Introduction
Cardiac fibrosis serves a key role in various forms
of chronic heart disease, resulting in reduced tissue compliance
and impaired heart function (1).
At present, there is no specific therapy to treat patients with
cardiac fibrosis. As closely associated to cardiac fibrosis and
heart remodeling, hyperhomocysteinemia (HHCY) is generally known as
a risk factor of cardiovascular diseases (2). Previous tudies have shown that HHCY
can lead to myocardial interstitial and perivascular fibrosis and
accelerate the progress of cardiac remodeling (2,3).
However, although the unfavorable effects of HHCY on cardiovascular
disease are well documented, the underlying mechanism of HHCY
leading to cardiac fibrosis remains unknown.
As one of the essential trace minerals, selenium
(Se) has been demonstrated to be crucial for cardiovascular health
(4). Se deficiency is associated
with a significantly increased risk of cardiovascular morbidity and
mortality (5). Previous studies
have confirmed the antioxidant properties of Se, and it has been
suggested that Se protection against inflammation or oxidative
stress is mainly exerted through Se-dependent glutathione
peroxidases and other selenoproteins (6,7).
Inflammation response is widely involved in most diseases, and to
some extent, is a normal process of body defense against certain
stimuli. However, excessive inflammation may lead to cell damage
and even tissue necrosis (8). HHCY
has been reported to serve an important role in pro-inflammatory
processes in numerous diseases, including intestinal inflammation
and aortic adventitial inflammation (9,10),
although the underlying mechanisms remain unclear. The present
study aimed therefore to determine whether Se could exert an
anti-inflammatory protection against HHCY-induced cardiac fibrosis
in mouse cardiac fibroblasts (CFs) and the potential underlying
mechanisms.
Recent studies reported that some non-coding RNAs
show great potential in the regulation of cardiac fibrosis
(11-13).
Long non-coding RNAs (lncRNAs) are a type of none-coding RNA of
>200 nucleotides in length, which participate in various
biological behavioral processes, including the onset and
development of some cardiac diseases (12-16).
The lncRNA scaffold attachment factor B interacting lncRNA (SAIL)
has been shown to improve cardiac fibrosis after myocardial
infarction by regulating the transcription of fibrosis-related
genes, such as α-SMA, collagen I and Ⅲ (12,14).
The lncRNA maternally expressed 3 (MEG3) has been reported to
regulate cardiomyocyte proliferation and apoptosis (15,16).
Inhibiting the expression of MEG3 can reverse hypoxia-induced
growth inhibition of heart progenitor cells (15). In addition, a previous study from
Wu et al (17) demonstrated
that MEG3 knockdown in cardiomyocytes can reduce cardiomyocyte
apoptosis and is associated with an improvement of heart function
post-myocardial infarction. Furthermore, Gong et al
(18) attributed the beneficial
effects of MEG3 silencing on suppressing cardiomyocyte apoptosis to
the increased expression of microRNA-183, which mediates the
inhibition of p27, by activating PI3K/AKT/FOXO3a signaling pathway
in hypoxic H9C2 cells. In addition, a previous study demonstrated
the protection effects of MEG3 inhibition against endoplasmic
reticulum stress-mediated cardiac apoptosis by targeting p53
protein (16). However, although
MEG3 was reported to be mainly enriched in CFs, study focusing on
the effects of MEG3 on CFs is rare. The present study aimed
therefore to determine the effect of MEG3 on HHCY-induced fibrosis
in CFs and to explore the possible links between Se and MEG3.
Materials and methods
Isolation and culture of mice CFs
The animal experimental protocols were approved by
the Animal Care and Use Committee of Renmin Hospital of Wuhan
University (approval no. WDRM-20200608). A total of 30 male C57BL/6
mice (age, 8-10 weeks; weight, 25-29 g) were purchased from the
China Three Gorges University (approval no. SCXK 2017-0012;
Yichang, China). The animals were maintained in
specific-pathogen-free and environmentally controlled isolation
conditions with free access to food and water (temperature,
20-25˚C; humidity, 45-55%; 12-h light/dark cycle) for 1 week prior
to the beginning of the study. Before collection of cardiac
tissues, mice were sacrificed with pentobarbital sodium (180 mg/kg)
injected intraperitoneally. Subsequently, hearts were removed
quickly from the chest and washed three times in PBS containing 1%
penicillin-streptomycin solution (100X) (19). Excess tissues were removed and the
majority of the left ventricle was collected for further shearing
into 1 mm3 pieces with microsurgical scissors. Once
broken tissue was digested by 0.05% trypsin for 3 min at 37˚C,
before subsequent digestion three times at 37˚C (~3 min each time)
was performed in DMEM/F12 medium (Gibco; Thermo Fisher Scientific,
Inc.) containing 0.018% collagenase II (cat. no. 2275; Biofroxx;
neoFroxx GmbH). After placing at room temperature for 1 min, the
supernatant was collected and neutralized in DMEM/F12 medium
containing 10% FBS (TICO Europe) after each digestion. Neutralized
supernatant was centrifuged at 1,000 x g for 9 min at 4˚C to
collect the cell pellet, which was further resuspended in DMEM/F12
medium containing 10% FBS and seeded in a six-well plate. CFs were
obtained after 1 h of adherence whereas the supernatant cells that
had not adhered were discarded. CFs were cultured in DMEM/F12
medium containing 10% FBS at 37˚C for 24 h. CFs at passage two were
used for subsequent experiments and were randomly divided into six
groups as follows: i) Control group (Ctrl); ii) Se group, only
treated with 100 nM sodium selenite (Sigma-Aldrich; Merck KGaA;
cat. no. S5261); iii) HCY (200 µM)-stimulated group; iv) HCY + Se
group (co-treated with HCY and Se); v) HCY + adenovirus (Ad)-shMEG3
group (HCY + Ad-shMEG3); and vi) HCY + Ad-scramble group (HCY +
Ad-scr). CFs in six-well plates were transfected with MOI of 100
for 12 h at 37˚C before subsequent drug treatment. The CFs were
treated with Se and HCY at the same time at 37˚C for 24 h.
Adenoviruses (constructed by GV119 plasmid vector) expressing short
hairpin RNA (shRNA) against MEG3 (5'-ACCCTCCTGGATTAGGCCAAA-3') and
negative control shRNA (5'-TTCTCCGAACGTGTCACGT-3') were generated
by Shanghai Genechem Co., Ltd.
