Introduction
Cardiac hypertrophy is an adaptive response to
hemodynamic stress and is associated with impaired cardiac
function, including an increase in cardiomyocyte size, higher
sarcomere organization and enhanced protein synthesis for
natriuretic peptide A (ANP), natriuretic peptide B (BNP) and
β-myosin heavy chain (β-MHC), and higher sarcomere organization
(1,2). Pathological cardiac hypertrophy is an
independent risk factor for myocardial infarction, arrhythmia and
heart failure (HF) (3). Without
effective intervention, prolonged HF can lead to sudden death
(3). The underlying mechanisms
remain unclear. Therefore, in-depth studies are urgently
required.
Transcription factor EC (TFEC) is a basic
helix-loop-helix transcription factor that is a member of the MITF
family (4,5). TFEC is highly enriched in caudal
endothelial cells when hematopoietic stem cells (HSCs) colonize the
caudal hematopoietic tissue (6). A
previous study demonstrated that TFEC facilitates HSC-derived
hematopoiesis in a non-cell-autonomous fashion (6). TFEC is also expressed in bone
marrow-derived macrophages following stimulation with the Th2
cytokines interleukin (IL)-4 and IL-13 or lipopolysaccharide
(7). However, TFEC is suggested to
be not ubiquitously expressed (8).
For example, TFEC RNA is not found in several cell types, including
fibroblasts, myoblasts, chondrosarcoma cells and myeloma cells
(8). Whether TFEC is expressed
during cardiac hypertrophy and involved in the progression of
cardiac hypertrophy has never been explored.
The present study aimed to determine whether TFEC is
dysregulated in mouse models of cardiac hypertrophy. In addition,
this study aimed to explore the mechanism by which TFEC might be
involved in the development of cardiac hypertrophy.
Materials and methods
Animal models
Eight-week-old male adult C57BL/6 mice weighing
20-25 g (n=45) were purchased from Charles River Laboratories. The
mice were housed under a 12-h light/dark cycle and pathogen-free
conditions and were given free access to standard mouse chow and
tap water. This study was approved by the research ethics committee
of Weapon Industry 521 Hospital (approval no. WI-J2018923A).
Pressure-overload cardiac
hypertrophy
The pressure-overload model of cardiac hypertrophy
was established by transverse aortic constriction (TAC) (9). Adult mice (C57BL/6, 8 weeks old),
weighing 20-25 g, were anaesthetized with pentobarbital sodium (50
mg/kg; intraperitoneal injection; n=12) (10). To confirm anesthesia, the absence
of withdrawal reflex in response to a tail pinch was observed. The
mice were placed in the supine position. Following successful
endotracheal intubation, the cannula was connected to a
volume-cycled rodent ventilator. To identify the thoracic aorta,
the chest was opened. Then, a 5-0 silk suture was placed below the
transverse aorta and tied around a 26-gauge blunt needle, which was
subsequently removed. The sham group underwent the aforementioned
procedure, but did not receive a 5-0 silk suture. The chest was
closed and the animals were maintained under ventilation until
autonomic breathing successfully recovered. After 4 weeks,
surviving animals (all but one mouse, which died during TAC
surgery, survived) were analyzed by echocardiography and killed by
cervical dislocation. The hearts were quickly harvested and
incubated in 4% paraformaldehyde at room temperature for 20 min or
dissected into the left ventricle (LV), right ventricle and
ventricular septum, which were rapidly frozen in liquid nitrogen
and stored at -80˚C for subsequent analyses.
The mice that survived 24 h after operation (n=10
mice per group) were observed for 4 weeks. The survival rate of 10
C57BL/6 mice in sham group was 100%. Only one of the 10 C57BL/6
animals undergoing TAC operation died (survival rate for TAC, 90%).
The heart and body weight (HW/BW) ratio was compared between the
TAC group and sham group.
Angiotensin II (AngII)
infusion-induced animal model of cardiac hypertrophy
A mouse model (C57BL/6, 8 weeks old, weighing 20-25
g) of AngII (1.46 mg/kg/day for 28 days) infusion-induced cardiac
hypertrophy was established (11).
Control group mice (C57BL/6; 8 weeks old; weight 20-25 g) were
injected daily via intraperitoneal administration with PBS (100 µl
for 28 days). Mice were anaesthetized via intraperitoneal
administration of sodium pentobarbital (50 mg/kg) (10). To confirm the anesthesia, the
absence of a reflex in response to a foot squeeze was observed.
Then, an AngII mini-osmotic pump (Alzet model 2002; Cupertino) was
used. The body temperature was maintained at 37±0.5˚C
during the surgery. All mice were euthanized by resection of the
heart and decapitation under deep isoflurane anesthesia (5%)
(12). Animal hearts were quickly
harvested and incubated in 4% paraformaldehyde at room temperature
for 20 min or dissected into the LV, right ventricle and
ventricular septum, which were rapidly frozen in liquid nitrogen
and stored at -80˚C for subsequent analyses. The ratio
of HW/BW was compared between the AngII treatment group and
controls.
For the control group, all mice (n=10) survived
during the experiment, equating to a 100% survival rate in the 10
C57BL/6 mice of the control group. Only one of the 10 C57BL/6
animals undergoing AngII treatment died (survival rate for AngII
treated mice, 90%).
Cell culture
One-day-old (n=4 in the same litter for each
experiment) mice were born from the wide type 8-10 week-old female
adult C57BL/6 mice (Charles River Laboratories). Pups were
decapitated using sterile scissors (straight) without anaesthesia
and the chest was open along the sternum to allow access to the
chest cavity and the heart as previously described (13). Briefly, murine hearts were
harvested, placed in ice-cold Hanks' medium and cut into pieces.
The tissues were digested with trypsin and type II collagenase at
37˚C for 15 min. The cells were passed through a cell
strainer (200 mesh), centrifuged at 1,000 x g for 10 min at room
temperature, seeded in Petri dishes and incubated for 2 h at
37˚C. The supernatants containing cardiomyocytes were
collected and mixed in DMEM (HyClone; Cytiva). Before treatment
with AngII (1 µmol/l) for 24 h, the cells were cultured in DMEM
containing 1% FBS for 12 h at 37˚C.
Measurement of cell surface area
Primary cardiomyocytes were isolated as described
above and previously (13) were
fixed with 4% paraformaldehyde for 20 min at room temperature,
permeabilized with 0.4% Triton X-100 (Beijing Solarbio Science
& Technology Co., Ltd.) for 1 h at room temperature and then
blocked with 5% goat serum (Beijing Solarbio Science &
Technology Co., Ltd.) at 37˚C for 1 h. Then, cells were
incubated with anti-sarcomeric alpha actin antibody (1:250; cat.
no. ab137346; Abcam) at 4˚C overnight and subsequently
with a DyLight 594-conjugated goat anti-mouse antibody (cat. no.
A23610; AmyJet Scientific, Inc.) at room temperature for 1 h. Cells
were subsequently observed under a fluorescence microscope
(magnification, x20).
