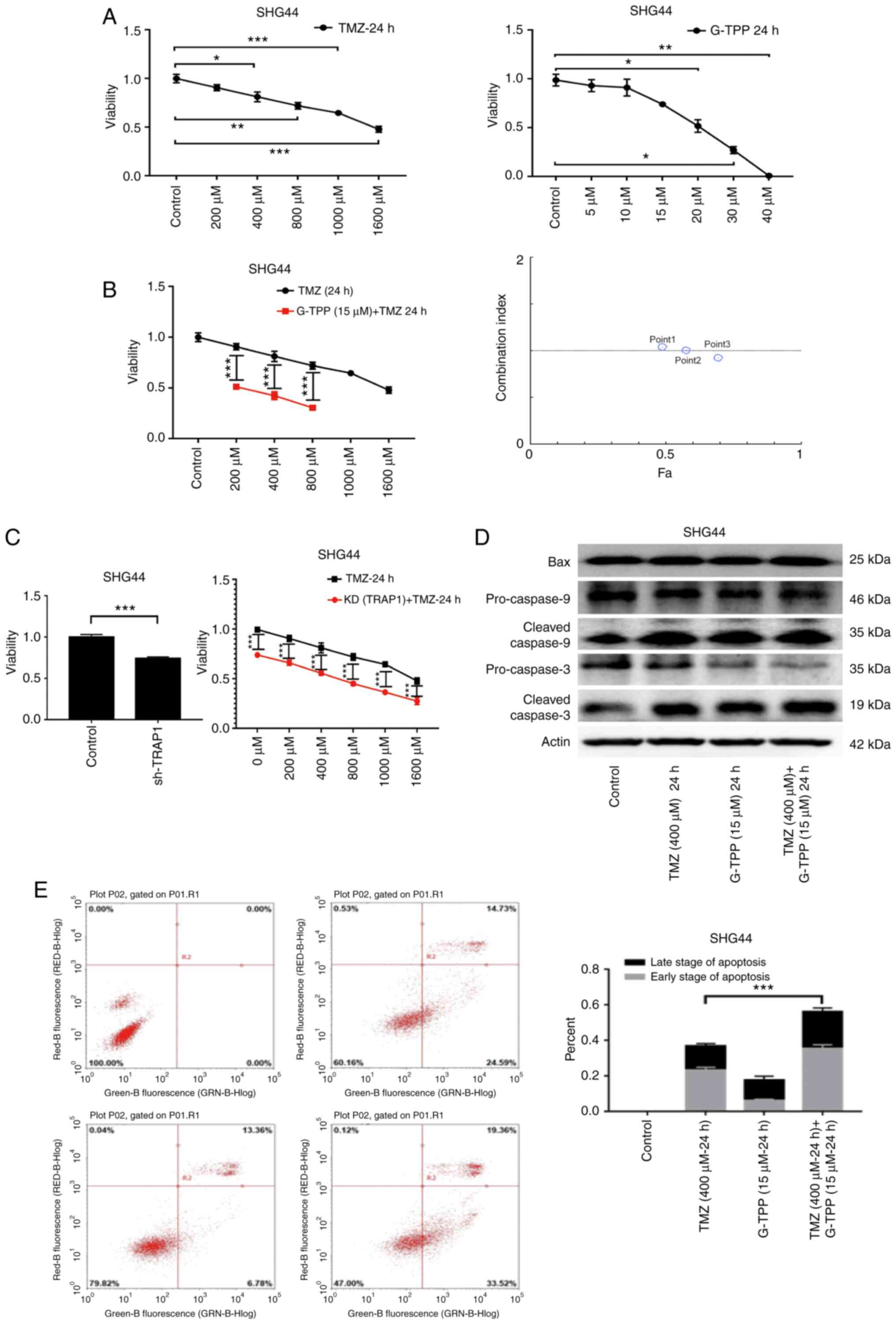
MTT assay, G-TPP and TMZ mediated a significant
concentration-dependent inhibitory effect on the viability of the
GBM cell line, SHG44 (Fig. 2A).
When the two agents were combined, cell viability was significantly
more inhibited, and the CI analysis indicated an additive effect
(Fig. 2B). When TRAP1 was knocked
down by shRNA, the GBM cells were sensitized to TMZ (Fig. 2C) and WB revealed that the levels of
expression of apoptosis-related proteins were increased (Fig. 2D). In the flow cytometric apoptosis
assay, the degree of apoptosis in the combination treatment group
was significantly increased (Fig.
2E). These results demonstrate that inhibition of TRAP1
sensitizes GBM cells to TMZ and indicate the existence of an
underlying complementary pathophysiological process that is
responsible.
TMZ induces apoptosis of GBM cells by
activating the p53 pathway and concurrently downregulates
PINK1
The mechanism of TMZ-induced apoptosis in GBM cells
was evaluated. After exposing GBM cell lines, SHG44 and U87 to
titrated concentrations of TMZ, a significant increase in the
expression of stress protein p53 was observed, which was confirmed
by WB and IF assays. The co-localization analysis indicated that
the p53 protein predominantly accumulated in the nucleus (Fig. 3A), which is consistent with the
known role of p53 as an important nuclear transcription factor.