Cell proliferation assessment
Cell Counting Kit-8 (CCK-8; Dojindo Molecular
Technologies, Inc.; cat. no. CK04) was used to evaluate CF
proliferation. Briefly, CFs were seeded in a 96-well plate at the
density of 5,000 cells/well and were treated with 100 nM Se and 200
µM HCY at 37˚C for 24 h. Subsequently, 10 µl CCK-8 reagent was
added to each well, and the cells were incubated at 37˚C for 90
min. The absorbance was read at a wavelength of 450 nm using a
microplate spectrophotometer.
Detection of reactive oxygen species
(ROS) production
Dihydroethidium (DHE; Beyotime Institute of
Biotechnology; cat. no. S0063) staining was used to detect the
cellular ROS production. DHE, after entering living cells, is
dehydrogenated by intracellular superoxide anions to produce
ethidium, which then binds to RNA or DNA to produce red
fluorescence. The stronger the red fluorescence, the higher levels
of intracellular superoxide production. In the present study, CFs
were first seeded into 24-well plates at a density of
1x105 cells/well. DHE (10 µM) was then added to CFs and
incubated at 37˚C for 30 min before treated being with 100 nM Se
and 200 µM HCY at 37˚C for 24 h. Cells were washed with PBS and the
fluorescence was observed under a fluorescence microscope using
x100 magnification.
ELISA assays
Cell culture supernatant from six-well plates in
each group was collected and the levels of tumor necrosis factor-α
(TNF-α: ELK Biotechnology, Co., Ltd.; cat. no. ELK1395) and
interleukin-1β (IL-1β; ELK Biotechnology, Co., Ltd.; cat. no.
ELK1271) were evaluated using ELISA kits according to the
manufacturer's protocols.
SOD and MDA detection
First, CFs in six-well plate were lysed by RIPA
lysis buffer (Beyotime Institute of Biotechnology; cat. no. P0013B)
at 4˚C. The lysate of each group was then collected, followed by
centrifugation at 2,000 x g at 4˚C for 10 min. SOD kit (cat. no.
A001-3; Nanjing Jiancheng Bioengineering Institute) and MDA kit
(cat. no. A003-1; Nanjing Jiancheng Bioengineering Institute) were
used to evaluate the levels of SOD and MDA, respectively, according
to the manufacturer's instruction.
Reverse transcription quantitative
(RT-q)PCR
Total RNA was extracted from CFs using RNAiso Plus
reagent (Takara Bio, Inc.; cat. no. 9108) and dissolved into
diethyl pyrocarbonate-treated water. After testing the
concentration of RNA, SweScript RT First Strand® cDNA
Synthesis Kit (Wuhan Servicebio Technology Co., Ltd.; cat. no.
G3330-50) was used to reverse transcribe RNA into cDNA according to
the manufacturer's instructions. The following thermocycling
conditions were used for PCR: Initial denaturation at 95˚C for 30
sec, followed by 40 cycles of 95˚C for 15 sec, 60˚C for 10 sec and
72˚C for 30 sec. The reactions were conducted in an ABI VIIa7
system (Applied Biosystems; Thermo Fisher Scientific, Inc.) using a
2x SYBR Green qPCR Master Mix (Low ROX; Wuhan Servicebio Technology
Co., Ltd.; cat. no. G3320-05). The sequences of the primers were as
follows: MEG3 forward, 3'-GCTCATCTTATTCTGGGCACCT-5' and reverse,
3'-TCGTGGACATTCCTCTTCCG-5'; and GAPDH forward,
3'-CCTCGTCCCGTAGACAAAATG-5' and reverse,
3'-TGAGGTCAATGAAGGGGTCGT-5'. The 2-ΔΔCq method was used
to analyze the result of qPCR (20).
Western blotting
Total protein was extracted from CFs lysed by RIPA
lysis buffer (Beyotime Institute of Biotechnology; cat. no. P0013B)
at 4˚C. Protein concentration was determined using a bicinchoninic
acid protein assay kit (Beyotime Institute of Biotechnology; cat.
no. P0012S). Proteins were separated by 10% SDS-PAGE (25 µg per
lane) and were transferred onto PVDF membranes. Membranes were
blocked in 5% non-fat milk at room temperature for 2 h, followed by
incubation with primary antibodies at 4˚C overnight. The next day,
membranes were washed three times with TBST (20 mM Tris, 137 mM
NaCl, 0.1% Tween) and were incubated with secondary antibodies for
1 h at room temperature. Membranes were washed three times with
TBST and protein bands were visualized using enhanced
chemiluminescence reagent (Beyotime Institute of Biotechnology;
cat. no. P0018FM). The relative protein expression levels were
normalized to endogenous control GAPDH using AlphaEaseFC software
(FluorChem8900; Alpha Innotech Corporation; ProteinSimple). The
present study used primary antibodies against GAPDH (Abcam; cat.
no. ab37168; 1:10,000), collagen I (Abcam; cat. no. ab34710;
1:1,000), collagen Ⅲ (Abcam; cat. no. ab7778; 1:1,000), α-SMA
(Abcam; cat. no. ab32575; 1:1,000), JAK2 (Abcam; cat. no. ab108596;
1:1,000), phosphorylated (p)-JAK2 (Cell Signaling Technology, Inc.;
cat. no. 3230; 1:1,000), STAT3 (Abcam; cat. no. ab68153; 1:1,000)
and p-STAT3 (Abcam; cat. no. ab76315; 1:1,000). The secondary
antibody used in the present study was the horseradish
peroxidase-conjugated goat anti rabbit antibody (ASPEN; cat. no.
AS1107; 1:10,000).
Statistical analysis
Statistical analysis was performed using SPSS
software (version 19.0; IBM Corp). All data were presented as the
means ± standard deviation. Comparisons between two groups were
evaluated using unpaired two-tailed Student's t-test. Multiple
comparisons were performed using one-way ANOVA followed by
Bonferroni's post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Se alleviates fibrosis induced by
HCY
The occurrence of cardiac fibrosis is closely
related to the activation and proliferation of cardiac fibroblasts.