Echocardiography study
Mice were anesthetized with isoflurane inhalation at
the concentration of 2.5% for anesthetic induction and at 1% for
anesthetic maintenance (14-16).
Echocardiography was performed to evaluate the function of the LV
using a MyLab™ Seven VET system (Esaote SpA) equipped with a 10-MHz
probe. Parasternal short axis images at the mid-papillary muscle
level were recorded in M-mode. The LV dimensions, including LV
end-systolic diameter (LVEDs), LV end-diastolic diameter (LVEDd),
posterior wall thickness, left ventricular ejection fractions
(LVEF), maximum change in left ventricular pressure over time
(dp/dtmax, mmHg/s) and minimum change in left ventricular pressure
over time (dp/dtmin, mmHg/s) were measured.
Histopathology staining
The mouse hearts were fixed in 4% formalin for 20
min at room temperature. The heart sections were embedded in
paraffin and sectioned to a thickness of 5 µm for staining with
hematoxylin and eosin (H&E). Slides were stained with
hematoxylin (Beijing Solarbio Science & Technology Co., Ltd.)
for 5 min and washed with tap water for 2 min at room temperature.
The sides were differentiated with 1% alcohol for 2 min and washed
with tap water for 5 min. Subsequently, the slides were stained
with eosin at room temperature for 2 min (Beijing Solarbio Science
& Technology Co., Ltd.). The slides were washed with tap water
for 2 min and 70% alcohol twice, after which the slides were sealed
and observed under a fluorescence microscope (magnification, x20;
BX43; Olympus Corporation). For wheat germ agglutinin (WGA)
staining of cardiac muscle cell membrane, the tissue sections were
stained with 1.0 mg/ml Alexa FluorVR 488-conjugated WGA (Molecular
Probes) at room temperature for 20 min to visualize the size of
cardiomyocytes using a fluorescence microscope (magnification, x20;
IX71; Olympus Corporation).
Construction of adenovirus
vectors
Adenovirus vectors carrying RNA for overexpression
of TFEC (rAd-TFEC) or a short hairpin (sh) RNA targeting TFEC
(rAd-sh-TFEC) or a negative control (rAd-NC) were constructed by
Shanghai GeneChem Co., Ltd. C57BL/6 mice were injected once with
vectors at a concentration of 1x109 U/ml (50 µl) through
the left ventricular chamber after 28 days of treatment with
AngII.
Reverse transcription quantitative
(RT-q) PCR
Total RNA was extracted from the heart tissues or
primary cardiomyocytes using TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) and reverse transcribed into cDNA using a
One Step PrimeScriptTM RT-PCR Kit (Takara Bio, Inc.)
according to the manufacturers' instructions. RT-qPCR analysis was
performed using LightCycler 480 SYBR Green 1 Master Mix (No.
04707516001; Roche Diagnostics) according to the manufacturers'
protocol. The relative mRNA expression was normalized to that of
GAPDH using the 2-∆∆Cq method (17). The sequences of the primers (Sangon
Biotech Co., Ltd.) were as follows: GAPDH forward,
5'-TCATCAACGGGAAGCCCATC-3', reverse, 5'-CTCGTGGTTCACACCCATCA-3';
ANP forward, 5'-ACCTGCTAGACCACCTGGAG-3', reverse,
5'-CCTTGGCTGTTATCTTCGGTACCGG-3'; BNP forward,
5'-GAGGTCACTCCTATCCTCTGG-3', reverse, 5'-GCCATTTCCTCCGACTTTTCTC-3';
β-MHC forward, 5'-CCGAGTCCCAGGTCAACAA-3', reverse,
5'-CTTCACGGGCACCCTTGGA-3'; AMP-activated protein kinase (AMPK)
forward 5'-TCAAGCCCAGGACAGGATTT-3', reverse,
5'-CTCTTGCGTCTCCCGACTTG-3'; acetyl-CoA carboxylase (ACC) forward,
5'-CCTGGTTCCCTGCTTACCTG-3', reverse, 5'-GTGGGATTGGACGTGCTGTA-3';
and mechanistic target of rapamycin (mTOR) forward
5'-AGAGGTCGGCACTCCACTAT-3' and reverse,
5'-TGGCCAGGCTTCTGAACAAA-3'.
Compound C treatment
Primary cardiomyocytes were treated with compound C
(cat. no. 62749-26-2; Sigma-Aldrich; Merck KGaA) at a concentration
of 20 µM or PBS for 24 h at 37˚C. Subsequently, rAd-NC
or rAd-sh-TFEC was transfected into primary caridomycytes for 24 h
and cells were collected for further study.
Western blotting
Proteins isolated from heart tissue or primary
cardiomyocytes were extracted using a total protein extraction kit
(Beijing Solarbio Science & Technology Co., Ltd.) at room
temperature. A BCA protein assay kit (Pierce; Thermo Fisher
Scientific, Inc.) was used to determine the protein concentration.
Proteins (40 µg) were separated by 12% SDS-PAGE and transferred
onto PVDF membranes. The membranes were blocked with 5% fat-free
milk (Pierce; Thermo Fisher Scientific, Inc.) at room temperature
for 2 h. Membranes were incubated with primary antibodies against
GAPDH (cat. no. ab8245; 1:3,000), phosphorylated (p)-AMPK (cat. no.
ab133448; 1:1,000), AMPK (cat. no. ab131357; 1:1,000), p-ACC (cat.
no. ab173583; 1:1,000), ACC (cat. no. ab222774; 1:1,000), p-mTOR
(cat. no. ab109268; 1:1,000), mTOR (cat. no. ab134903; 1:1,000)
(all from Abcam) and TFEC (cat. no. ab116167; 1:1,000; Abcam)
overnight at 4˚C. The membranes were then washed with
0.1% TBST and incubated with horseradish peroxidase-conjugated goat
anti-rabbit IgG (1:5,000; cat. no. ZB-2301; Beijing Zhongshan
Golden Bridge Biotechnology Co., Ltd.) for 2 h at room temperature.
Enhanced chemiluminescence reagent (GE Healthcare) was used to
detect the signal on the membrane. Signals were detected using a
Super ECL Plus kit (Nanjing KeyGen Biotech Co., Ltd.) and
quantitative analysis was performed using UVP 7.0 software (UVP,
LLC). Relative protein expressions were normalized to GAPDH. All
experiments were repeated three times. ImageJ 1.43b software
(National Institutes of Health) was used for densitometry
analysis.
Statistical analysis
Prism 7.0 (GraphPad Software, Inc.) was used for the
quantification of all data. In all experiments, each measurement
was performed at least in triplicate. The data were presented as
the means ± the standard error of the mean. Two-tailed unpaired
Student's t-test was used for comparisons between two groups.
Comparisons between more than three groups were made by one-way
ANOVA followed by Bonferroni post hoc test for multiple
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
TFEC is upregulated in the
hypertrophic myocardium of mice subjected to TAC
Compared with the sham control, TAC increased the
size of mouse hearts, but no significant changes in body weight
were observed (Fig. 1A and
B). Furthermore, the HW/BW ratio
was significantly increased in TAC mice compared with those of the
sham control (Fig. 1B).
