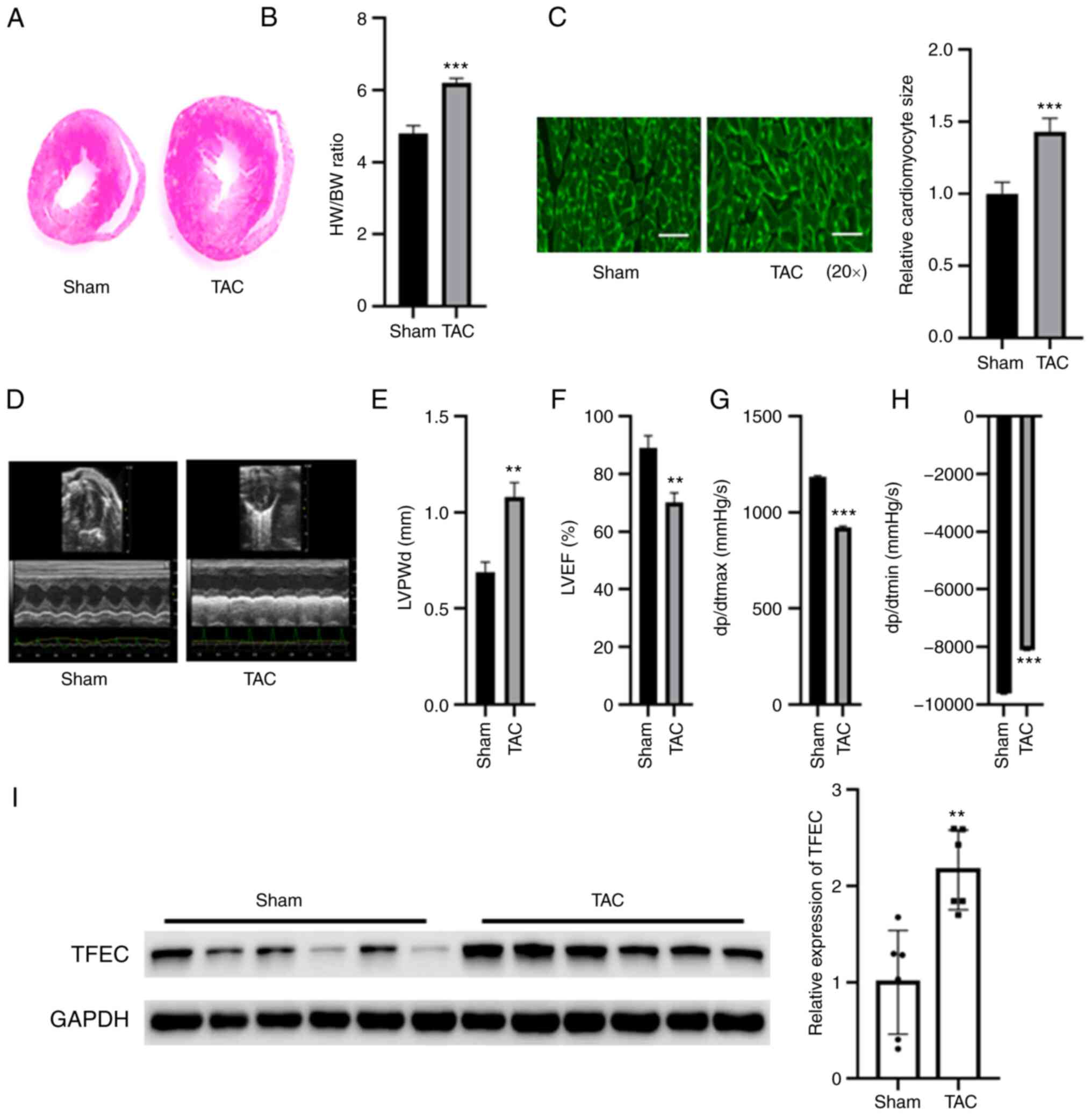
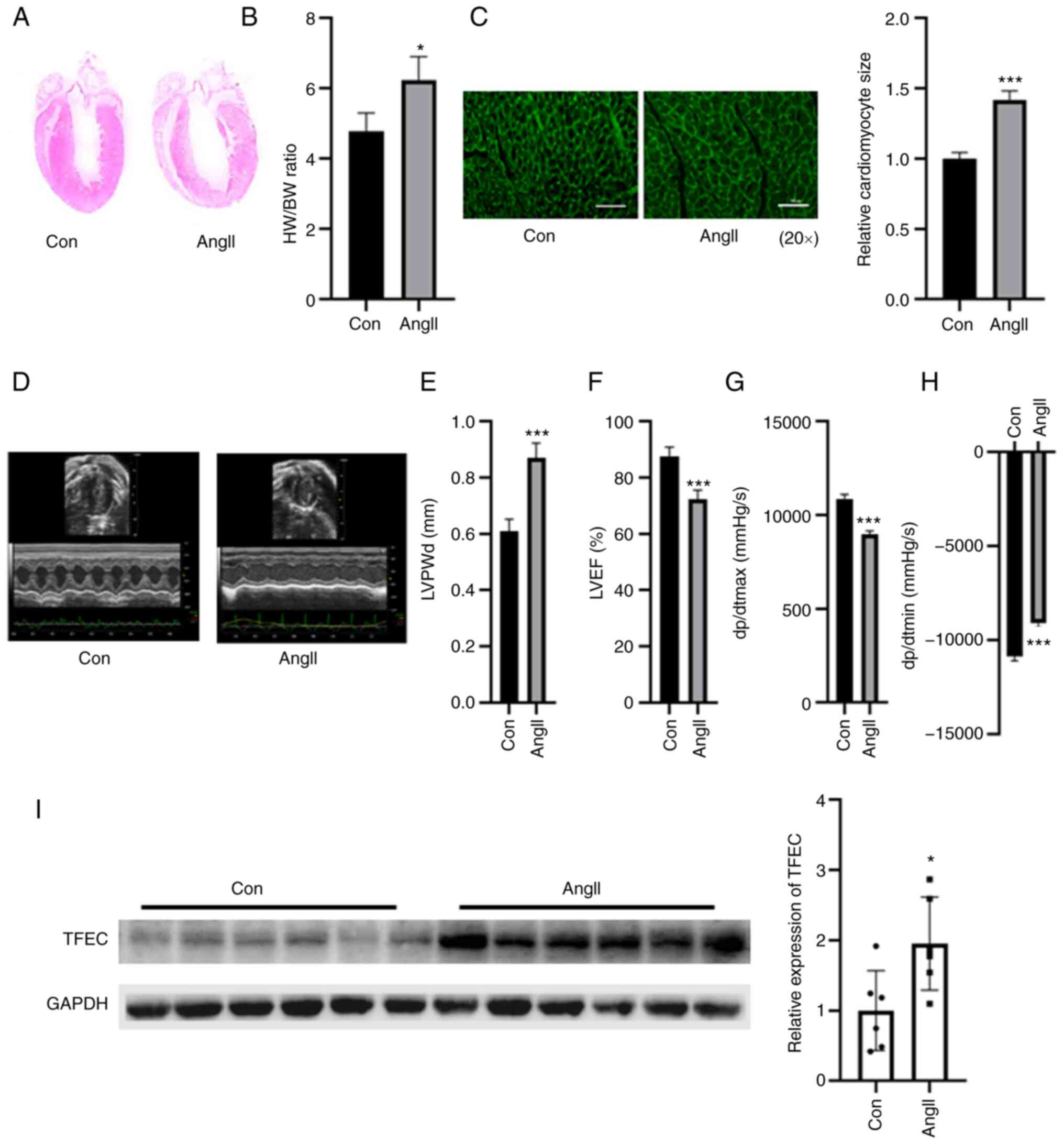
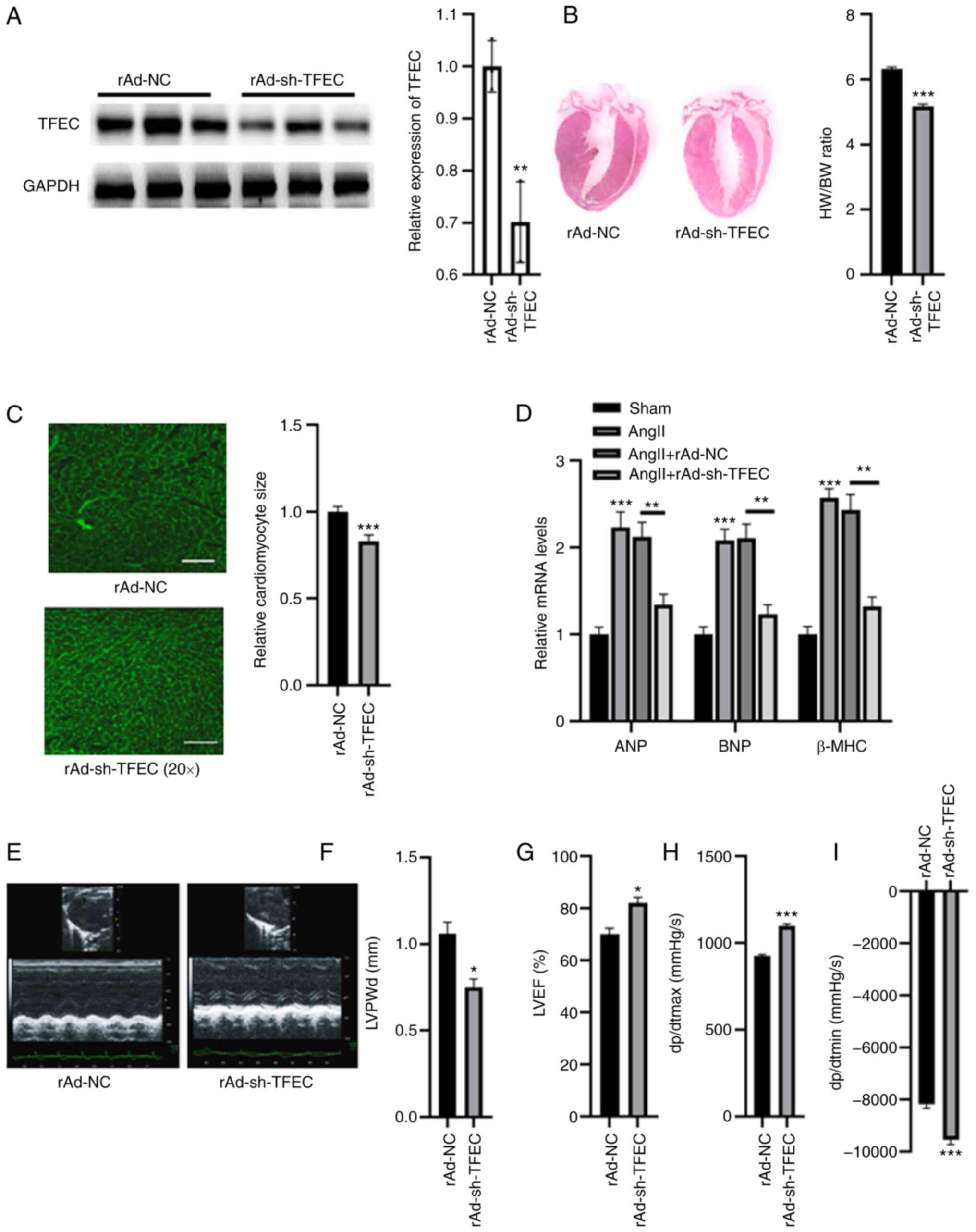
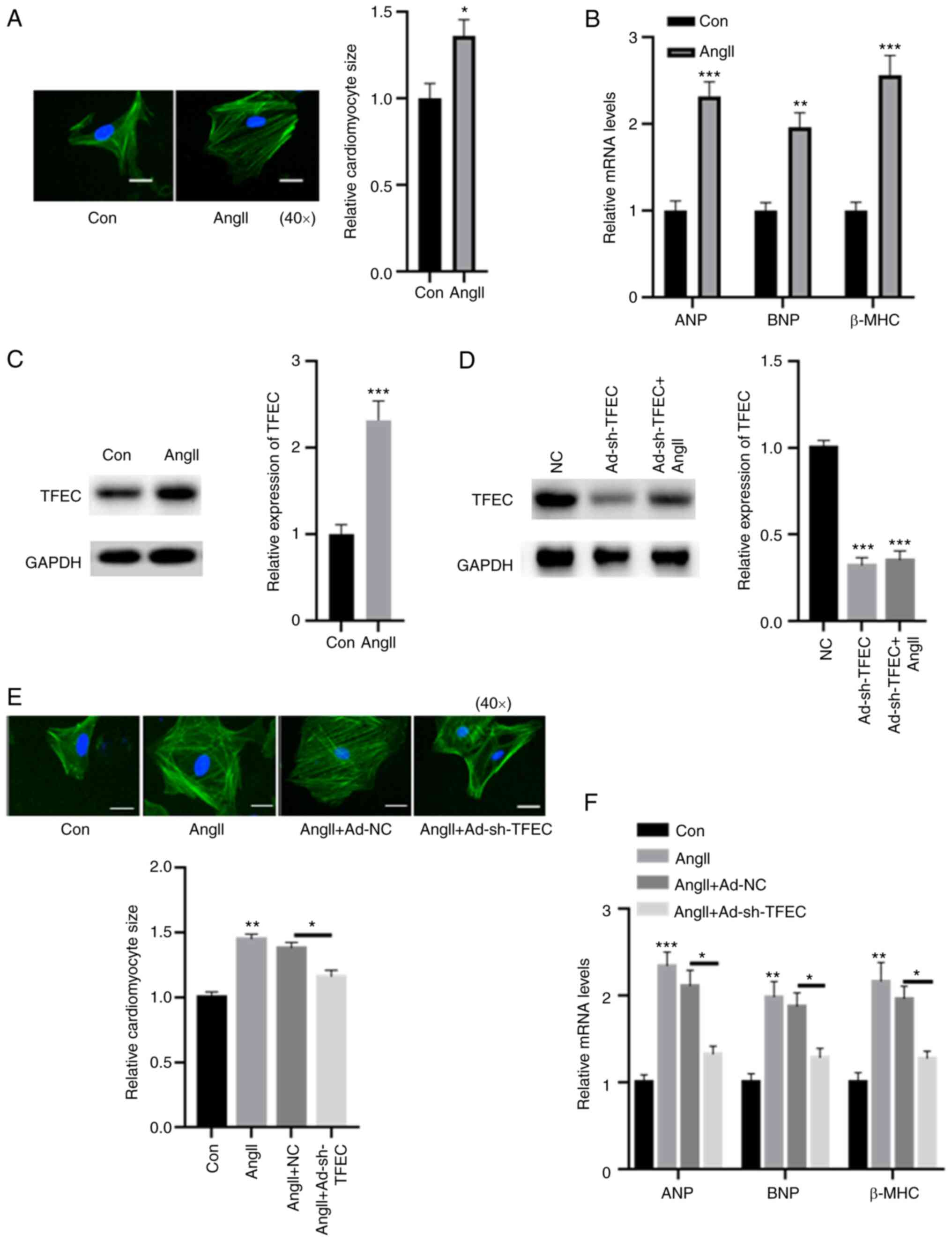
|
1
|
Nakamura M and Sadoshima J: Mechanisms of
physiological and pathological cardiac hypertrophy. Nat Rev
Cardiol. 15:387–407. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Gallo S, Vitacolonna A, Bonzano A,
Comoglio P and Crepaldi T: ERK: A key player in the pathophysiology
of cardiac hypertrophy. Int J Mol Sci. 20(2164)2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Meng R, Pei Z, Zhang A, Zhou Y, Cai X,
Chen B, Liu G, Mai W, Wei J and Dong Y: AMPK activation enhances
PPARα activity to inhibit cardiac hypertrophy via ERK1/2 MAPK
signaling pathway. Arch Biochem Biophys. 511:1–7. 2011.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Petratou K, Spencer SA, Kelsh RN and
Lister JA: The MITF paralog tfec is required in neural crest
development for fate specification of the iridophore lineage from a
multipotent pigment cell progenitor. PLoS One.