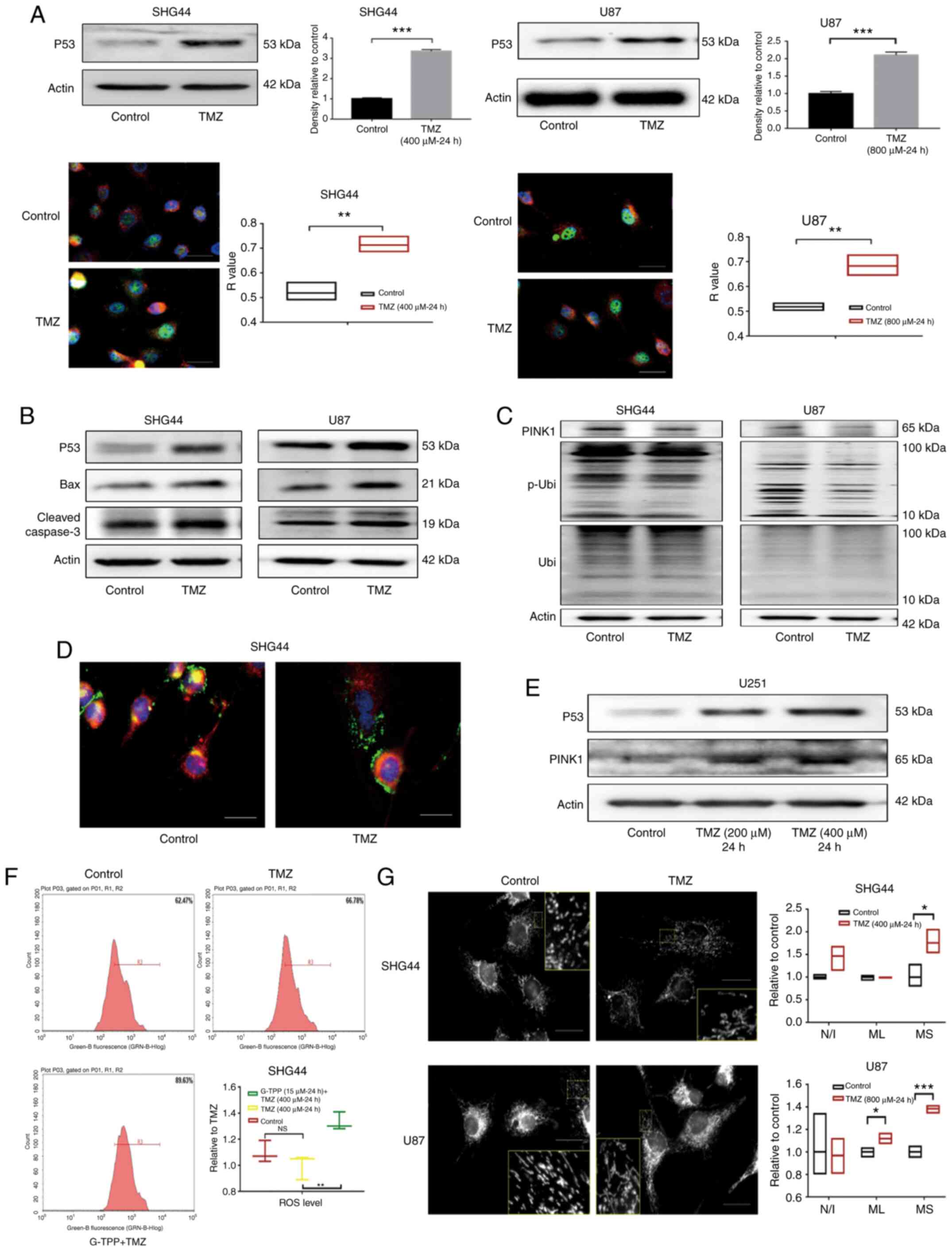 | Figure 3(A) The expression of p53 in
glioblastoma cell lines SHG44 and U87 was significantly upregulated
following TMZ treatment. In the cell immunofluorescence assay, p53,
nucleus and mitochondria were stained green, blue and red,
respectively, which indicated that the upregulated p53
predominantly accumulated in the nucleus following TMZ treatment
(scale bar, 80 µm). (B) Following treatment with TMZ, the
upregulation of p53 and the activation of its downstream apoptotic
pathways mediated by the Bcl-2 family was observed. (C) In GBM
cells stimulated by TMZ, downregulation of PINK1 was observed, as
well as a decreased level of Ser-65 site phosphorylated ubiquitin
that indicated the impaired function of PINK1. (D) Following
treatment with TMZ, the co-localization of lysosome (green) and
mitochondria (red) decreased, which indicated the downregulation of
mitophagy. The nucleus was stained blue (scale bar, 80 µm). (E) In
the p53 mutant U251 cells, TMZ treatment increased the level of p53
and PINK1. (F) A total of 62.47% cells of the control group were
included in the R3 gate, while 66.78% of the TMZ treatment group
and 89.63% of the TMZ + G-TPP group were in the same R3 gate. The
level of ROS in GBM cells in the TMZ treatment group was not
statistically different from that in the control group (P=0.2892),
while the level of ROS in the G-TPP and TMZ combined treatment
group was significantly higher than the TMZ group (P=0.0041). (G)
Following TMZ treatment, the level of mitochondrial fusion of
glioblastoma cells was increased. The mean size of the MS was
elevated in SHG44 and U87 cells, and the mean ML in U87 cells was
also increased, although the difference in the number of N/I was
not significantly different between the two groups (scale bar, 80
µm). *P<0.05; **P<0.01;
***P<0.001. GBM, glioblastoma multiforme; G-TPP,
Gamitrinib triphenylphosphonium; TMZ, temozolomide; CI, combination
index; TRAP1, TNF receptor-associated protein 1; PINK1,
PTEN-induced kinase 1; p, phosphorylated; Ubi, ubiquitin; ROS,
reactive oxygen species. MS, mitochondrial network; ML, length of
mitochondria; N/I, mitochondrial nets/individual mitochondria. |
Downstream, upregulation of the pro-apoptotic Bcl-2
family member Bax was observed. Previous studies have reported that
p53 induces apoptosis by promoting the transcription of Bax
(23,24). Downstream of Bax the activation of
the apoptosis-executing protein caspase-3 was observed (Fig. 3B). In addition, PINK1 expression was
markedly downregulated by TMZ in GBM cells. The function of PINK1
was evaluated with an antibody specific for
phosphorylated-ubiquitin (Ser65). The phosphorylating activity of
PINK1 was identified to be downregulated by TMZ (Fig. 3C). Furthermore, staining of
lysosomes and mitochondria demonstrated a decreased co-localization
of the two organelles in the TMZ treatment group, which indicated
the downregulation of mitophagy (Fig.