Furthermore, the cardiomyocyte size was significantly increased in
the hypertrophic myocardia of mice subjected to TAC compared with
sham control mice (Fig. 1C). An
echocardiography study indicated that the left ventricular
posterior wall diameter (LVPWd) and dp/dtmin were increased in mice
subjected to TAC compared with the sham control mice (Fig. 1D, E and H);
however, LVEF (%) and dp/dtmax were decreased in mice subjected to
TAC compared with sham control mice (Fig. 1F and G). The results from western blotting
demonstrated that TFEC expression was significantly increased in
the hypertrophic myocardia of mice subjected to TAC compared with
the sham control mice (Fig.
1I).
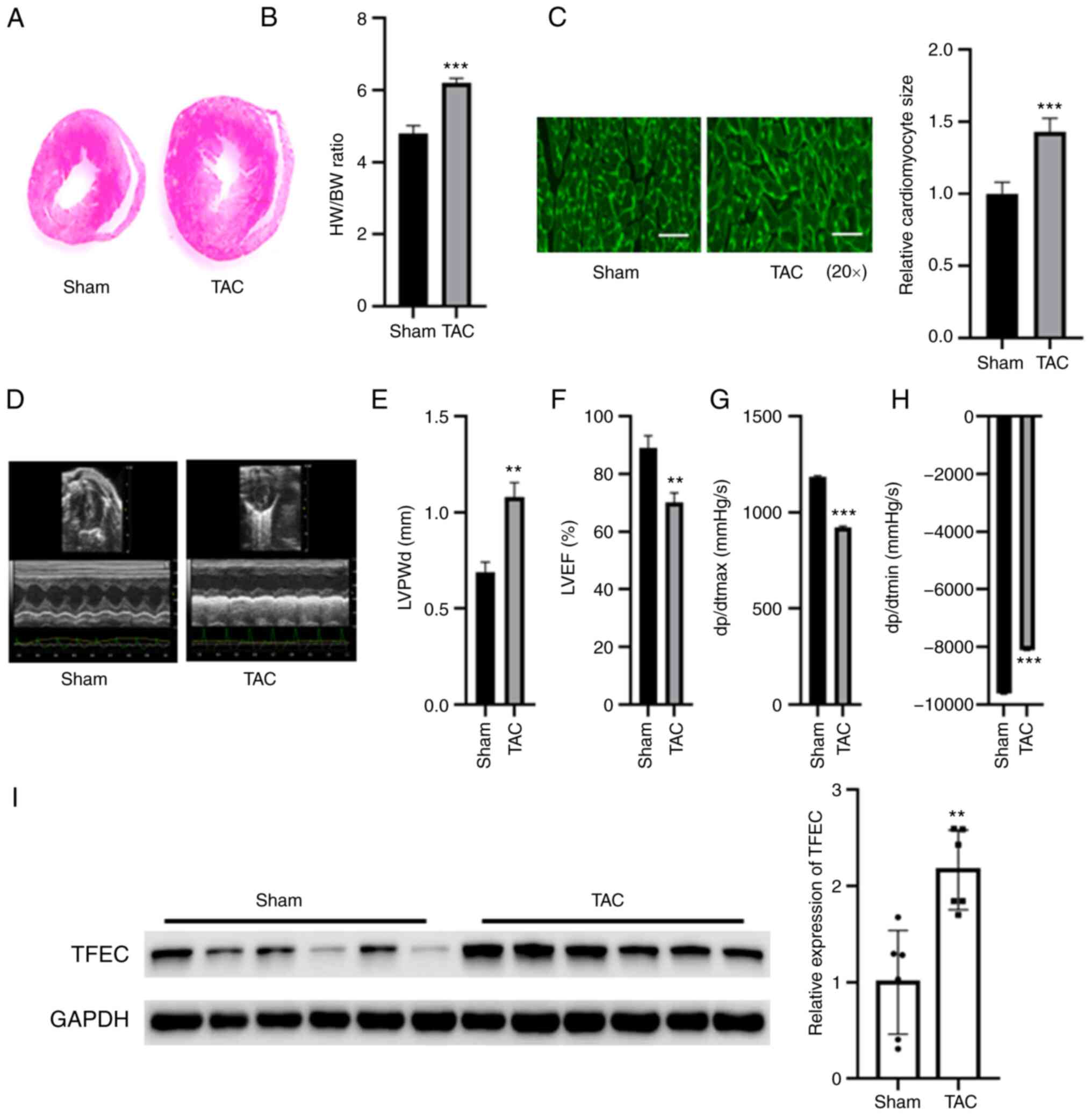 | Figure 1TFEC expression is increased in the
hypertrophic myocardia of mice subjected to TAC. (A) Hematoxylin
and eosin staining of heart tissues. (B) HW/BW was evaluated in the
sham and TAC groups. (C) Wheat germ agglutinin staining showed that
the cardiomyocyte size was significantly increased in the
hypertrophic myocardia of mice subjected to TAC compared with
control mice (magnification, x20). (D) Representative
echocardiographic images. (E) LVPWd, (F) LVEF (%), (G) dp/dtmax and
(H) dp/dtmin were quantified in the hearts of TAC and sham control
groups. (I) Western blotting showed that TFEC expression was
increased in the hypertrophic myocardia of mice subjected to TAC
compared with control mice. **P<0.01 and
***P<0.001. TAC, transverse aortic constriction;
TFEC, transcription factor EC; HW/BW, heart weight and body weight
ratio; LVEF, left ventricular ejection fractions; LVPWd, left
ventricular posterior wall diameter; dp/dtmax, maximum change in
left ventricular pressure over time; dp/dtmin, minimum change in
left ventricular pressure over time. |
TFEC is upregulated in the
hypertrophic myocardia of mice treated with AngII
The weight of the hypertrophic myocardia was
significantly increased in the AngII-treated group compared with
the control group, but no significant changes were observed in the
body weight of mice between the two groups (Fig. 2A and B). As presented in Fig. 2B, the HW/BW ratio was increased in
the AngII treatment and control groups. The relative cardiomyocyte
size was increased compared with that of the control group
(Fig. 2C). Echocardiographic
assays indicated that cardiac function was impaired in the hearts
of AngII-treated mice, as indicated by elevated LVPWd and dp/dtmin
and decreased LVEF (%) and dp/dtmax (Fig. 2D-H). The results from western
blotting demonstrated that TFEC expression was significantly
increased in the hearts of AngII-treated mice compared with those
of the control mice (Fig. 2I).