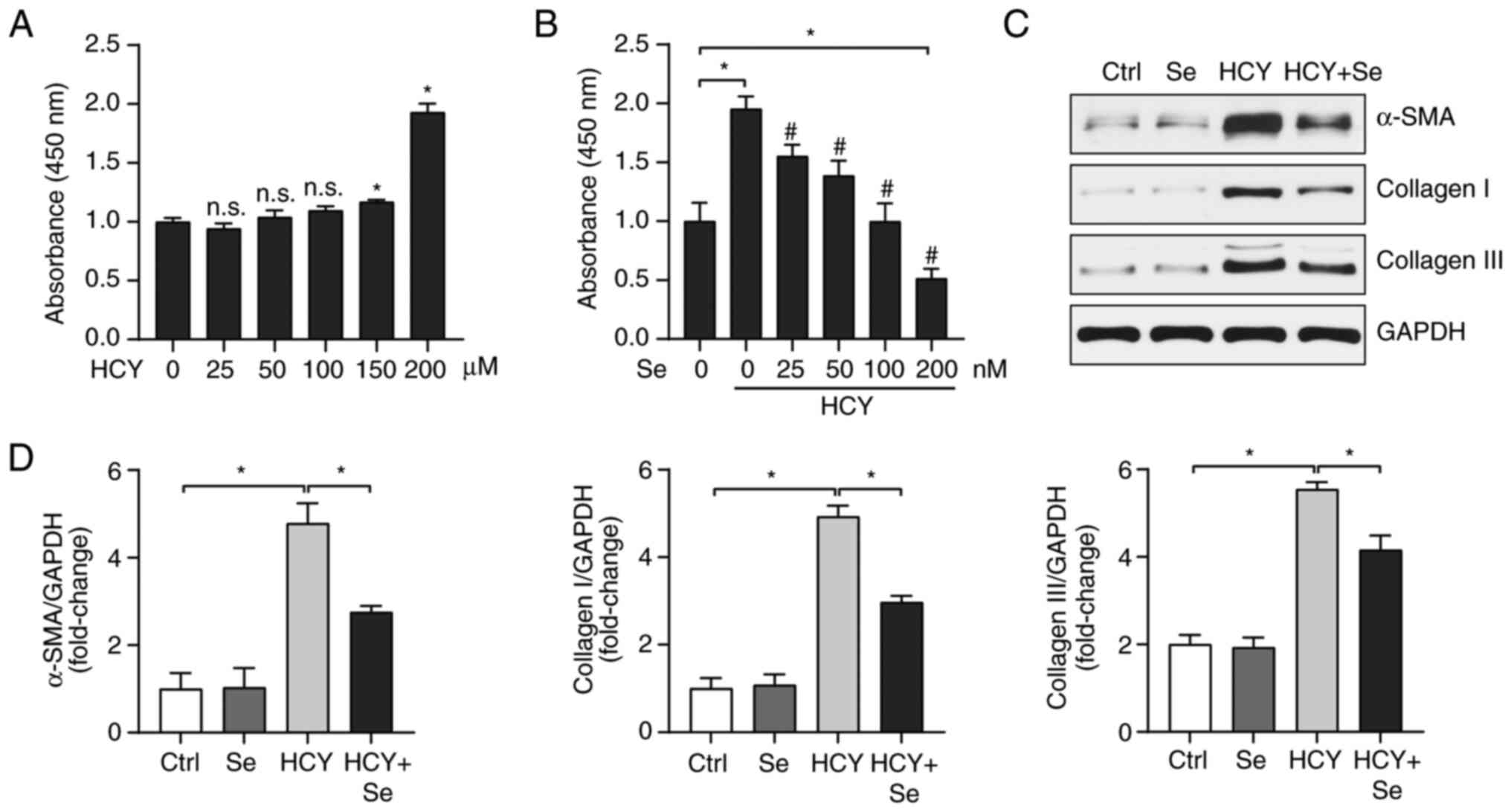
As presented in Fig. 1A, when HCY
concentration was <200 µM, HCY ability to stimulate the
proliferation of CFs was null, therefore the 200 µM concentration
was selected for subsequent experiment. To clarify the effect of Se
on cardiac fibrosis, we first examined the effect of Se on CF
proliferation. Different concentrations of Se were used and the
results demonstrated that 100 nM Se had a significant inhibitory
effect on CF proliferation following treatment with HCY, and 200 nM
Se resulted in significant cytotoxicity (Fig. 1B). Furthermore, 100 nM Se showed a
significant inhibitory effect on the expression of the fibrosis
related proteins α-SMA, collagen I and Ⅲ following treatment with
HCY (Fig. 1C). These results
demonstrate that Se may effectively improve HCY-induced CF
fibrosis.
Se downregulates MEG3 and MEG3
knockdown decreases HCY-induced fibrosis
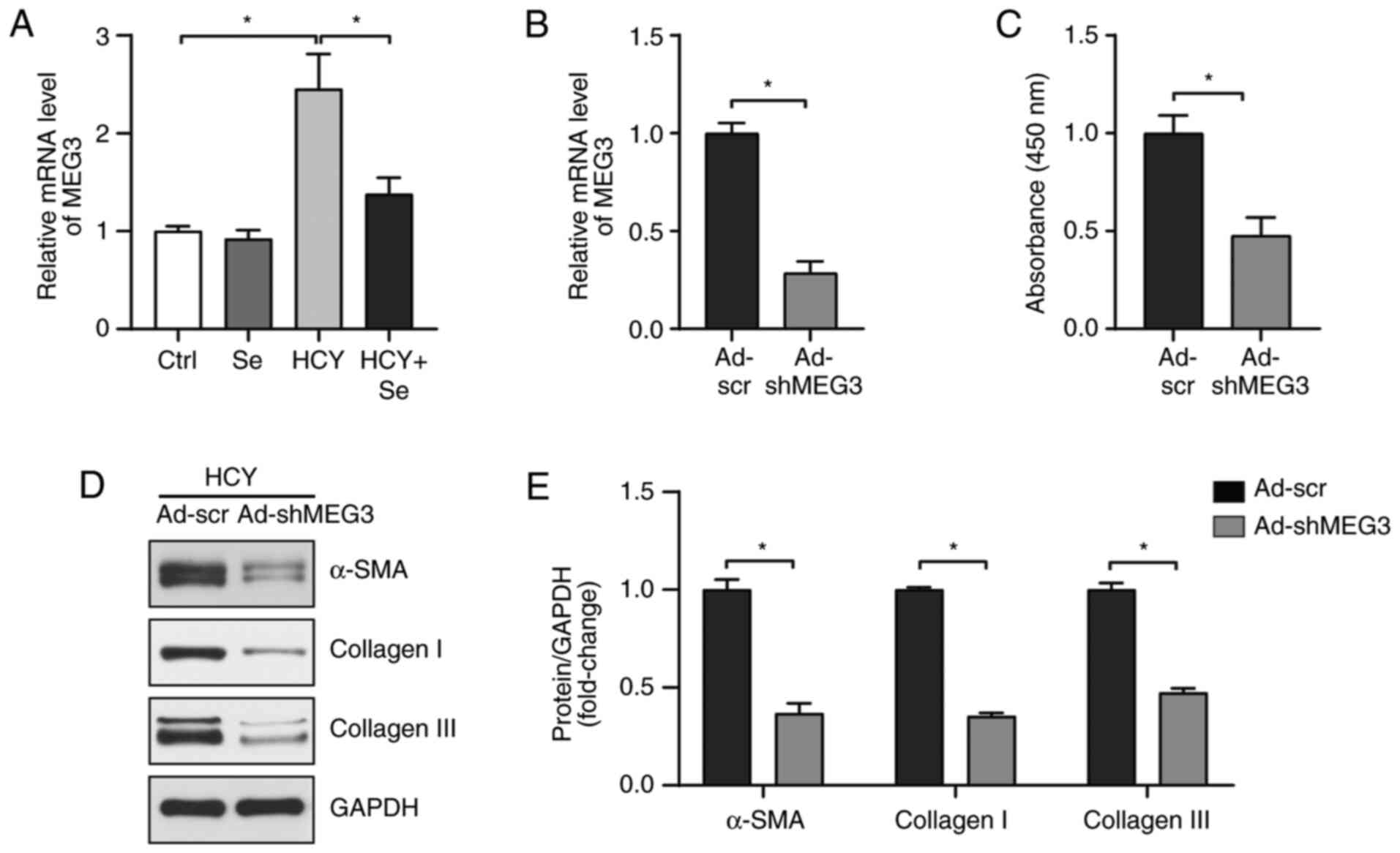
Since MEG3 has been reported to serve a role in
cardiac fibrosis (21), the
expression of MEG3 was determined in different experimental groups.