3D). Notably, when the expression levels of p53 and PINK1 were
evaluated in the p53 mutant GBM cell line, U251, the upregulation
of p53 and PINK1 was observed under the TMZ treatment, which
indicated that p53-PINK1 regulation may only exist in cell line
with wild-type p53 (Fig. 3E).
Mitophagy is an important aspect of the
mitochondrial quality control system. To evaluate mitochondrial
function under the condition of downregulated mitophagy,
intracellular ROS levels following TMZ treatment were assessed, but
no difference was identified when compared with the control
(Fig. 3F). Furthermore,
mitochondrial morphology was assessed and the level of
mitochondrial fusion in SHG44 and U87 cells was observed to be
elevated following TMZ treatment (Fig.
3G). Currently, it is considered that mitochondrial fusion is a
compensatory mechanism for impaired mitochondrial function, which
can achieve functional complementation between dysfunctional
mitochondria. This restores mitochondrial function to a certain
extent (25) and reduces ROS
generated by mitochondrial dysfunction and the subsequent damaging
reactions.
TRAP1 inhibition and TMZ treatment
induce an exacerbated mitochondrial unfolded protein response
(mtUPR) in GBM cells
The mtUPR was evaluated following TRAP1 knockdown
using shRNA (Fig. 4A). The results
indicated that TRAP1 plays an important role in the protein quality
control system in mitochondria. SHG44 cells were then treated with
G-TPP and a typical mtUPR was observed (Fig. 4B). IF assays to evaluate the level
of Hsp60 expression also confirmed this effect (Fig. 4C). These findings are consistent
with previous studies (26,27). Treatment with a combination of G-TPP
and TMZ resulted in a higher mtUPR when compared to treatment with
G-TPP alone, although no definite mtUPR occurred in cells that were
treated with TMZ alone (Fig. 4D).
This indicates that G-TPP and TMZ act together in aggravating the
mtUPR in GBM cells.
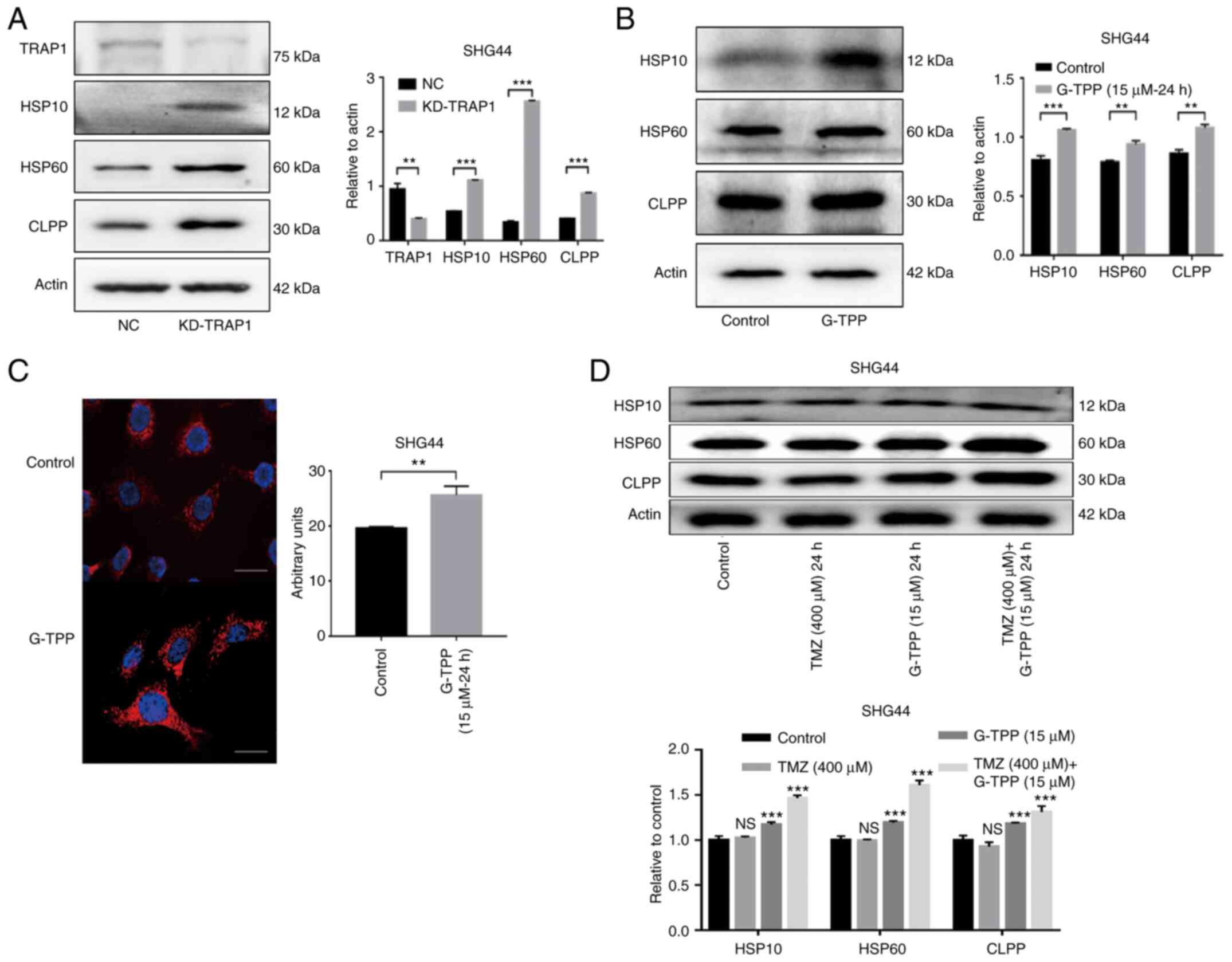 | Figure 4(A) The increased expression of
Hsp10, Hsp60 and CLPP indicated that knocking down TRAP1 induced
mtUPR. The efficiency of TRAP1 knockdown was ~60%. (B) G-TPP
treatment of glioblastoma cells induced mtUPR. (C) Cell
immunofluorescence assay confirmed that the expression level of
Hsp60 (red) in glioblastoma cells was significantly increased