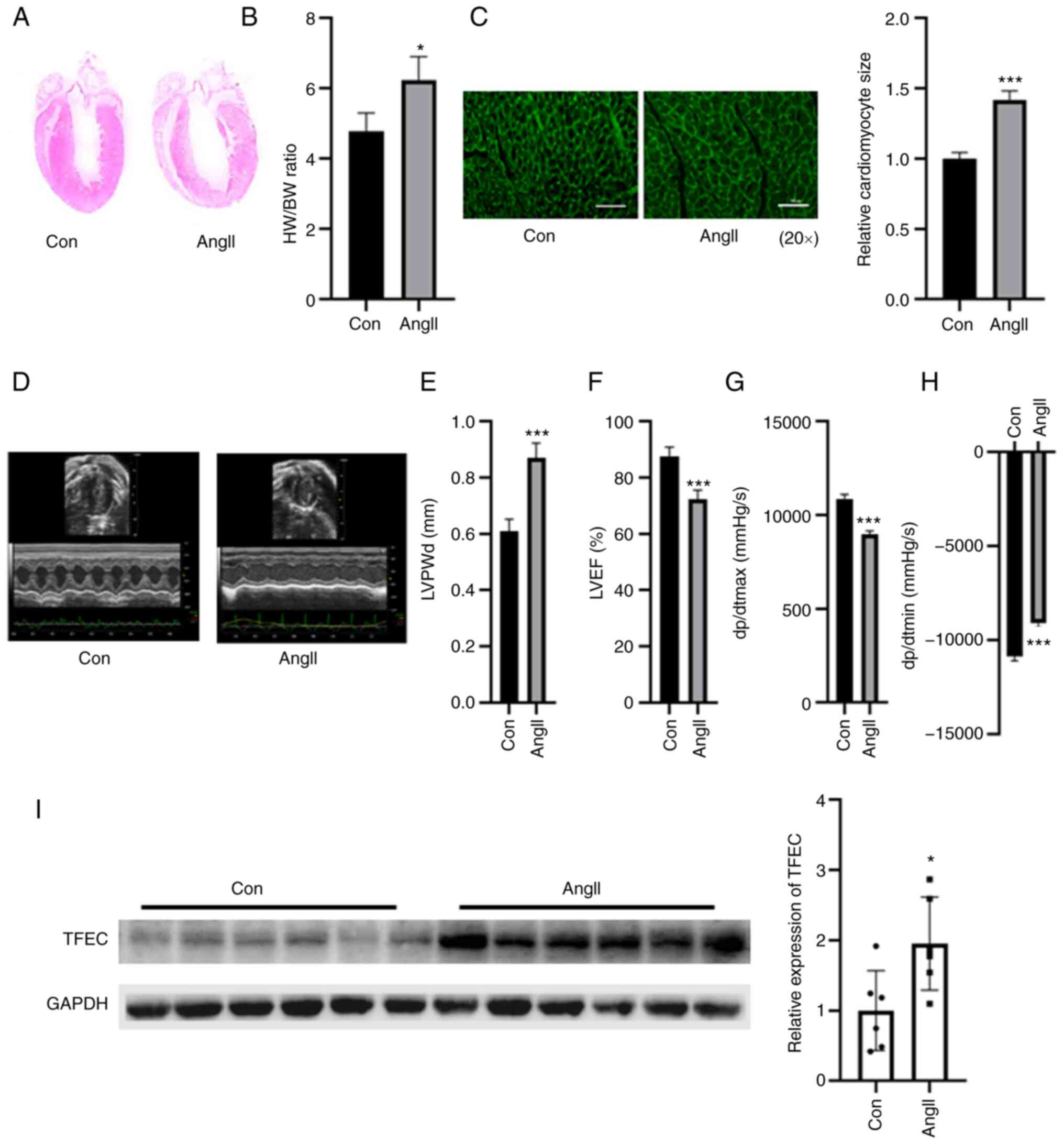 | Figure 2TFEC expression is elevated in the
hearts of AngII-treated mice compared with control mice. (A)
Hematoxylin and eosin staining of heart tissues. (B) HW/BW was
evaluated in control and Ang-treated mice. (C) Wheat germ
agglutinin staining was performed to evaluate the relative
cardiomyocyte size (magnification, x20). (D) Representative
echocardiographic images. (E) LVPWd, (F) LVEF (%), (G) dp/dtmax and
(H) dp/dtmin were quantified in the hearts of AngII-treated mice
and sham control mice. (I) Western blotting assay demonstrated that
TFEC expression was increased in the hearts of AngII-treated mice
compared with those of control mice. *P<0.05 and
***P<0.001. AngII, angiotensin II; TFEC,
transcription factor EC; HW/BW, heart weight and body weight ratio;
LVEF, left ventricular ejection fractions; LVPWd, left ventricular
posterior wall diameter; dp/dtmax, maximum change in left
ventricular pressure over time; dp/dtmin, minimum change in left
ventricular pressure over time. |
TFEC knockdown improves cardiac
function in AngII-induced mice
The expression of TFEC was silenced in AngII-treated
mice. As presented in Fig. 3A,
compared with rAd-NC, injection with rAd-sh-TFEC significantly
decreased the expression of TFEC in heart tissues. Furthermore, the
HW/BW ratio and relative cardiomyocyte size were significantly
decreased in mice injected with rAd-sh-TFEC compared with mice
injected with rAd-NC (Fig. 3B and
C). The expression levels of ANP,
BNP and β-MHC were significantly increased in the hearts of
AngII-treated mice; however, compared with transfection with
rAd-NC, transfection with rAd-sh-TFEC decreased the expression
levels of ANP, BNP and β-MHC (Fig.
3D). Echocardiographic assays indicated that transfection with
rAd-sh-TFEC improved cardiac function in the hearts of
AngII-treated mice compared with transfection with rAd-NC, as
indicated by decreased LVPWd and dp/dtmin and increased LVEF (%)
and dp/dtmax (Fig. 3E-I).
