Introduction
The endothelium, which is an important part of the
vasculature, forms the inner cell lining of all blood and lymphatic
vessels in the body (1,2). Furthermore, mounting evidence suggests
that endothelial dysfunction contributes to the development of
various diseases, including cancers and cardiovascular diseases
(3-5).
Endothelial function is known to be impaired in hyperlipidemic
patients (6), and elevated levels
of plasma lipids, particularly oxidized low-density lipoprotein
(ox-LDL), have been shown to induce endothelial injuries, such as
senescence and cell death, resulting in endothelial dysfunction
(7). The mechanisms underlying
ox-LDL-induced endothelial injury have been reported in several
studies. In endothelial cells, ox-LDL primarily signals through
lectin-like ox-LDL receptor 1 (LOX-1) (8). The binding of ox-LDL to LOX-1
upregulates cytoplasmic adaptor protein TRAF3IP2 expression, which
further activates downstream signaling pathway of IKK/NF-κB p65,
resulting in apoptotic cell death (7,9,10).
Ferroptosis is a newly identified form of
non-apoptotic cell death (11)
characterized by the iron-dependent accumulation of lipid
peroxides. Glutathione peroxidase (GPx)4, one of the GPxs present
in mammals, has been shown to play an important role in reducing
lipid peroxide levels (12).
Wortmann et al (13)
observed that the loss of endothelial GPx4 combined with limited
access to vitamin E induces endothelial cell death in multiple
organs and promotes thrombus formation. Although ferroptosis has
been suggested as a possible pathway that mediates endothelial cell
death and dysfunction, whether ox-LDL plays a pathophysiological
role in endothelial cell ferroptosis has not been elucidated.
Statins, which are inhibitors of HMG-CoA reductase,
have beneficial effects in reducing serum cholesterol levels
(14). The results of recent
studies suggest that statins exert pleiotropic effects apart from
their serum lipid-lowering effects. One of the most extensively
investigated cholesterol-independent functions of statins is
cardiovascular protection (15),
where the specific cardiovascular protective effects of statins are
dose-dependent. Low doses of statins promote angiogenesis through
activation of AKT, while high doses may exert anti-angiogenic
effects and therefore inhibit tumor growth (16,17).
Moreover, statins have been reported as inducers or sensitizers of
ferroptosis in various cancer cells, including melanoma, colorectal
and prostate cancer cells (18).
However, whether statins have any effect on endothelial cell
ferroptosis remains unclear, and there is no experimental evidence
demonstrating a link between statins and the development of
endothelial cell ferroptosis.
The present study investigated whether ox-LDL
induces human endothelial cell ferroptosis and whether this effect
is mediated through affecting the expression of GPx4 and
cystine-glutamate antiporter (xCT) as well as the migration and
angiogenesis of human endothelial cells. It was also investigated
whether deferoxamine mesylate (DFOM) treatment could inhibit
ferroptosis by reversing the effects of ox-LDL on the migration and
angiogenesis of human endothelial cells and the expression of GPx4
and xCT. Furthermore, the protective effects of fluvastatin against
ox-LDL-induced ferroptosis were investigated, hoping to uncover a
novel function for fluvastatin in attenuating ox-LDL-induced
ferroptosis and improving endothelial cell dysfunction.
Materials and methods
Cell culture
Human umbilical vein cells (HUVECs) were purchased
from ScienCell Research Laboratory and cultured in endothelial
culture medium [C-22010; Miaotong (Shanghai) Biotech Co., Ltd.] at
37˚C in a humidified incubator with 5% CO2. HUVECs
cultured for 3-6 passages were used in the present study.
Tube formation assay
Before tube formation assays, HUVECs transfected
with Scramble control siRNA or siRNA specific for GPx4 or xCT (JTS
Scientific Ltd.) along with the transfection reagents of
DharmaFECT-3 (DharmaconFECT3; T-2003; Thermo Fisher Scientific,
Inc.) in reduced-serum medium (Gibco; Thermo Fisher Scientific,
Inc.; 31985070) were transfected and incubated for 24 h in
accordance with the manufacturer's protocol. HUVECs were incubated
with reagents containing scramble control siRNA or siRNA specific
for GPx4 or xCT at 37°C in 5% CO2 for 24 h.