As presented in Fig. 2A, HCY
elevated the expression level of MEG3, whereas Se treatment
reversed this increase. To verify that the regulation of Se on
fibrosis was MEG3-independent, an MEG3-knockdown adenovirus
(Ad-shMEG3) was used to infect CFs (Fig. 2B) and the impact of MEG3 knockdown
on HCY-induced CF fibrosis was explored. Consistent with the effect
of Se, MEG3 knockdown significantly inhibited the increased
proliferation of CFs induced by HCY (Fig. 2C). In addition, the expression of
α-SMA, collagen I and III was significantly decreased in CFs
infected by Ad-shMEG3 compared with the Ad-scramble group (Fig. 2D and E). These results indicated that Se could
alleviate CF fibrosis through partly downregulating MEG3.
Se or MEG3 knockdown decreases
HCY-induced increase in oxidative stress
Since inflammation or oxidative stress are widely
involved in progression of cardiac fibrosis (22-24),
we investigated the production of ROS in the experimental groups.
The results demonstrated that Se significantly inhibited the
accumulation of ROS in CFs following treatment with HCY (Fig. 3A). Furthermore, Se significantly
increased SOD activity and decreased MDA contents in HCY-treated
CFs (Fig. 3B and C). In addition, MEG knockdown had the
same anti-oxidative stress effect (Fig. 3D and F). These findings suggested that the
anti-oxidative effects of Se may be mediated by MEG3
regulation.
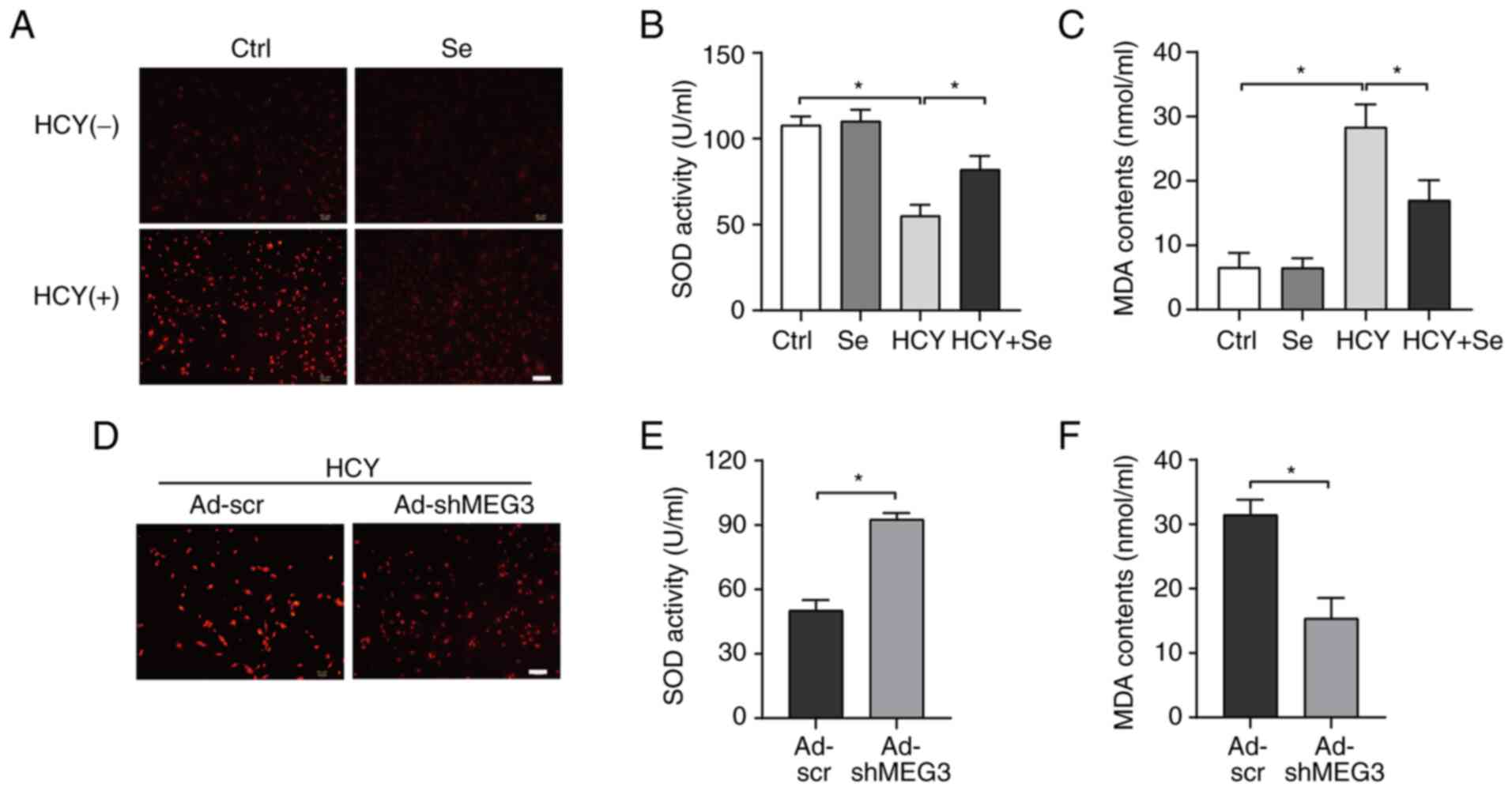 | Figure 3Se or MEG3 knockdown decreases
HCY-induced oxidative stress. (A) Representative images of
dihydroethidium staining in CFs stimulated with the indicated
treatments. Scale bar, 100 µm. (B and C) (B) SOD activity and (C)
MDA content in CFs stimulated with the indicated treatments (n=3).