16(e0244794)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ballesteros-Alvarez J, Dilshat R, Fock V,
Möller K, Karl L, Larue L, Ögmundsdóttir MH and Steingrímsson E:
MITF and TFEB cross-regulation in melanoma cells. PLoS One.
15(e0238546)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Mahony CB, Fish RJ, Pasche C and Bertrand
JY: Tfec controls the hematopoietic stem cell vascular niche during
zebrafish embryogenesis. Blood. 128:1336–1345. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Rehli M, Sulzbacher S, Pape S, Ravasi T,
Wells CA, Heinz S, Söllner L, El Chartouni C, Krause SW,
Steingrimsson E, et al: Transcription factor Tfec contributes to
the IL-4-inducible expression of a small group of genes in mouse
macrophages including the granulocyte colony-stimulating factor
receptor. J Immunol. 174:7111–7122. 2005.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zhao GQ, Zhao Q, Zhou X, Mattei MG and de
Crombrugghe B: TFEC, a basic helix-loop-helix protein, forms
heterodimers with TFE3 and inhibits TFE3-dependent transcription
activation. Mol Cell Biol. 13:4505–4512. 1993.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Sun S, Kee HJ, Jin L, Ryu Y, Choi SY, Kim
GR and Jeong MH: Gentisic acid attenuates pressure overload-induced
cardiac hypertrophy and fibrosis in mice through inhibition of the
ERK1/2 pathway. J Cell Mol Med. 22:5964–5977. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Dutton JW III, Artwohl JE, Huang X and
Fortman JD: Assessment of pain associated with the injection of
sodium pentobarbital in laboratory mice (Mus musculus). J Am Assoc
Lab Anim Sci. 58:373–379. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Takayanagi T, Kawai T, Forrester SJ, Obama
T, Tsuji T, Fukuda Y, Elliott KJ, Tilley DG, Davisson RL, Park JY
and Eguchi S: Role of epidermal growth factor receptor and
endoplasmic reticulum stress in vascular remodeling induced by
angiotensin II. Hypertension. 65:1349–1355. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kopechek JA, McTiernan CF, Chen X, Zhu J,
Mburu M, Feroze R, Whitehurst DA, Lavery L, Cyriac J and Villanueva
FS: Ultrasound and Microbubble-targeted delivery of a microRNA
inhibitor to the heart suppresses cardiac hypertrophy and preserves
cardiac function. Theranostics. 9:7088–7098. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Tachibana S, Chen C, Zhang OR, Schurr SV,
Hill C, Li R, Manso AM, Zhang J, Andreyev A, Murphy AN, et al:
Analyzing oxygen consumption rate in primary cultured mouse
neonatal cardiomyocytes using an extracellular flux analyzer. J Vis
Exp: Feb 13, 2019 (Epub ahead of print). doi: 10.3791/59052.