following G-TPP treatment. Nuclei were stained blue (scale bar, 80
µm). (D) Increased expression of Hsp10, Hsp60 and CLPP was observed
in the combined TMZ and G-TPP treatment group.
**P<0.01; ***P<0.001 vs. the indicated
groups or control. TRAP1, TNF receptor-associated protein 1; mtUPR,
mitochondrial unfolded protein response; Hsp, heat shock protein;
CLPP, caseinolytic mitochondrial matrix peptidase proteolytic
subunit; G-TPP, Gamitrinib triphenylphosphonium; TMZ, temozolomide;
NC, negative control; NS, not significant. |
Combined treatment with G-TPP and TMZ
significantly increases the level of ROS in glioblastoma cells
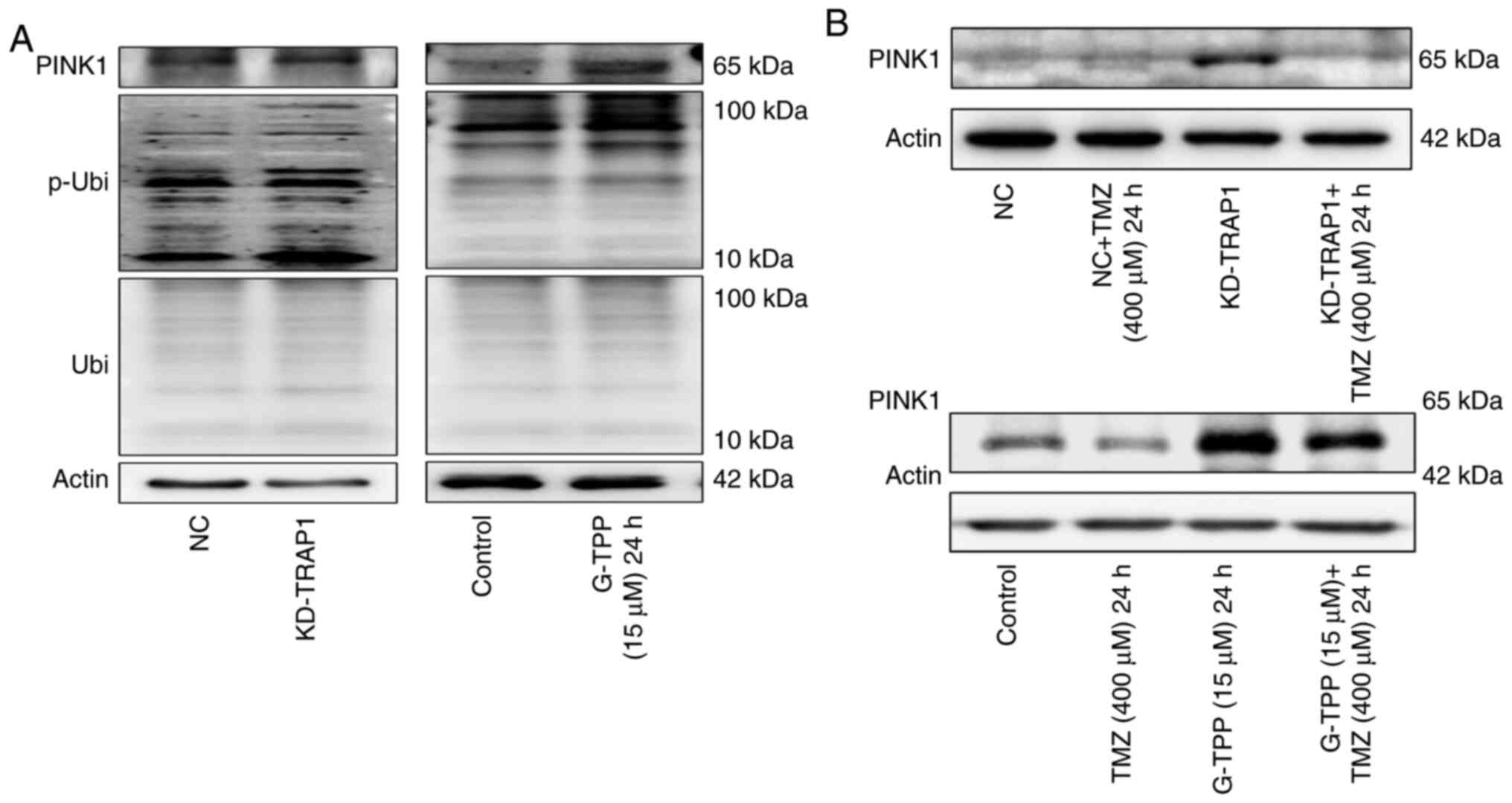
It is generally hypothesized that upon exposure to
mtUPR-inducing factors, cells will increase mitophagy to remove
damaged mitochondria, reduce the production of harmful metabolites
and recycle material. This is exactly what was observed when TRAP1
was knocked down or G-TPP was administered in the present study,
particularly, elevated PINK1 expression and phosphorylated
ubiquitin at the Ser65 site (Fig.
5A). However, due to the inhibitory effect of TMZ on the
expression of PINK1, mitophagy (an essential aspect of the
mitochondrial quality control system) could not take place in the
presence of a combination treatment with G-TPP and TMZ (Fig. 5B). When the extent of mtUPR exceeds
the capacity of the compensatory mechanisms to maintain homeostasis
in mitochondria, dysfunction of the mitochondrial system, such as
fusion, occurs. This may be the underlying mechanism responsible
for the increased level of mtUPR in cells treated with the
combination of agents. Numerous damaged mitochondria with mtUPR
that cannot be normally removed will affect mitochondrial
metabolism and result in the production of increased quantities of
deleterious factors, such as ROS (28). Therefore, the ROS level was
evaluated and TMZ treatment alone did not affect the level of
intracellular ROS, although this increased significantly when TMZ
was combined with G-TPP (Fig. 3F).
This is consistent with the above-mentioned inference. It is
believed that a large quantity of ROS damages mitochondria and
nuclear DNA and induces apoptosis (29) and that there may also be a cyclic
amplification effect consisting of: ROS, DNA damage, ROS (29). Aggravated mtUPR and the following
ROS burst may be the mechanism by which TRAP1 inhibition sensitizes