(D) Representative images of dihydroethidium staining in CFs
infected with Ad-scr or Ad-shMEG3. Scale bar, 100 µm. (E and F) (E)
SOD activity and (F) MDA content in CFs infected with Ad-scr or
Ad-shMEG3 (n=3). *P<0.05. CFs, cardiac fibroblasts;
HCY, homocysteine; SOD, superoxide dismutase; MDA, malondialdehyde;
Se, selenium; Ctrl, control; Ad, adenovirus; sh, short hairpin;
MEG3, maternally expressed gene 3. |
Se or MEG3 knockdown decreases the
pro-inflammatory cytokines secretion
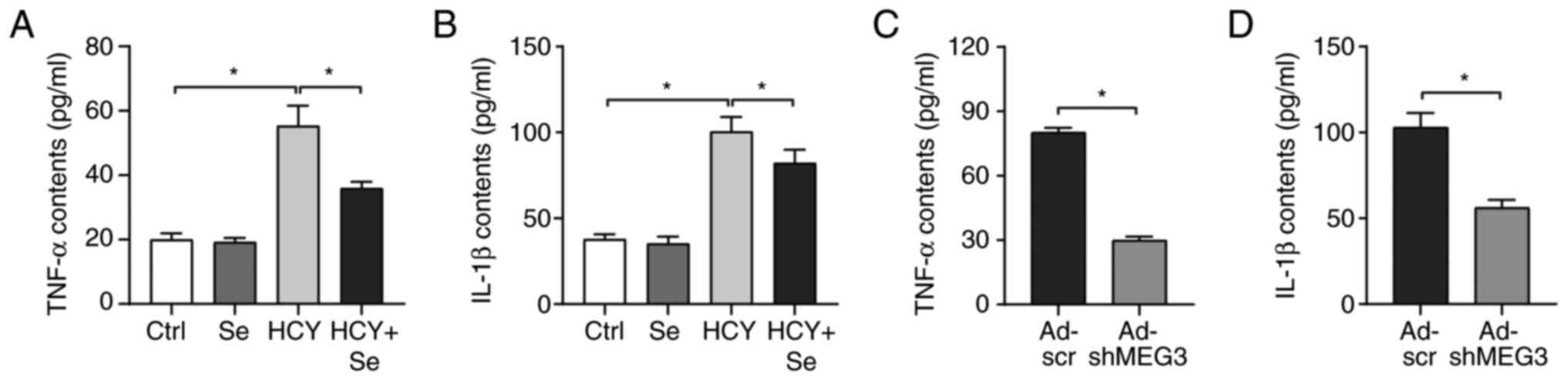
The effect of Se on inflammation was determined in
the present study. The results from ELISA demonstrated that
HCY-induced increase in TNF-α and IL-1β secretion was reversed
following treatment with Se compared with control group (Fig. 4A and B). Furthermore, MEG3 knockdown alleviated
HCY-induced inflammation in CFs (Fig.
4C and D). These results
suggested that Se may alleviate the inflammatory response caused by
HCY tby regulating the expression of MEG3.
Activation of JAK2/STAT3 is mitigated
following Se treatment or MEG3 knockdown
Increasing evidence suggests that JAK2/STAT3 pathway
is activated during the progress of fibrosis (25,26).
The phosphorylation levels of JAK2 and STAT3 were therefore
determined in the present study. As presented in Fig. 5, the activation of JAK2 and STAT3
was amplified in CFs following HCY treatment. Conversely, treatment
with Se or MEG3 knockdown significantly decreased p-JAK2 and
p-STAT3 expression. These findings suggested that the inhibitory
effect of Se on inflammation and oxidative stress may be mediated
by a negative regulation of JAK2/STAT3 pathway activation.
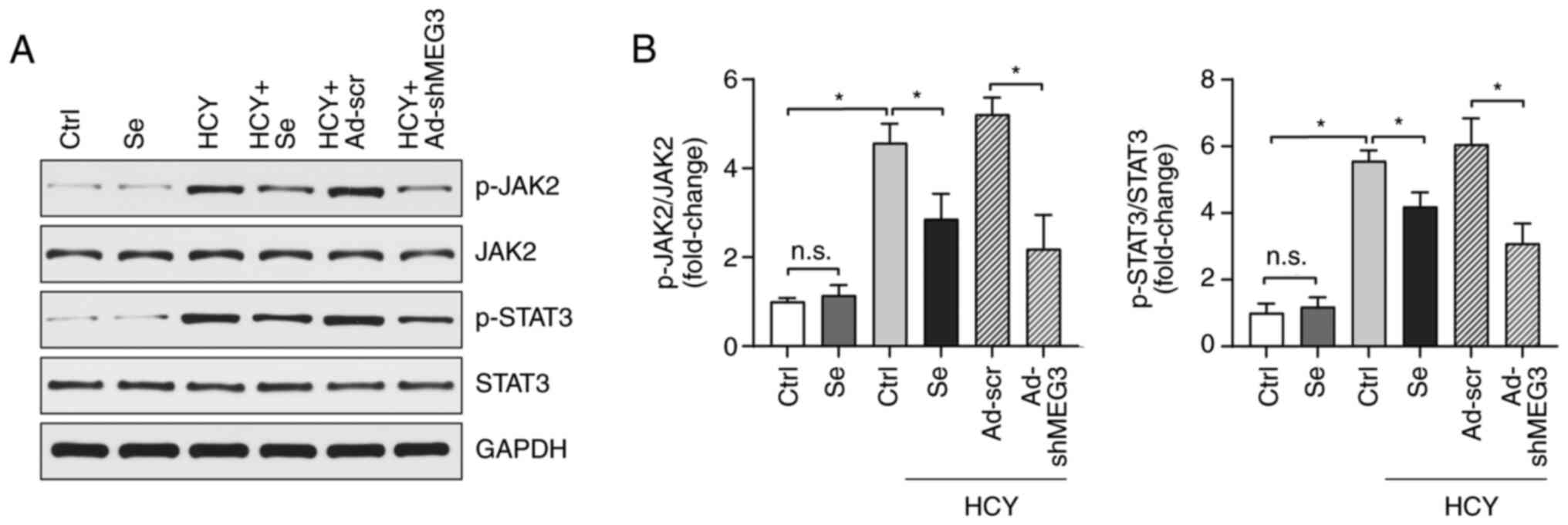 | Figure 5Activation of JAK2/STAT3 is mitigated
by Se intervention or MEG3 knockdown. (A) Expression of p-JAK2,
JAK2, p-STAT3 and STAT3 was evaluated by western blotting in CFs.