The sequences were as follows: xCT siRNA-1, forward,
5'-GGAGGUCAUUACACAUAUATT-3' and reverse,
5'-UAUAUGUGUAAUGACCUCCTT-3'; xCT siRNA-2 forward,
5'-GCCUACUUUACGACCAUUATT-3' and reverse,
5'-UAAUGGUCGUAAAGUAGGCTT-3'; xCT siRNA-3 forward,
5'-CACCCUUUGACAAUGAUAATT-3' and reverse,
5'-UUAUCAUUGUCAAAGGGUGTT-3'; GPX4 siRNA-1 forward,
5'-CAGGGAGUAACGAAGAGAUTT-3' and reverse,
5'-AUCUCUUCGUUACUCCCUGTT-3'; GPX4 siRNA-2 forward,
5'-GACCGAAGUAAACUACACUTT-3' and reverse,
5'-AGUGUAGUUUACUUCGGUCTT-3'; GPX4 siRNA-3 forward,
5'-UGGUGAUAGAGAAGGACCUTT-3' and reverse,
5'-AGGUCCUUCUCUAUCACCATT-3'; scramble control siRNA, forward,
5'-UUCUCCGAACGUGUCACGUTT-3' and reverse,
5'-ACGUGACACGUUCGGAGAATT-3'; These siRNA were all purchased from
JTS Scientific Ltd. The transfection efficiencies of siRNA were
estimated by RT-PCR after 24 h and Western blotting after 48 h. The
effective siRNA delivery was >90% knockdown. the mRNA expression
of GPX4/xCT was reduced to <10% of its previous level after
transfecting GPX4/xCT siRNA for 24 h. Additionally, the protein
level of GPX4/xCT was reduced to <10% of its previous level
after transfecting GPX4/xCT siRNA for 48 h. GPX4 siRNA-1 and xCT
siRNA-2 have the appropriate transfection efficiency and were
carefully selected as the candidates. The mRNA expression of GPX4
was reduced to 5% of its previous level after transfecting GPX4
siRNA-1 for 24 h. Furthermore, the protein level of GPX4 was
reduced to 7% of its previous level after transfecting GPX4 siRNA-1
for 48 h. Similarly, the mRNA expression of xCT was reduced to 4%
of its previous level after transfecting xCT siRNA-2 for 24 h.
Finally, the protein level of xCT reduced to ~7% of its previous
level after transfecting xCT siRNA-2 for 48 h.
Then, 4x105 HUVECs/well were seeded in a
48-well plate that was pre-coated at 37°C for 12 h with
growth factor-reduced Matrigel (BD Biosciences; 354230). The cells
were assayed 24 h after incubation, with images acquired randomly
from three different fields per well using microscope [SOPTOP EX21;
infinity color corrected optical system with light splitting ratio
R: T=8:2; Sunny Optical Technology (Group) Company Limited;
magnification, x100]. The level of tube formation was analyzed
using ImageJ software 1.46r (National Institutes of Health). HUVEs
were divided into the following groups: i) Control; ii) ox-LDL;
iii) si-GPx4 + ox-LDL; iv) si-GPx4 + ox-LDL + DFOM; v) si-xCT +
ox-LDL; vi) si-xCT + ox-LDL + DFOM. Each group contained 8 wells
and each experiment was repeated 5 times. In separate experiments,
HUVECs were seeded in Matrigel with 100 µg/ml of ox-LDL or 125 µM
DFOM. Tube formation was observed after 24 h of incubation. In the
fluvastatin rescue experiment, HUVECs were seeded in Matrigel
supplemented with 2.5,5 or 10 µmol/l fluvastatin. Tube formation
was observed after 24 h of incubation.
MTT assay
Cell viability was assessed using the MTT assay.
Briefly, HUVECs were seeded in 96-well plates at a density of
4x105 cells per well and were incubated at
37°C for 48 h. After being treated with ox-LDL, DFOM, or
fluvastatin for 24 h, 20 µl of 5 mg/ml MTT (Sangon Biotech Co.,
Ltd.) was added to each well. Then, after incubating at 37˚C for 4
h, 100 µl of dimethyl sulfoxide was added and mixed thoroughly to
dissolve the formazan crystals, after which time the absorbance was
measured at 490 nm using a microplate reader (Awareness
Technologies, Inc.; Stat 2100).