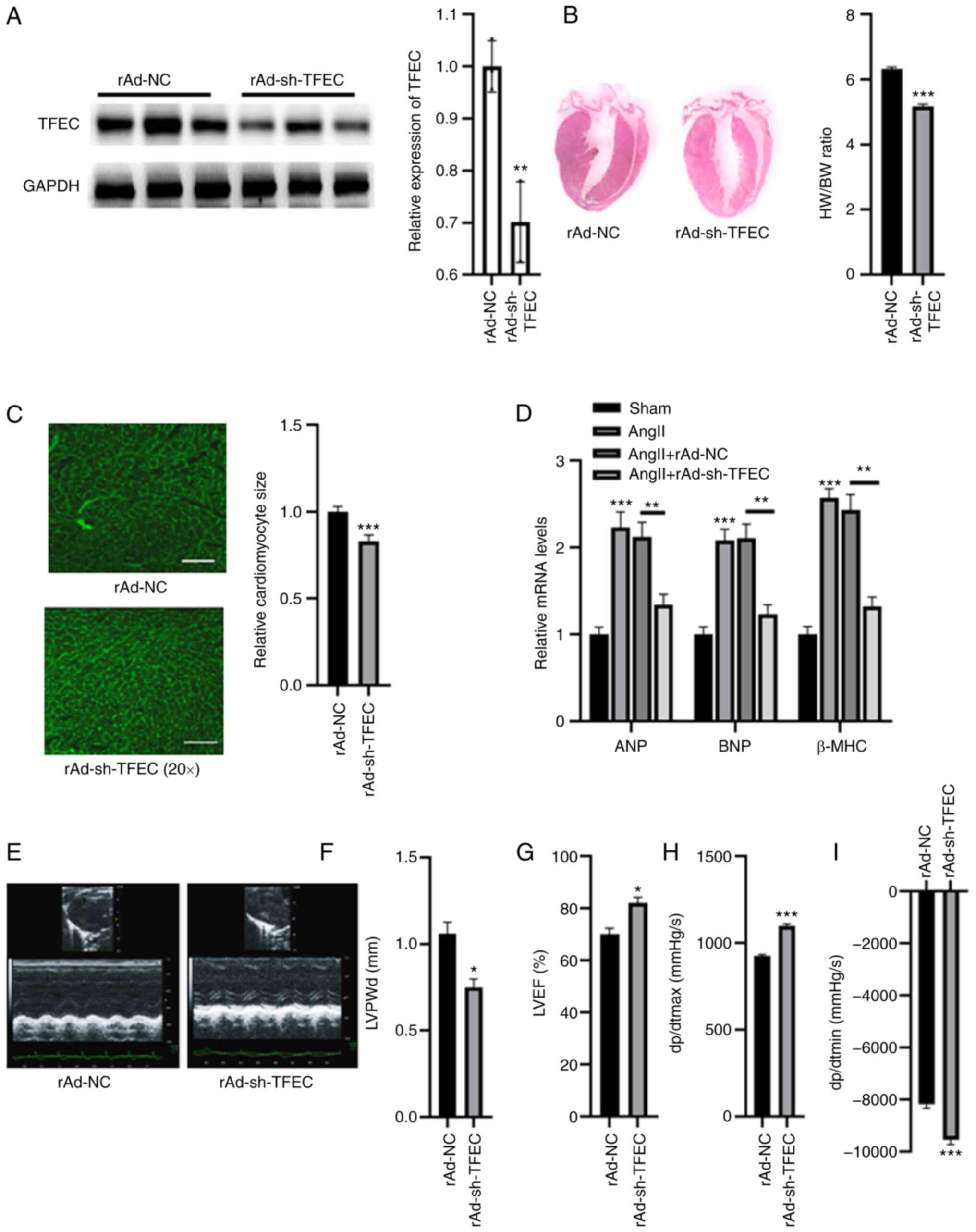 | Figure 3TFEC knockdown improves cardiac
function in AngII-treated mice compared with NC mice. (A) Western
blotting analysis showed that injection with rAd-sh-TFEC
significantly decreased the expression of TFEC in heart tissues
compared with injection with rAd-NC. (B) Knockdown of TFEC reduced
the HW/BW compared with NC group. (C) Relative cardiomyocyte size
was decreased in mice injected with rAd-sh-TFEC compared with those
injected with rAd-NC (magnification, x20). (D) Transfection with
rAd-sh-TFEC reduced the relative mRNA levels of ANP, BNP and β-MHC
compared with transfection with rAd-NC. (E) Representative
echocardiographic images. (F) LVPWd, (G) LVEF (%), (H) dp/dtmax and
(I) dp/dtmin were quantified in the hearts of rAd-sh-TFEC and
rAd-NC-treated mice. *P<0.05, **P<0.01
and ***P<0.001. TFEC, transcription factor EC; sh,
short hairpin; NC, negative control; HW/BW, heart weight and body
weight ratio; ANP, atrial natriuretic peptide; BNP, brain
natriuretic peptide; β-MHC, β-myosin heavy chain; LVEF, left
ventricular ejection fractions; LVPWd, left ventricular posterior
wall diameter; dp/dtmax, maximum change in left ventricular
pressure over time; dp/dtmin, minimum change in left ventricular
pressure over time. |
Silencing TFEC abolishes AngII-induced
cardiomyocyte hypertrophy
A cellular cardiomyocyte model was established. As
demonstrated in Fig. 4A, treatment
with AngII increased the relative cardiomyocyte size. The
expression levels of ANP, BNP and β-MHC were significantly
increased in primary cardiomyocytes treated with AngII compared
with control cardiomyocytes (Fig.
4B). In primary cardiomyocytes, treatment with AngII
significantly increased the expression of TFEC compared with the
control (Fig. 4C). Furthermore,
transfection with rAd-sh-TFEC significantly reduced the expression
of TFEC, even in primary cardiomyocytes treated with AngII
(Fig. 4D). In addition, the
AngII-induced increase in cardiomyocyte size could be reversed
following TFEC knockdown in primary cardiomyocytes (Fig. 4E). The elevated expression levels
of ANP, BNP and β-MHC induced by AngII could be partially abolished
after TFEC knockdown (Fig.
4F).
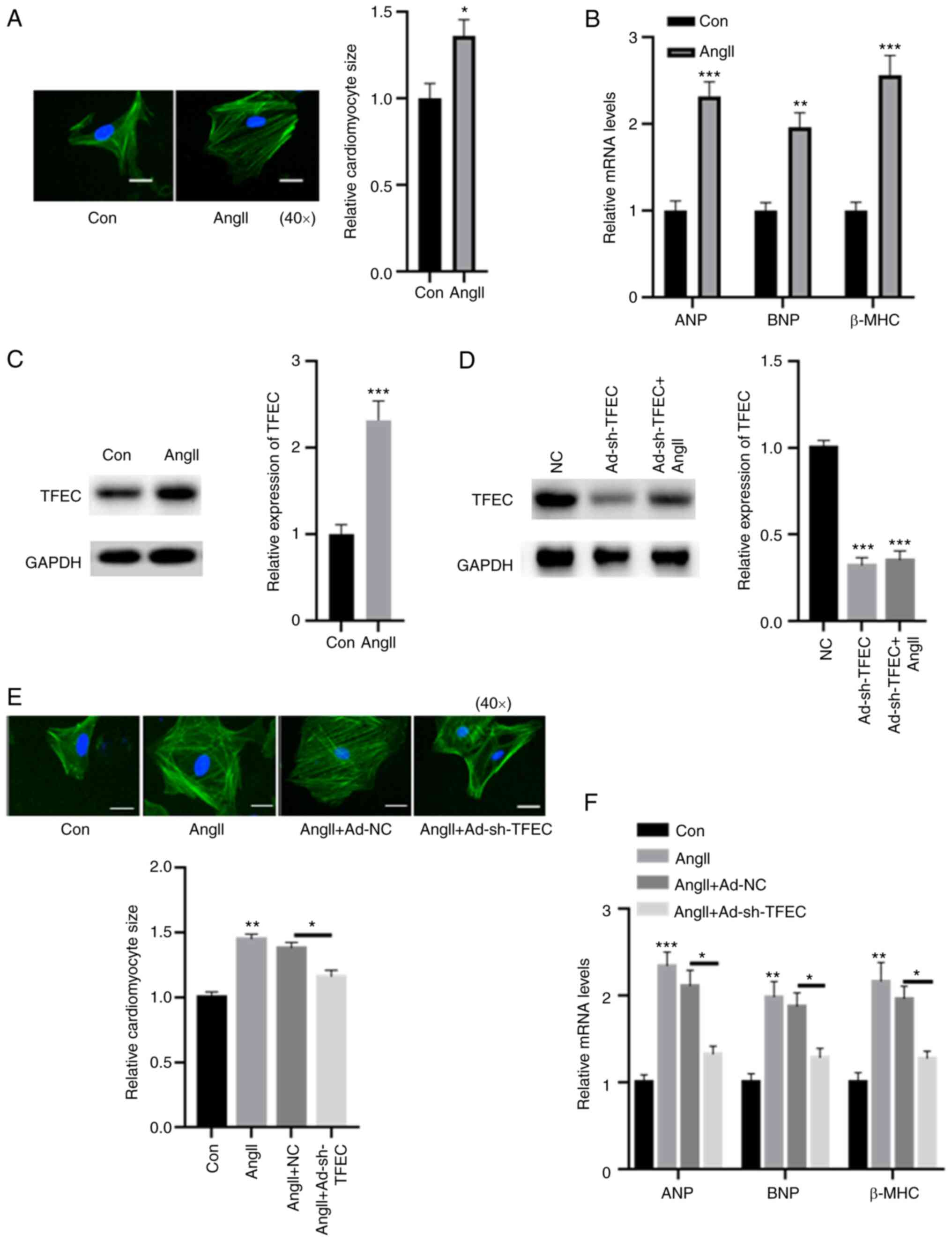 | Figure 4Silencing TFEC abolishes AngII-induced
cardiomyocyte hypertrophy. (A) Compared with the control, treatment
with AngII increased the relative cardiomyocyte size
(magnification, x40). (B) mRNA levels of ANP, BNP and β-MHC were
significantly increased in primary cardiomyocytes treated with
AngII compared with the control. (C) In primary cardiomyocytes,
treatment with AngII significantly increased the expression of TFEC
compared with treatment with the control. (D) Transfection with
rAd-sh-TFEC significantly decreased the expression of TFEC even in
primary cardiomyocytes treated with AngII. (E) AngII-induced
increase in cardiomyocyte size could be reversed by TFEC knockdown
in primary cardiomyocytes (magnification, x40). (F) Elevated mRNA
levels of ANP, BNP and β-MHC induced by AngII could be partially
abolished following TFEC knockdown in primary cardiomyocytes.
*P<0.05, **P<0.01 and
***P<0.001. AngII, angiotensin II; Con, control; NC,
negative control; ANP, atrial natriuretic peptide; BNP, brain
natriuretic peptide; β-MHC, β-myosin heavy chain; TFEC,
transcription factor EC; sh, short hairpin. |
TFEC inactivates AMPK/mTOR
signaling
AMPK/mTOR signaling is suggested to play a key role
in the AngII-induced cardiac hypertrophy model (18). Hence, we evaluated the effect of
TFEC on AMPK/mTOR signaling. The results from western blotting
demonstrated that TFEC expression was significantly increased in