(B) Relative quantitative results of protein levels (n=3).
*P<0.05. HCY, homocysteine; p, phosphorylated; JAK2,
janus kinase 2; STAT3, signal transducer and activator of
transcription 3; CFs, cardiac fibroblasts; Se, selenium; Ctrl,
control; Ad, adenovirus; sh, short hairpin; MEG3, maternally
expressed gene 3; NS, non-significant. |
Discussion
Cardiovascular diseases represent the leading causes
of mortality worldwide (27).
Almost all cardiovascular diseases are related to the pathological
myocardial remodeling process that is characterized by tissue
fibrosis (28-30).
It is therefore crucial to clarify the specific molecular mechanism
of cardiac fibrosis and to determine some targets for the treatment
of cardiovascular diseases. HHCY is characterized by an abnormally
high level of homocysteine in the blood, which has been reported as
an independent risk factor for several cardiovascular diseases
(31,32). Previous studies have reported that
HHCY accelerates the pathological cardiac remodeling by promoting
myocardial interstitial and perivascular fibrosis (2,33).
During pathological cardiac remodeling and cardiac fibrosis, CFs
activate and secrete various matrix protein and proinflammatory
cytokines including α-SMA, collagen I and Ⅲ, which can promote CF
migration and proliferation (34).
In the present study, we demonstrated that the secretion of the
pro-inflammatory cytokines TNF-α and IL-1β and the expression of
the fibrosis related genes α-SMA, collagen I and Ⅲ were
significantly increased in HCY-treated CFs. Furthermore, HCY
significantly promoted the proliferation of CFs. These results
indicated that the effect of HHCY on cardiac fibrosis may be
mediated by the promotion of CF proliferation and inflammation
activation.
Se is an important trace element with good
antioxidant effect. Se is an essential micronutrient element found
in mammals and an important antioxidant of food origin (35,36).
Se exists in various selenoproteins and selenate in the form of
selenocysteine, which serves an essential role in various diseases
through inhibiting oxidative stress and inflammatory reaction
(37). Previous studies have
demonstrated that Se can protect cells against oxidative damage and
endoplasmic reticulum stress-induced apoptosis, and can serve an
important role in the reverse transport pathway during endoplasmic
reticulum-related protein degradation (38). In addition, increased inflammation
and oxidative stress are involved in the occurrence and development
of most cardiovascular diseases (4). Previous studies have reported that Se
alleviates the pathological progress of various cardiovascular
diseases, such as heart failure and atherosclerosis, by negatively
regulating inflammation and oxidative stress (39,40).
However, there are only a few studies about the effect of selenium
on cardiac fibrosis. The present study demonstrated that Se could
effectively alleviate HCY-induced cardiac fibrosis in CFs. In
addition, Se significantly inhibited the activation and
proliferation of CFs, and suppressed the secretion of
proinflammatory cytokines and ROS. Regarding the mechanism, Se
played an anti-fibrotic effect through downregulating of MEG3. MEG3
is a lncRNA that possesses multiple biological functions and plays
an important role in numerous diseases. Previous studies have
demonstrated that MEG3 plays essential roles in cardiovascular
diseases, such as inducing cardiomyocyte apoptosis, contributing to
myocardial ischemia-reperfusion injury and regulating cardiac
remodeling in cardiac hypertrophy (41-43).
In addition, it has been reported that knocking down MEG3
expression can prevent the induction of myocardial matrix
metalloproteinase-2, reduce myocardial fibrosis and improve
diastolic function following transverse aortic coarctation in mice
(21). The present study
demonstrated that MEG3 knockdown can reduce HCY-induced CF
inflammation, and that Se can effectively reduce HCY-induced
inflammation by regulating the expression of MEG3.
As the most conservative members of the JAK/STAT
pathway family, JAK2/STAT3 are closely related to the pathogenesis,
prevention and treatment of a various types of disease, especially
heart diseases such as myocardial hypertrophy, myocardial ischemia,
heart failure, ischemic preconditioning and ischemia-reperfusion
(44-46).
A previous study demonstrated that Se deficiency in cardiomyocytes
can lead to a decrease in the expression of potassium channels,
mitochondrial STAT3 activity and mitochondrial function, thus
promoting cardiomyocyte apoptosis (47). We therefore speculated that the
effect of Se on cardiac fibrosis may be mediated by the JAK2/STAT3
pathway. The present study showed that Se or MEG3 knockdown can
significantly inhibit the activation of JAK2/STAT3 signal pathway
induced by HCY.
JAK2/STAT3 signal pathway is one of the important
pathways of cell signal transduction, which plays an important role
in cell growth, activation, differentiation and apoptosis. The main
substrate of JAK kinase is a cytokine receptor, which binds to
ligand in the form of dimer or monomer. However, JAK kinase has
been known to activate STAT family members. Once STAT factors are
phosphorylated by JAKs, they become homodimers or heterodimers,
which are transferred to the nucleus, where they show activation or
inhibition of transcription. It has been reported that STAT1 and
STAT3 members can be activated by JAK2 in the absence of new
protein synthesis and hydrogen peroxide treatment (48). At present, some studies have shown
that collagen synthesis in cardiac fibroblasts can be stimulated by
regulating JAK/STAT pathway, which is closely related to the
pathological changes of cardiac fibrosis (49,50).
In addition, a previous study reported that hydrogen sulfide can
alleviate cardiac fibrosis in diabetic rats by downregulating
JAK/STAT pathway to inhibit oxidative stress and ER stress,
inflammatory response and apoptosis (51). The results from the present study
demonstrated that the pro-inflammatory and oxidative stress
responses of cardiac fibroblasts induced by HCY was activated,
which were inhibited by Se. These findings suggested that Se may
not only have and antioxidant effect, but may also regulate
JAK/STAT signal pathway. Since JAK2/STAT3 signal pathway can affect
oxidative stress and myocardial fibrosis, and because oxidative
stress leads to myocardial fibrosis and myocardial remodeling in
mice (23,24), we speculated that Se could inhibit
cardiomyocyte apoptosis and improve myocardial fibrosis by
inhibiting the activation of JAK2/STAT3 signal pathway.