GBM cells to TMZ treatment.
Discussion
As the most malignant type of primary tumor of the
central nervous system, glioblastoma is associated with a
particularly poor prognosis (30).
In the current regime for comprehensive therapy, chemotherapy plays
an indispensable role and TMZ, as an effective chemotherapeutic
agent verified by large-scale clinical trials, remains the dominant
drug for GBM chemotherapy (30,31).
However, prolonged TMZ chemotherapy results in the development of
GBM resistance, which is an important factor affecting the efficacy
of the drug (8,10). Therefore, there is an urgent need to
develop appropriate adjuvant drugs for combination with TMZ
chemotherapy to increase sensitivity and decrease resistance. TRAP1
became the focus of the present study as it is a molecule with an
important role in the physiological and pathological processes of
tumor cell biology. Studies have shown that TRAP1 is highly
expressed in numerous types of cancer, indicating that this
molecule may influence various shared behaviors of tumor cells
(32,33).
Previous studies have shown that TRAP1 is involved
in the protein quality control system in mitochondria. Knocking
down TRAP1 causes a typical mtUPR (17), which is consistent with the findings
of the present study. Furthermore, studies have reported that TRAP1
reduces the accumulation of ROS in cells (34,35).
These characteristics indicate that TRAP1 may represent an
alternative target for sensitizing GBM to TMZ chemotherapy. In the
present study, it was found that TMZ induced apoptosis by
activating p53 and its downstream pathways. Moreover, two other
effects induced by TMZ are of note.
Firstly, on treatment with TMZ, tumor cells
downregulate PINK1 expression and activity, which may in turn
downregulate mitophagy. As one of the main pathways for initiating
mitophagy, PINK1 anchors to the surface of mitochondria,
phosphorylates the ubiquitin at Ser65 and recruits Parkin to
conduct mitophagy (36). The
observed downregulation of PINK1 expression and phosphorylating
ability implies that the effect of TMZ downregulates mitophagy.
Studies have shown that p53 binds to the PINK1 promoter region and
blocks its transcription, which results in the downregulation of
mitophagy (37). This is consistent
with the TMZ-induced upregulation of p53 expression and aggregation
in the nucleus in present study.