primary cardiomyocytes transfected with Ad-TFEC compared with that
transfected with Ad-NC (Fig. 5A),
indicating that transfection with rAd-TFEC overexpression vector
was successful. The results from RT-qPCR demonstrated that
transfection with rAd-TFEC did not change the expression levels of
AMPK, ACC and mTOR, compared with transfection with rAd-NC
(Fig. 5B). The results from
western blotting showed that TFEC overexpression decreased the
expression of p-AMPK and p-ACC but increased the expression of
p-mTOR in primary cardiomyocytes (Fig.
5C). Conversely, silencing TFEC increased the expression of
p-AMPK and p-ACC but decreased the level of p-mTOR (Fig. 5D). Furthermore, treatment with
Compound C, which is an AMPK inhibitor, significantly inhibited the
activation of AMPK/ACC but increased the activation of mTOR, even
in primary cardiomyocytes transfected with rAd-TFEC (Fig. 5D).
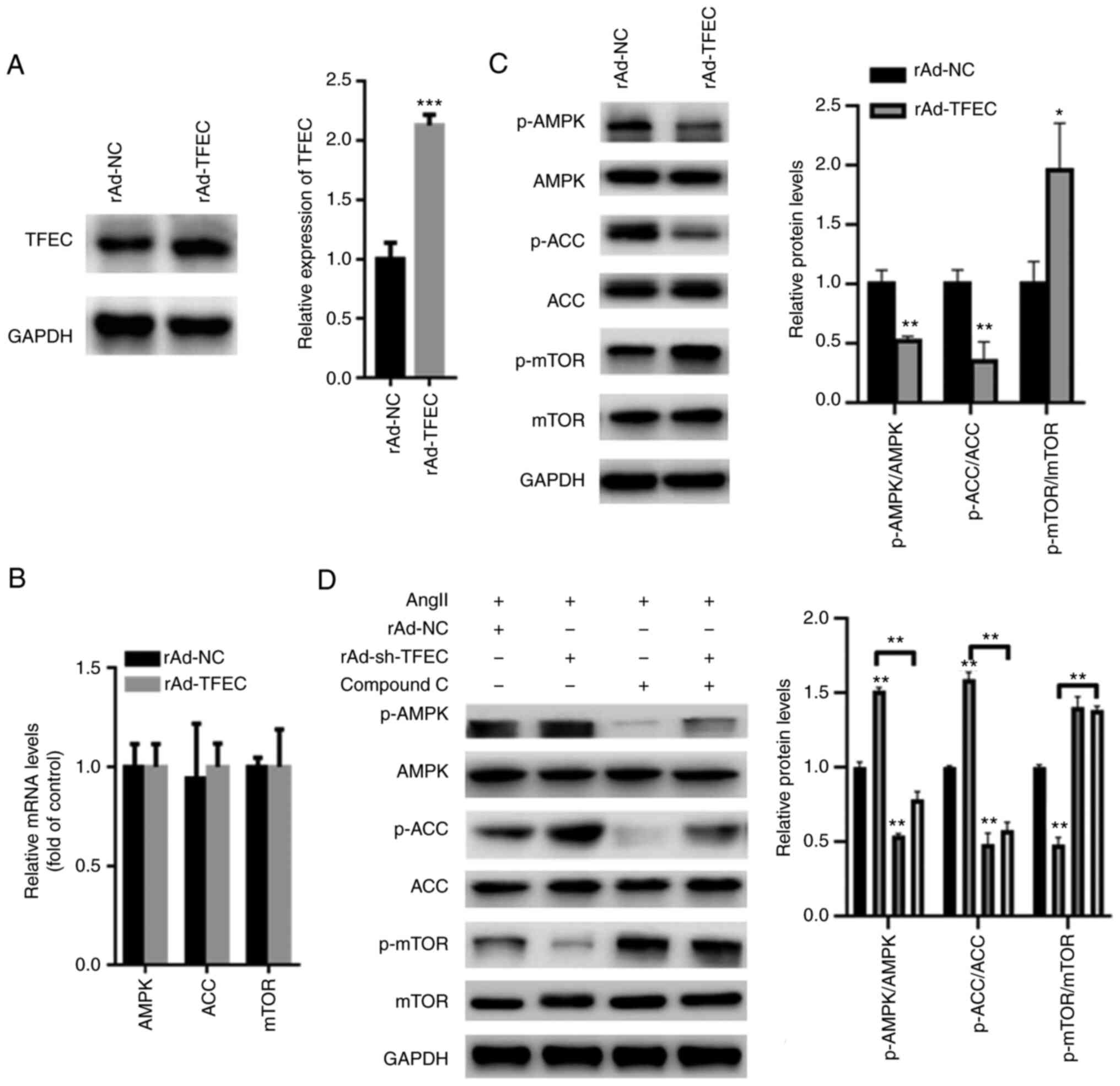 | Figure 5TFEC activates AMPK/mTOR signaling in
primary cardiomyocytes. (A) Western blotting showed that TFEC
expression was significantly increased in primary cardiomyocytes
transfected with Ad-TFEC compared with those transfected with
Ad-NC. (B) Reverse transcription quantitative PCR showed that
transfection with rAd-TFEC did not change the mRNA levels of AMPK,
ACC and mTOR, compared with transfection with rAd-NC. (C) Western
blotting showed that TFEC overexpression decreased the expression
of p-AMPK and p-ACC but increased the level of p-mTOR. (D) Compound
C significantly suppressed the activation of AMPK/ACC but increased
the activation of mTOR, even in primary cardiomyocytes transfected
with rAd-sh-TFEC. *P<0.05, **P<0.01 and
***P<0.001. AMPK, AMP-activated protein kinase; mTOR,
mechanistic target of rapamycin; ACC, acetyl-CoA carboxylase; NC,
negative control; p, phosphorylated; sh, short hairpin; TFEC,
transcription factor EC. |
Discussion
Cardiac hypertrophy and fibrosis can be induced by
many factors, including mechanical pressure overload and AngII
(19). The present study explored
therefore the expression of TFEC in a TAC-induced mouse model and a
AngII-induced cardiac hypertrophy model. Elevated expression of
TFEC was found in the hearts of mice with pressure overload- and
AngII-induced hypertrophy. These observations suggested a potential
detrimental role of TFEC in the pathology of cardiac
hypertrophy.
AngII is the most important constituent of the
renin-angiotensin aldosterone system, which serves a key role in
regulating cardiomyocyte growth, cardiac hypertrophy and
extracellular matrix accumulation (20-22).
Thus, TFEC expression was silenced in the mouse model of
AngII-induced cardiac hypertrophy. The results demonstrated that
cardiac TFEC expression was decreased in rAd-sh-TFEC-transfected
mice pretreated with AngII. In the process of cardiac remodeling,
enlargement of the heart is a major and clear change that is
tightly associated with hypertrophy and fibrosis (23). In the mouse model of AngII-induced
cardiac remodeling, TFEC knockdown ameliorated cardiac hypertrophy
by reducing cardiomyocyte enlargement and enhancing the HW/BW
ratio. In addition, mice transfection with rAd-sh-TFEC protected
the hearts from the pathological hypertrophy induced by AngII,
since TFEC knockdown partially abolished the effects of AngII on
cardiac hypertrophy.
AMPK signaling is suggested to be an important
regulator of different physiological and pathological cellular
events that occur in cardiovascular diseases (24-26).
AMPK is a major checkpoint of cellular metabolism that enhances
autophagy by inhibiting mTOR signaling (18,27).