Transwell migration assay
Cell migration was evaluated using the Transwell
migration assay. Briefly, HUVECs were seeded at 2x105
cells per well in the upper chamber of the Transwell plate with
DMEM (Jianshunbio; 66001-077) containing 1% FBS (Shanghai ExCell
Biology, Inc.; FCS500), and the lower chamber was filled with
endothelial culture medium(MT-Bio, C-22010) with 20% fetal bovine
serum (Excell Bio, FCS500). After incubating for 24 h, the
non-migrated cells were removed from the top of the filter using
cotton swabs, and cells that migrated to the bottom were fixed in
4% paraformaldehyde at room temperature for 15 min on a mild shaker
(RCK 2D 200C; Dam industry Co., Ltd.). After three washes with PBS
on the mild shaker, cells were stained with 0.01% crystal violet
(Hzhxbio; HCY124) and incubated at room temperature for 20 min.
Cells were then counted using microscope [SOPTOP EX21; infinity
color corrected optical system with light splitting ratio R:T=8:2;
Sunny Optical Technology (Group) Company Limited; magnification,
x100]. As mentioned above, each group contains 8 wells and the
experiment was repeated for 5 times.
Western blot analysis
Total protein from treated HUVECs was isolated using
RIPA lysis buffer (Beyotime Institute of Biotechnology, P0013) and
the protein concentration was determined with a Bio-Rad Protein
Assay kit (Bio-Rad Laboratories, Inc.). Then, equal amounts of
protein (50 µg) were separated by 12% SDS-PAGE. The separated
proteins were then transferred to PVDF membranes (Merck KGaA,
Germany) and blocked for 1 h at room temperature in PBS with 0.05%
Tween-20 containing 3% non-fat powdered milk. The membranes were
then incubated with primary antibodies overnight at 4˚C, followed
by an incubation with HRP-conjugated Goat Anti-rabbit IgG secondary
antibodies (1:3,000; A0181; Beyotime Institute of Biotechnology) at
room temperature for 2 h. Subsequently, the protein bands were
visualized with an enhanced chemiluminescence detection system
(Amersham Cytiva, RPN2232). The following primary antibodies were
used: 4-hydroxynonenal (4-HNE; 1:2,000; ab46544; Abcam), TNF-α
(1:500; WL01581; Wanleibio; Co., Ltd.), IL-1α (1:2,000; ab134908;
Abcam), GPx4 (1:1,000; 14432-1-AP; ProteinTech Group, Inc.), xCT
(1:1,000; 26864-1-AP; ProteinTech Group, Inc.);
Caspase-3/cleaved-caspase3 (1:500; WL02117; Wanleibio; Co., Ltd.),
Bax (1:1,000; WL01637; Wanleibio; Co., Ltd.), Bcl-2 (1:1,000;
WL01556; Wanleibio; Co., Ltd.), β-actin (1:2,000; WL01845
Wanleibio; Co., Ltd.).
Statistical analysis
All quantitative data are presented as the means ±
standard deviation (SD). Statistical analyses were performed using
SPSS 22.0 (IBM Corp.) using the unpaired Student's t-test or
one-way ANOVA followed by a Fisher's post hoc comparison test.
Two-sided tests were used throughout this study, and a P<0.05
was considered to indicate statistically significant
difference.
Results
Ox-LDL promotes endothelial
dysfunction by induction of ferroptosis
Accumulating evidence has demonstrated that
endothelial dysfunction is a crucial and early step in the
development of cardiovascular diseases, including atherosclerosis
(5). Increased local and systemic
levels of ox-LDL have been shown to induce endothelial injury and
contribute to the development and progression of atherosclerosis
(19). Wortmann et al
observed that GPx4 deficiency combined with a vitamin E-depleted
diet caused vascular dysfunction and early death in mice (13). The present study investigated
whether ox-LDL-induced endothelial cell ferroptosis occurs through
the regulation of GPx4 activity (Fig.
1). It was observed that treatment with ox-LDL (100 µg/ml) for
24 h significantly induced the expression of pro-inflammatory
cytokines, including TNF-α and IL-1α (Fig. 1A, E
and F). In addition, the migration
and angiogenesis of human endothelial cells were examined after
ox-LDL treatment. The data revealed that ox-LDL strongly decreased
cell migration in Transwell assays (Fig. 1B) and disrupted tube formation in
vitro (Fig. 1C), indicating
that ox-LDL induces endothelial cell dysfunction.