Secondly, the ROS level of tumor cells did not
increase significantly while the level of mitochondrial fusion did
increase in present study. Typically, decreased mitophagy will be
accompanied by the reciprocal accumulation of damaged mitochondria,
thus increasing the level of harmful metabolites, such as ROS in
cells (38). However, in the case
of TMZ treatment, it is possible that the elevated level of
mitochondrial fusion compensates for the downregulation of
mitophagy in sustaining a healthy mitochondrial system. Suppressing
the function of TRAP1 in the present study, either by G-TPP
treatment or by shRNA, may have overloaded this compensatory effect
of mitochondrial fusion by inducing the mtUPR, which further
exacerbated mitochondrial dysfunction and damage. The accumulation
of dysfunctional or damaged mitochondria is always accompanied by
the production of deleterious metabolites, such as ROS (39), which damage cells and induce
apoptosis, as reported by a large number of earlier studies
(40,41). In addition to inducing mitochondrial
damage and nuclear DNA damage (42), ROS also activates the downstream JNK
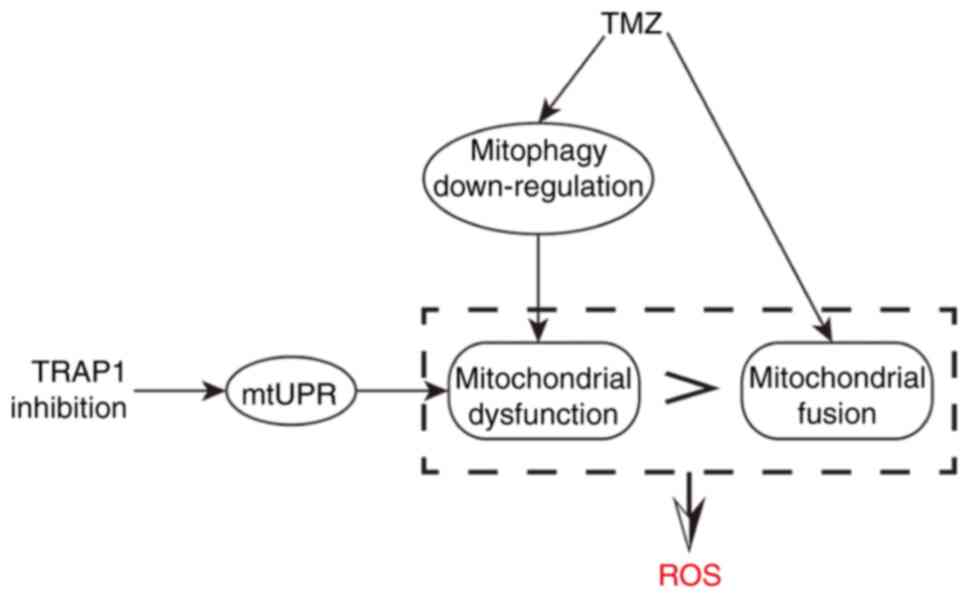
pathway and other pathways, thereby inducing apoptosis (43). It is hypothesized that this is the
underlying mechanism for sensitizing GBM cells with wild-type p53
to TMZ, that is to say, by TRAP1 inhibition and the additive effect
of TMZ and G-TPP treatment (Fig.
6). However, as p53 is frequently mutated in GBM, the present
study aimed to establish whether this mechanism continues to work
in p53 mutant GBM. U251, a cell line with mutant p53, was treated
with TMZ and concentration-dependent, increased expression levels
of p53 and PINK1 were observed. The point mutation in the DNA
binding domain of p53 in U251 cells impairs the affinity of p53 to
DNA, which weakens the downregulation of PINK1 by p53. This result
strengthens the hypothesis that p53 downregulates the transcription
of PINK1 in the form of nucleus transcriptional factor and also
indicates that the above-mentioned mechanism may be ineffective in
p53 mutant cell lines.
The connections between the various pathways in
tumor cells are complex. The present study cannot reveal all of the
changes caused by TMZ treatment or interference of TRAP1, however,
as a key molecule linking the mitochondrial protein quality control
system and mitochondrial metabolism, TRAP1 plays important roles in
many physiological and pathophysiological cellular processes. This
in turn indicates that the present study on TRAP1 has great
potential and practical value in the diagnosis and treatment of
tumors.
Inhibiting the function of TRAP1 by G-TPP treatment
or shRNA induces the mtUPR, while TMZ treatment downregulates
mitophagy in addition to inducing apoptosis of GBM cells via the
p53 pathway. The combination of these two effects aggravates the
damage to the mitochondrial system and increases the accumulation
of harmful metabolites and ROS in GBM cells, thus inducing adverse
events, even apoptosis. Therefore, TRAP1, as a key molecule that
affects the mitochondrial protein quality control system and
mitochondrial metabolism, represents a promising target in the
treatment of GBM. However, certain limitations exist to the present
study. The mechanism revealed in the current study requires
confirmation in additional cell lines and in vivo studies,
and the mechanism of TMZ-induced mitochondrial fusion will be
investigated in future studies.
Acknowledgements
Not applicable.
Funding
Funding: The present study was financially supported by the
Department of Science and Technology of Jilin Province (grant no.