In summary, the present study demonstrated that Se
can inhibit the expression of MEG3 and negatively regulate the
activation of JAK2/STAT3 pathway, thus effectively reducing
inflammation and oxidative stress caused by HCY. These results may
provide promising targets for the prevention and treatment of
cardiac fibrosis.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by the Hubei Province Health
and Family Planning Scientific Research Project (grant no.
WJ2015MA021).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WL and YL performed the experiments and participated
in drafting the manuscript. SC, JL and LT performed data analysis.
HX and CZ designed and supervised the studies. WL and HX confirmed
the authenticity of all the raw data. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
All animal experimental protocols were approved by
the Animal Care and Use Committee of Renmin Hospital of Wuhan
University (approval no. WDRM-20200608).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bacmeister L, Schwarzl M, Warnke S,
Stoffers B, Blankenberg S, Westermann D and Lindner D: Inflammation
and fibrosis in murine models of heart failure. Basic Res Cardiol.
114(19)2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Herrmann M, Taban-Shomal O, Hübner U, Böhm
M and Herrmann W: A review of homocysteine and heart failure. Eur J
Heart Fail. 8:571–576. 2006.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zhao Q, Song W, Huang J, Wang D and Xu C:
Metformin decreased myocardial fibrosis and apoptosis in
hyperhomocysteinemia-induced cardiac hypertrophy. Curr Res Transl
Med. 69(103270)2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Triposkiadis F, Xanthopoulos A and Butler
J: Cardiovascular aging and heart failure: JACC review topic of the
week. J Am Coll Cardiol. 74:804–813. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Schomburg L, Orho-Melander M, Struck J,
Bergmann A and Melander O: Selenoprotein-P deficiency predicts
cardiovascular disease and death. Nutrients.
11(1852)2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Huang JQ, Zhou JC, Wu YY, Ren FZ and Lei
XG: Role of glutathione peroxidase 1 in glucose and lipid
metabolism-related diseases. Free Radic Biol Med. 127:108–115.
2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Hariharan S and Dharmaraj S: Selenium and
selenoproteins: It's role in regulation of inflammation.
Inflammopharmacology. 28:667–695. 2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Liu X, Xia S, Zhang Z, Wu H and Lieberman
J: Channelling inflammation: Gasdermins in physiology and disease.
Nat Rev Drug Discov. 20:384–405. 2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Zhu S, Li J, Bing Y, Yan W, Zhu Y, Xia B
and Chen M: Diet-induced hyperhomocysteinaemia increases intestinal
inflammation in an animal model of colitis. J Crohns Colitis.
9:708–719. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Liu Z, Luo H, Zhang L, Huang Y, Liu B, Ma
K, Feng J, Xie J, Zheng J, Hu J, et al: Hyperhomocysteinemia
exaggerates adventitial inflammation and angiotensin II-induced
abdominal aortic aneurysm in mice. Circ Res. 111:1261–1273.
2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Li Y, Duan JZ, He Q and Wang CQ: miR-155
modulates high glucose-induced cardiac fibrosis via the Nrf2/HO-1
signaling pathway. Mol Med Rep. 22:4003–4016. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Luo S, Zhang M, Wu H, Ding X, Li D, Dong
X, Hu X, Su S, Shang W, Wu J, et al: SAIL: A new conserved
anti-fibrotic lncRNA in the heart. Basic Res Cardiol.
116(15)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Zhang F, Fu X, Kataoka M, Liu N, Wang Y,
Gao F, Liang T, Dong X, Pei J, Hu X, et al: Long noncoding RNA
Cfast regulates cardiac fibrosis. Mol Ther Nucleic Acids.
23:377–392. 2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Statello L, Guo CJ, Chen LL and Huarte M:
Gene regulation by long non-coding RNAs and its biological
functions. Nat Rev Mol Cell Biol. 22:96–118. 2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Su J, Fang M, Tian B, Luo J, Jin C, Wang
X, Ning Z and Li X: Atorvastatin protects cardiac progenitor cells
from hypoxia-induced cell growth inhibition via MEG3/miR-22/HMGB1
pathway. Acta Biochim Biophys Sin (Shanghai). 50:1257–1265.
2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li X, Zhao J, Geng J, Chen F, Wei Z, Liu
C, Zhang X, Li Q, Zhang J, Gao L, et al: Long non-coding RNA MEG3
knockdown attenuates endoplasmic reticulum stress-mediated
apoptosis by targeting p53 following myocardial infarction. J Cell
Mol Med. 23:8369–8380. 2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Wu H, Zhao ZA, Liu J, Hao K, Yu Y, Han X,
Li J, Wang Y, Lei W, Dong N, et al: Long noncoding RNA Meg3
regulates cardiomyocyte apoptosis in myocardial infarction. Gene
Ther. 25:511–523. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Gong L, Xu H, Chang H, Tong Y, Zhang T and
Guo G: Knockdown of long non-coding RNA MEG3 protects H9c2 cells
from hypoxia-induced injury by targeting microRNA-183. J Cell
Biochem. 119:1429–1440. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Qu C, Liu X, Ye T, Wang L, Liu S, Zhou X,
Wu G, Lin J, Shi S and Yang B: miR-216a exacerbates TGF-β-induced
myofibroblast transdifferentiation via PTEN/AKT signaling. Mol Med
Rep. 19:5345–5352. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Piccoli MT, Gupta SK, Viereck J,
Foinquinos A, Samolovac S, Kramer FL, Garg A, Remke J, Zimmer K,
Batkai S and Thum T: Inhibition of the cardiac fibroblast-enriched
lncRNA Meg3 prevents cardiac fibrosis and diastolic dysfunction.
Circ Res. 121:575–583. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Purnomo Y, Piccart Y, Coenen T, Prihadi JS
and Lijnen PJ: Oxidative stress and transforming growth
factor-β1-induced cardiac fibrosis. Cardiovasc Hematol Disord Drug
Targets. 13:165–172. 2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wu J, Xia S, Kalionis B, Wan W and Sun T:
The role of oxidative stress and inflammation in cardiovascular
aging. Biomed Res Int. 2014(615312)2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Tsutsui H, Kinugawa S and Matsushima S:
Mitochondrial oxidative stress and dysfunction in myocardial
remodelling. Cardiovasc Res. 81:449–456. 2009.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhang Y, Dees C, Beyer C, Lin NY, Distler
A, Zerr P, Palumbo K, Susok L, Kreuter A, Distler O, et al:
Inhibition of casein kinase II reduces TGFβ induced fibroblast
activation and ameliorates experimental fibrosis. Ann Rheum Dis.