AMPK/mTOR-mediated autophagy protects diabetic rats from myocardial
damage (24). The present study
demonstrated that TFEC overexpression inhibited AMPK/mTOR signaling
in AngII-treated primary cardiomyocytes. Conversely, silencing TFEC
induced an increase in p-AMPK and p-ACC expression and a decrease
in p-mTOR expression. However, following cotreatment with Compound
C, the effect of sh-TFEC on AMPK/mTOR signaling was abolished. It
is possible that TFEC may induce myocardial injury and dysfunction
at least in part via inhibition of AMPK/mTOR signaling. Previous
studies have also reported that AMPK activation protects against
the pathological progression of cardiac hypertrophy, ventricular
remodeling and heart failure (28,29).
In the present study, we therefore hypothesized that the
TFEC-mediated inactivation of AMPK/mTOR signaling may contribute to
cardiac hypertrophy. However, the precise mechanisms by which
TFEC-inactivated AMPK signaling could affect cardiac hypertrophy
are not yet fully understood and require further investigation.
This study presented some limitations. It would be
interesting to evaluate the serum concentration of cholesterol,
triglyceride, HDL and LDL in mice, since these markers are known to
be associated with heart function. In a future study, these
biochemical parameters will be determined in the serum of
pressure-overload cardiac hypertrophy and AngII infusion-induced
cardiac hypertrophy animal models.
In conclusion, the findings from the present study
demonstrated that TFEC was overexpressed in the hearts of mice with
cardiac hypertrophy, and that silencing TFEC could improve
AngII-induced cardiac hypertrophy and dysfunction by activating
AMPK/mTOR signaling. These results suggested that TFEC may serve
important roles in cardiac hypertrophy and dysfunction, providing
potential therapeutic targets for heart failure.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by a grant from the
Weapon Industry 521 Hospital (grant no. WI-20180952).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TZ and ZW performed the experiments, analyzed the
data and wrote the paper. YC and CN performed some of the RT-qPCR
experiments. XZ designed the experiments, analyzed the data and
gave final approval of the version to be published. All authors
read and approved the final manuscript. TZ and XZ confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
The present study was approved by the Research
Ethics Committee of Weapon Industry 521 Hospital (approval no.
WI-J2018923A).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nakamura M and Sadoshima J: Mechanisms of
physiological and pathological cardiac hypertrophy. Nat Rev
Cardiol. 15:387–407. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Gallo S, Vitacolonna A, Bonzano A,
Comoglio P and Crepaldi T: ERK: A key player in the pathophysiology
of cardiac hypertrophy. Int J Mol Sci. 20(2164)2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Meng R, Pei Z, Zhang A, Zhou Y, Cai X,
Chen B, Liu G, Mai W, Wei J and Dong Y: AMPK activation enhances
PPARα activity to inhibit cardiac hypertrophy via ERK1/2 MAPK
signaling pathway. Arch Biochem Biophys. 511:1–7. 2011.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Petratou K, Spencer SA, Kelsh RN and
Lister JA: The MITF paralog tfec is required in neural crest
development for fate specification of the iridophore lineage from a
multipotent pigment cell progenitor. PLoS One.
16(e0244794)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ballesteros-Alvarez J, Dilshat R, Fock V,
Möller K, Karl L, Larue L, Ögmundsdóttir MH and Steingrímsson E:
MITF and TFEB cross-regulation in melanoma cells. PLoS One.