20180101136JC), the Department of Finance of Jilin Province (grant
no. 2018SCZ030), the Education Department of Jilin Province (grant
no. JJKH20190005KJ), Development and Reform Commission Engineering
Laboratory Project of Jilin Province (grant no. 2019C031), the
Norman Bethune Program of Jilin University (grant no. 2015218), the
Lateral Research Funds of Jilin University (grant no. 2015377), the
Excellent Talents Training Plan of China-Japan Union Hospital
(grant no. YXZN-201803), the National Natural Science Foundation of
China (grant nos. 81772794 and 81472419) and the Jilin Provincial
Industrial Innovation Project (grant no. 2018C052-7).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
NW conducted experiments and wrote the original
draft of the manuscript. PZ and RH assisted with the experiments
and figures. LS and DD reviewed and edited the manuscript and
participated in the design of the study. YG conceptualized the
project, performed project administration and acquired the funding.
YG and DD confirmed the authenticity of all the raw data. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ostrom QT, Gittleman H, Liao P, Rouse C,
Chen Y, Dowling J, Wolinsky Y, Kruchko C and Barnholtz-Sloan J:
CBTRUS statistical report: Primary brain and central nervous system
tumors diagnosed in the United States in 2007-2011. Neuro Oncol. 16
(Suppl 4):iv1–iv63. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Wirsching HG, Galanis E and Weller M:
Glioblastoma. Handb Clin Neurol. 134:381–397. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: Radiotherapy plus concomitant and adjuvant temozolomide
for glioblastoma. N Engl J Med. 352:987–996. 2005.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Gilbert MR, Dignam JJ, Armstrong TS, Wefel
JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S,
Won M, et al: A randomized trial of bevacizumab for newly diagnosed
glioblastoma. N Engl J Med. 370:699–708. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Gilbert MR, Wang M, Aldape KD, Stupp R,
Hegi ME, Jaeckle KA, Armstrong TS, Wefel JS, Won M, Blumenthal DT,
et al: Dose-dense temozolomide for newly diagnosed glioblastoma: A
randomized phase III clinical trial. J Clin Oncol. 31:4085–4091.
2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Stupp R, Hegi ME, Mason WP, van den Bent
MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B,
Belanger K, et al: Effects of radiotherapy with concomitant and
adjuvant temozolomide versus radiotherapy alone on survival in
glioblastoma in a randomised phase III study: 5-year analysis of
the EORTC-NCIC trial. Lancet Oncol. 10:459–466. 2009.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Liu P, Li P, Lei T, Qu L, Huang H and Mu
Q: Acute lymphoblastic leukemia following temozolomide treatment in
a patient with glioblastoma: A case report and review of the
literature. Oncol Lett. 15:8663–8668. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Strobel H, Baisch T, Fitzel R, Schilberg
K, Siegelin MD, Karpel-Massler G, Debatin KM and Westhoff MA:
Temozolomide and other alkylating agents in glioblastoma therapy.
Biomedicines. 7(69)2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Oike T, Suzuki Y, Sugawara K, Shirai K,
Noda SE, Tamaki T, Nagaishi M, Yokoo H, Nakazato Y and Nakano T:
Radiotherapy plus concomitant adjuvant temozolomide for
glioblastoma: Japanese mono-institutional results. PLoS One.
8(e78943)2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Lee SY: Temozolomide resistance in
glioblastoma multiforme. Genes Dis. 3:198–210. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Hoter A and El-Sabban ME: The HSP90
family: Structure, regulation, function, and implications in health
and disease. Int J Mol Sci. 19(2560)2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Altieri DC: Hsp90 regulation of
mitochondrial protein folding: From organelle integrity to cellular
homeostasis. Cell Mol Life Sci. 70:2463–2472. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Altieri DC, Stein GS and Lian JB: TRAP-1,
the mitochondrial Hsp90. Biochim Biophys Acta. 1823:767–773.
2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Dutta R and Inouye M: GHKL, an emergent
ATPase/kinase superfamily. Trends Biochem Sci. 25:24–28.
2000.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Meyer P, Prodromou C, Hu B, Vaughan C, Roe
SM, Panaretou B, Piper PW and Pearl LH: Structural and functional
analysis of the middle segment of hsp90: Implications for ATP
hydrolysis and client protein and cochaperone interactions. Mol
Cell. 11:647–658. 2003.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Soti C, Vermes Á, Haystead TA and Csermely
P: Comparative analysis of the ATP-binding sites of Hsp90 by
nucleotide affinity cleavage: A distinct nucleotide specificity of
the C-terminal ATP-binding site. Eur J Biochem. 270:2421–2428.
2003.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Siegelin MD, Dohi T, Raskett CM, Orlowski
GM, Powers CM, Gilbert CA, Ross AH, Plescia J and Altieri DC:
Exploiting the mitochondrial unfolded protein response for cancer
therapy in mice and human cells. J Clin Invest. 121:1349–1360.
2011.PubMed/NCBI View
Article : Google Scholar
|
|
18
|
Kang BH and Altieri DC: Compartmentalized
cancer drug discovery targeting mitochondrial Hsp90 chaperones.
Oncogene. 28:3681–3688. 2009.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Masgras I, Sanchez-Martin C and Colombo G:
The chaperone TRAP1 as a modulator of the mitochondrial adaptations
in cancer cells. Front Oncol. 7(58)2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhang D, Dai D, Zhou M, Li Z, Wang C, Lu
Y, Li Y and Wang J: Inhibition of Cyclin D1 expression in human
glioblastoma cells is associated with increased temozolomide
chemosensitivity. Cell Physiol Biochem. 51:2496–2508.