|
|
14
|
Baird RC, Li S, Wang H, Naga Prasad SV,
Majdalany D, Perni U and Wu Q: Pregnancy-associated cardiac
hypertrophy in Corin-deficient mice: Observations in a transgenic
model of preeclampsia. Can J Cardiol. 35:68–76. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Sun J, Hao W, Fillmore N, Ma H, Springer
D, Yu ZX, Sadowska A, Garcia A, Chen R, Muniz-Medina V, et al:
Human Relaxin-2 fusion protein treatment prevents and reverses
isoproterenol-induced hypertrophy and fibrosis in mouse heart. J Am
Heart Assoc. 8(e013465)2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Sundaresan NR, Gupta M, Kim G, Rajamohan
SB, Isbatan A and Gupta MP: Sirt3 blocks the cardiac hypertrophic
response by augmenting Foxo3a-dependent antioxidant defense
mechanisms in mice. J Clin Invest. 119:2758–2771. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yan J, Yan JY, Wang YX, Ling YN, Song XD,
Wang SY, Liu HQ, Liu QC, Zhang Y, Yang PZ, et al:
Spermidine-enhanced autophagic flux improves cardiac dysfunction
following myocardial infarction by targeting the AMPK/mTOR
signalling pathway. Br J Pharmacol. 176:3126–3142. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhai CG, Xu YY, Tie YY, Zhang Y, Chen WQ,
Ji XP, Mao Y, Qiao L, Cheng J, Xu QB and Zhang C: DKK3
overexpression attenuates cardiac hypertrophy and fibrosis in an
angiotensin-perfused animal model by regulating the ADAM17/ACE2 and
GSK-3β/β-catenin pathways. J Mol Cell Cardiol. 114:243–252.
2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Hu S, Cheng M, Guo X, Wang S, Liu B, Jiang
H, Huang C and Wu G: Down-regulation of miR-200c attenuates
AngII-induced cardiac hypertrophy via targeting the MLCK-mediated
pathway. J Cell Mol Med. 23:2505–2516. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Xiao YF, Zeng ZX, Guan XH, Wang LF, Wang
CJ, Shi H, Shou W, Deng KY and Xin HB: FKBP12.6 protects heart from
AngII-induced hypertrophy through inhibiting
Ca2+/calmodulin-mediated signalling pathways in vivo and
in vitro. J Cell Mol Med. 22:3638–3651. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Dai W, Chen H, Jiang J, Kong W and Wang Y:
Silencing MR-1 attenuates inflammatory damage in mice heart induced
by AngII. Biochem Biophys Res Commun. 391:1573–1578.
2010.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Liang J, Huang B, Yuan G, Chen Y, Liang F,
Zeng H, Zheng S, Cao L, Geng D and Zhou S: Stretch-activated
channel Piezo1 is up-regulated in failure heart and cardiomyocyte
stimulated by AngII. Am J Transl Res. 9:2945–2955. 2017.PubMed/NCBI
|
|
24
|
Ren PH, Zhang ZM, Wang P, Zhu HP and Li
ZQ: Yangxinkang tablet protects against cardiac dysfunction and
remodelling after myocardial infarction in rats through inhibition
of AMPK/mTOR-mediated autophagy. Pharm Biol. 58:321–327.
2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wang Y, Zhu S, Liu H, Wei W, Tu Y, Chen C,
Song J, Li J, Sun S, Wang C and Xu Z: Thyroxine alleviates energy
failure, prevents myocardial cell apoptosis, and protects against
doxorubicin-induced cardiac injury and cardiac dysfunction via the
LKB1/AMPK/mTOR axis in mice. Dis Markers.
2019(7420196)2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhao J, Chen D and Feng C: Troxerutin
attenuates isoproterenol-induced cardiac hypertrophy via the
LKB1/AMPK/mTOR pathway. Panminerva Med. 63:233–234. 2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Zhang J, Zhao P, Quan N, Wang L, Chen X,
Cates C, Rousselle T and Li J: The endotoxemia cardiac dysfunction
is attenuated by AMPK/mTOR signaling pathway regulating autophagy.
Biochem Biophys Res Commun. 492:520–527. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Majd S, Power JHT, Chataway TK and
Grantham HJM: A comparison of LKB1/AMPK/mTOR metabolic axis
response to global ischaemia in brain, heart, liver and kidney in a
rat model of cardiac arrest. BMC Cell Biol. 19(7)2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Qi H, Ren J, Ba L, Song C, Zhang Q, Cao Y,
Shi P, Fu B, Liu Y and Sun H: MSTN attenuates cardiac hypertrophy
through inhibition of excessive cardiac autophagy by blocking
AMPK/mTOR and miR-128/PPARγ/NF-κB. Mol Ther Nucleic Acids.
19:507–522. 2020.PubMed/NCBI View Article : Google Scholar
|