74:936–943. 2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Milara J, Hernandez G, Ballester B, Morell
A, Roger I, Montero P, Escrivá J, Lloris JM, Molina-Molina M,
Morcillo E and Cortijo J: The JAK2 pathway is activated in
idiopathic pulmonary fibrosis. Respir Res. 19(24)2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Yusuf S, Joseph P, Rangarajan S, Islam S,
Mente A, Hystad P, Brauer M, Kutty VR, Gupta R, Wielgosz A, et al:
Modifiable risk factors, cardiovascular disease, and mortality in
155 722 individuals from 21 high-income, middle-income, and
low-income countries (PURE): A prospective cohort study. Lancet.
395:795–808. 2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
El-Baz FK, Aly HF and Abd-Alla HI: The
ameliorating effect of carotenoid rich fraction extracted from
Dunaliella salina microalga against inflammation-associated cardiac
dysfunction in obese rats. Toxicol Rep. 7:118–124. 2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Xu H, Shen Y, Liang C, Wang H, Huang J,
Xue P and Luo M: Inhibition of the mevalonate pathway improves
myocardial fibrosis. Exp Ther Med. 21(224)2021.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Hu F, Li M, Han F, Zhang Q, Zeng Y, Zhang
W and Cheng X: Role of TRPM7 in cardiac fibrosis: A potential
therapeutic target (review). Exp Ther Med. 21(173)2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Nasir K, Tsai M, Rosen BD, Fernandes V,
Bluemke DA, Folsom AR and Lima JA: Elevated homocysteine is
associated with reduced regional left ventricular function: The
multi-ethnic study of atherosclerosis. Circulation. 115:180–187.
2007.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Stampfer MJ, Malinow MR, Willett WC,
Newcomer LM, Upson B, Ullmann D, Tishler PV and Hennekens CH: A
prospective study of plasma homocyst(e)ine and risk of myocardial
infarction in US physicians. JAMA. 268:877–881. 1992.PubMed/NCBI
|
|
33
|
Zhao Q, Song W, Huang J, Wang D and Xu C:
Metformin decreased myocardial fibrosis and apoptosis in
hyperhomocysteinemia-induced cardiac hypertrophy. Curr Res Transl
Med. 69(103270)2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Perbellini F, Watson SA, Scigliano M,
Alayoubi S, Tkach S, Bardi I, Quaife N, Kane C, Dufton NP, Simon A,
et al: Investigation of cardiac fibroblasts using myocardial
slices. Cardiovasc Res. 114:77–89. 2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Moghadaszadeh B and Beggs AH:
Selenoproteins and their impact on human health through diverse
physiological pathways. Physiology (Bethesda). 21:307–315.
2006.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Papp LV, Lu J, Holmgren A and Khanna KK:
From selenium to selenoproteins: Synthesis, identity, and their
role in human health. Antioxid Redox Signal. 9:775–806.
2007.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Rayman MP: Selenium and human health.
Lancet. 379:1256–1268. 2012.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Gao Y, Feng HC, Walder K, Bolton K,
Sunderland T, Bishara N, Quick M, Kantham L and Collier GR:
Regulation of the selenoprotein SelS by glucose deprivation and
endoplasmic reticulum stress-SelS is a novel glucose-regulated
protein. FEBS Lett. 563:185–190. 2004.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Bomer N, Grote Beverborg N, Hoes MF,
Streng KW, Vermeer M, Dokter MM, IJmker J, Anker SD, Cleland JGF,
Hillege HL, et al: Selenium and outcome in heart failure. Eur J
Heart Fail. 22:1415–1423. 2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Liu H, Xu H and Huang K: Selenium in the
prevention of atherosclerosis and its underlying mechanisms.
Metallomics. 9:21–37. 2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Chen Y, Zhang Z, Zhu D, Zhao W and Li F:
Long non-coding RNA MEG3 serves as a ceRNA for microRNA-145 to
induce apoptosis of AC16 cardiomyocytes under high glucose
condition. Biosci Rep. 39(BSR20190444)2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Zou L, Ma X, Lin S, Wu B, Chen Y and Peng
C: Long noncoding RNA-MEG3 contributes to myocardial
ischemia-reperfusion injury through suppression of miR-7-5p
expression. Biosci Rep. 39(BSR20190210)2019.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Uchida S: Besides imprinting: Meg3
regulates cardiac remodeling in cardiac hypertrophy. Circ res.
121:486–487. 2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Suzuki S, Tanaka K and Suzuki N:
Ambivalent aspects of interleukin-6 in cerebral ischemia:
Inflammatory versus neurotrophic aspects. J Cereb Blood Flow Metab.
29:464–479. 2009.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Bujak M, Dobaczewski M, Chatila K, Mendoza
LH, Li N, Reddy A and Frangogiannis NG: Interleukin-1 receptor type
I signaling critically regulates infarct healing and cardiac
remodeling. Am J Pathol. 173:57–67. 2008.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Takahashi K, Fukushima S, Yamahara K,
Yashiro K, Shintani Y, Coppen SR, Salem HK, Brouilette SW, Yacoub
MH and Suzuki K: Modulated inflammation by injection of
high-mobility group box 1 recovers post-infarction chronically
failing heart. Circulation. 118 (Suppl 14):S106–S114.
2008.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Zhang C, Deng Y, Lei Y, Zhao J, Wei W and
Li Y: Effects of selenium on myocardial apoptosis by modifying the
activity of mitochondrial STAT3 and regulating potassium channel
expression. Exp Ther Med. 14:2201–2205. 2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Kucinski I, Dinan M, Kolahgar G and
Piddini E: Chronic activation of JNK JAK/STAT and oxidative stress
signalling causes the loser cell status. Nat Commun.
8(136)2017.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Magaye RR, Savira F, Hua Y, Xiong X, Huang
L, Reid C, Flynn B, Kaye D, Liew D and Wang BH: Exogenous
dihydrosphingosine 1 phosphate mediates collagen synthesis in
cardiac fibroblasts through JAK/STAT signalling and regulation of
TIMP1. Cell Signal. 72(109629)2020.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Su SA, Yang D, Wu Y, Xie Y, Zhu W, Cai Z,
Shen J, Fu Z, Wang Y, Jia L, et al: EphrinB2 regulates cardiac
fibrosis through modulating the interaction of Stat3 and
TGF-β/Smad3 signaling. Circ Res. 121:617–627. 2017.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Liu M, Li Y, Liang B, Li Z, Jiang Z, Chu C
and Yang J: Hydrogen sulfide attenuates myocardial fibrosis in
diabetic rats through the JAK/STAT signaling pathway. Int J Mol
Med. 41:1867–1876. 2018.PubMed/NCBI View Article : Google Scholar
|