2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Im CN: Past, present, and emerging roles
of mitochondrial heat shock protein TRAP1 in the metabolism and
regulation of cancer stem cells. Cell Stress Chaperones.
21:553–562. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Marchenko ND: Mitochondrial death
functions of p53. Mol Cell Oncol. 1(e955995)2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Chipuk JE, Kuwana T, Bouchier-Hayes L,
Droin NM, Newmeyer DD, Schuler M and Green DR: Direct activation of
Bax by p53 mediates mitochondrial membrane permeabilization and
apoptosis. Science. 303:1010–1014. 2004.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kulek AR, Anzell A, Wider JM, Sanderson TH
and Przyklenk K: Mitochondrial quality control: Role in cardiac
models of lethal ischemia-reperfusion injury. Cells.
9(214)2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Baqri RM, Pietron AV, Gokhale RH, Turner
BA, Kaguni LS, Shingleton AW, Kunes S and Miller KE: Mitochondrial
chaperone TRAP1 activates the mitochondrial UPR and extends
healthspan in Drosophila. Mech Ageing Dev. 141-142:35–45.
2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Takemoto K, Miyata S, Takamura H, Katayama
T and Tohyama M: Mitochondrial TRAP1 regulates the unfolded protein
response in the endoplasmic reticulum. Neurochem Int. 58:880–887.
2011.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhang L, Wang X, Cueto R, Effi C, Zhang Y,
Tan H, Qin X, Ji Y, Yang X and Wang H: Biochemical basis and
metabolic interplay of redox regulation. Redox Biol.
26(101284)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Perillo B, Di Donato M, Pezone A, Di Zazzo
E, Giovannelli P, Galasso G, Castoria G and Migliaccio A: ROS in
cancer therapy: The bright side of the moon. Exp Mol Med.
52:192–203. 2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Marenco-Hillembrand L, Wijesekera O,
Suarez-Meade P, Mampre D, Jackson C, Peterson J, Trifiletti D,
Hammack J, Ortiz K, Lesser E, et al: Trends in glioblastoma:
Outcomes over time and type of intervention: A systematic evidence
based analysis. J Neurooncol. 147:297–307. 2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Weller M, Le Rhun E, Preusser M, Tonn JC
and Roth P: How we treat glioblastoma. ESMO Open. 4 (Suppl
2)(e000520)2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Pietrafesa M, Maddalena F, Possidente L,
Condelli V, Zoppoli P, Li Bergolis V, Rodriquenz MG, Aieta M, Vita
G, Esposito F and Landriscina M: Gene copy number and
Post-transductional mechanisms regulate TRAP1 expression in human
colorectal carcinomas. Int J Mol Sci. 21(145)2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Lettini G, Maddalena F, Sisinni L,
Condelli V, Matassa DS, Costi MP, Simoni D, Esposito F and
Landriscina M: TRAP1: A viable therapeutic target for future cancer
treatments? Expert Opin Ther Targets. 21:805–815. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Hua G and Zhang Q: Heat shock protein 75
(TRAP1) antagonizes reactive oxygen species generation and protects
cells from granzyme M-mediated apoptosis. J Biol Chem.
282:20553–20560. 2007.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhang P, Lu Y, Yu D, Zhang D and Hu W:
TRAP1 provides protection against myocardial ischemia-reperfusion
injury by ameliorating mitochondrial dysfunction. Cell Physiol
Biochem. 36:2072–2082. 2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Kazlauskaite A, Kondapalli C, Gourlay R,
Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M
and Muqit MM: Parkin is activated by PINK1-dependent
phosphorylation of ubiquitin at Ser65. Biochem J. 460:127–139.
2014.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Goiran T, Duplan E, Rouland L, El Manaa W,
Lauritzen I, Dunys J, You H, Checler F and Alves da Costa C:
Nuclear p53-mediated repression of autophagy involves PINK1
transcriptional down-regulation. Cell Death Differ. 25:873–884.
2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Lin Q, Li S, Jiang N, Shao X, Zhang M, Jin
H, Zhang Z, Shen J, Zhou Y, Zhou W, et al: PINK1-parkin pathway of
mitophagy protects against contrast-induced acute kidney injury via
decreasing mitochondrial ROS and NLRP3 inflammasome activation.
Redox Biol. 26(101254)2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Singh A, Kukreti R, Saso L and Kukreti S:
Oxidative stress: A key modulator in neurodegenerative diseases.
Molecules. 24(1583)2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Ozben T: Oxidative stress and apoptosis:
Impact on cancer therapy. J Pharm Sci. 96:2181–2196.
2007.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Simon HU, Haj-Yehia A and Levi-Schaffer F:
Role of reactive oxygen species (ROS) in apoptosis induction.
Apoptosis. 5:415–418. 2000.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Poetsch AR: The genomics of oxidative DNA
damage, repair, and resulting mutagenesis. Comput Struct Biotechnol
J. 18:207–219. 2020.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Shan F, Shao Z and Jiang S: Erlotinib
induces the human non-small-cell lung cancer cells apoptosis via
activating ROS-dependent JNK pathways. Cancer Med. 5:3166–3175.
2016.PubMed/NCBI View Article : Google Scholar
|