15(e0238546)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Mahony CB, Fish RJ, Pasche C and Bertrand
JY: Tfec controls the hematopoietic stem cell vascular niche during
zebrafish embryogenesis. Blood. 128:1336–1345. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Rehli M, Sulzbacher S, Pape S, Ravasi T,
Wells CA, Heinz S, Söllner L, El Chartouni C, Krause SW,
Steingrimsson E, et al: Transcription factor Tfec contributes to
the IL-4-inducible expression of a small group of genes in mouse
macrophages including the granulocyte colony-stimulating factor
receptor. J Immunol. 174:7111–7122. 2005.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zhao GQ, Zhao Q, Zhou X, Mattei MG and de
Crombrugghe B: TFEC, a basic helix-loop-helix protein, forms
heterodimers with TFE3 and inhibits TFE3-dependent transcription
activation. Mol Cell Biol. 13:4505–4512. 1993.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Sun S, Kee HJ, Jin L, Ryu Y, Choi SY, Kim
GR and Jeong MH: Gentisic acid attenuates pressure overload-induced
cardiac hypertrophy and fibrosis in mice through inhibition of the
ERK1/2 pathway. J Cell Mol Med. 22:5964–5977. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Dutton JW III, Artwohl JE, Huang X and
Fortman JD: Assessment of pain associated with the injection of
sodium pentobarbital in laboratory mice (Mus musculus). J Am Assoc
Lab Anim Sci. 58:373–379. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Takayanagi T, Kawai T, Forrester SJ, Obama
T, Tsuji T, Fukuda Y, Elliott KJ, Tilley DG, Davisson RL, Park JY
and Eguchi S: Role of epidermal growth factor receptor and
endoplasmic reticulum stress in vascular remodeling induced by
angiotensin II. Hypertension. 65:1349–1355. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kopechek JA, McTiernan CF, Chen X, Zhu J,
Mburu M, Feroze R, Whitehurst DA, Lavery L, Cyriac J and Villanueva
FS: Ultrasound and Microbubble-targeted delivery of a microRNA
inhibitor to the heart suppresses cardiac hypertrophy and preserves
cardiac function. Theranostics. 9:7088–7098. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Tachibana S, Chen C, Zhang OR, Schurr SV,
Hill C, Li R, Manso AM, Zhang J, Andreyev A, Murphy AN, et al:
Analyzing oxygen consumption rate in primary cultured mouse
neonatal cardiomyocytes using an extracellular flux analyzer. J Vis
Exp: Feb 13, 2019 (Epub ahead of print). doi: 10.3791/59052.
|
|
14
|
Baird RC, Li S, Wang H, Naga Prasad SV,
Majdalany D, Perni U and Wu Q: Pregnancy-associated cardiac
hypertrophy in Corin-deficient mice: Observations in a transgenic
model of preeclampsia. Can J Cardiol. 35:68–76. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Sun J, Hao W, Fillmore N, Ma H, Springer
D, Yu ZX, Sadowska A, Garcia A, Chen R, Muniz-Medina V, et al:
Human Relaxin-2 fusion protein treatment prevents and reverses
isoproterenol-induced hypertrophy and fibrosis in mouse heart. J Am
Heart Assoc. 8(e013465)2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Sundaresan NR, Gupta M, Kim G, Rajamohan
SB, Isbatan A and Gupta MP: Sirt3 blocks the cardiac hypertrophic
response by augmenting Foxo3a-dependent antioxidant defense
mechanisms in mice. J Clin Invest. 119:2758–2771. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yan J, Yan JY, Wang YX, Ling YN, Song XD,
Wang SY, Liu HQ, Liu QC, Zhang Y, Yang PZ, et al:
Spermidine-enhanced autophagic flux improves cardiac dysfunction
following myocardial infarction by targeting the AMPK/mTOR
signalling pathway. Br J Pharmacol. 176:3126–3142. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhai CG, Xu YY, Tie YY, Zhang Y, Chen WQ,
Ji XP, Mao Y, Qiao L, Cheng J, Xu QB and Zhang C: DKK3
overexpression attenuates cardiac hypertrophy and fibrosis in an
angiotensin-perfused animal model by regulating the ADAM17/ACE2 and
GSK-3β/β-catenin pathways. J Mol Cell Cardiol. 114:243–252.
2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Hu S, Cheng M, Guo X, Wang S, Liu B, Jiang
H, Huang C and Wu G: Down-regulation of miR-200c attenuates
AngII-induced cardiac hypertrophy via targeting the MLCK-mediated
pathway. J Cell Mol Med. 23:2505–2516. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Xiao YF, Zeng ZX, Guan XH, Wang LF, Wang
CJ, Shi H, Shou W, Deng KY and Xin HB: FKBP12.6 protects heart from
AngII-induced hypertrophy through inhibiting
Ca2+/calmodulin-mediated signalling pathways in vivo and
in vitro. J Cell Mol Med. 22:3638–3651. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Dai W, Chen H, Jiang J, Kong W and Wang Y:
Silencing MR-1 attenuates inflammatory damage in mice heart induced
by AngII. Biochem Biophys Res Commun. 391:1573–1578.
2010.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Liang J, Huang B, Yuan G, Chen Y, Liang F,
Zeng H, Zheng S, Cao L, Geng D and Zhou S: Stretch-activated
channel Piezo1 is up-regulated in failure heart and cardiomyocyte
stimulated by AngII. Am J Transl Res. 9:2945–2955. 2017.PubMed/NCBI
|
|
24
|
Ren PH, Zhang ZM, Wang P, Zhu HP and Li
ZQ: Yangxinkang tablet protects against cardiac dysfunction and
remodelling after myocardial infarction in rats through inhibition
of AMPK/mTOR-mediated autophagy. Pharm Biol. 58:321–327.
2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wang Y, Zhu S, Liu H, Wei W, Tu Y, Chen C,
Song J, Li J, Sun S, Wang C and Xu Z: Thyroxine alleviates energy
failure, prevents myocardial cell apoptosis, and protects against
doxorubicin-induced cardiac injury and cardiac dysfunction via the
LKB1/AMPK/mTOR axis in mice. Dis Markers.
2019(7420196)2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhao J, Chen D and Feng C: Troxerutin
attenuates isoproterenol-induced cardiac hypertrophy via the
LKB1/AMPK/mTOR pathway. Panminerva Med. 63:233–234. 2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Zhang J, Zhao P, Quan N, Wang L, Chen X,
Cates C, Rousselle T and Li J: The endotoxemia cardiac dysfunction
is attenuated by AMPK/mTOR signaling pathway regulating autophagy.
Biochem Biophys Res Commun. 492:520–527. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Majd S, Power JHT, Chataway TK and
Grantham HJM: A comparison of LKB1/AMPK/mTOR metabolic axis
response to global ischaemia in brain, heart, liver and kidney in a
rat model of cardiac arrest. BMC Cell Biol. 19(7)2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Qi H, Ren J, Ba L, Song C, Zhang Q, Cao Y,
Shi P, Fu B, Liu Y and Sun H: MSTN attenuates cardiac hypertrophy
through inhibition of excessive cardiac autophagy by blocking
AMPK/mTOR and miR-128/PPARγ/NF-κB. Mol Ther Nucleic Acids.
19:507–522. 2020.PubMed/NCBI View Article : Google Scholar
|