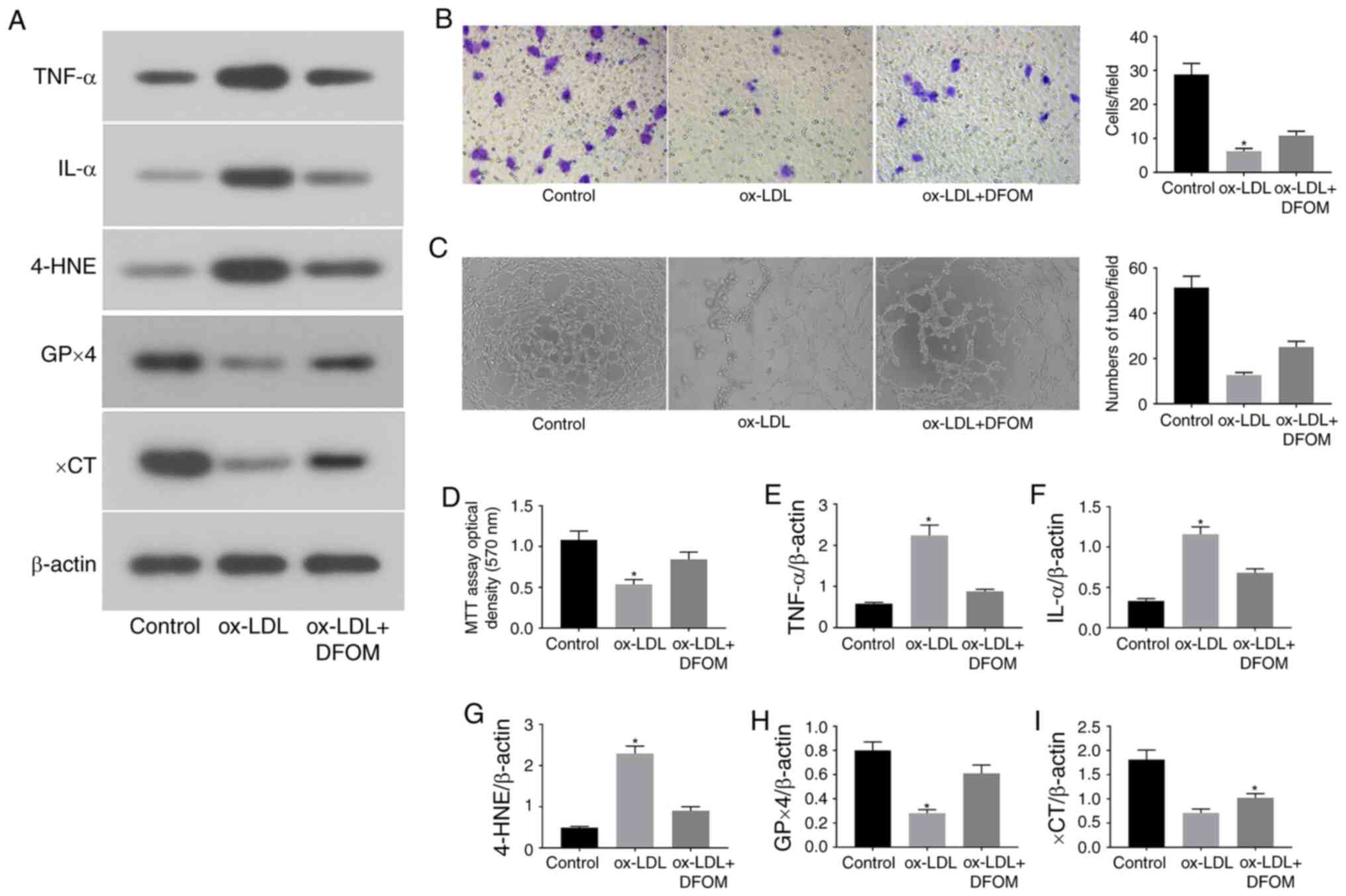 | Figure 1ox-LDL promotes endothelial cell
dysfunction via induction of ferroptosis. (A) Immunoblots of TNF-α,
IL-α, 4-HNE, GPx4 and xCT in the normal (control), ox-LDL and
ox-LDL + DFOM groups. (B) Representative Transwell migration assay
images of HUVECs treated with ox-LDL and ox-LDL + DFOM
(magnification, x10). (C) Representative tube formation assay
images of HUVECs treated with PBS, 100 µg/ml ox-LDL and 125 µM DFOM
(magnification, x10). (D) Analysis of HUVEC viability using the MTT
assay after ox-LDL treatment. Columns, means of three experiments;
bars, SD. Histograms of (E) relative TNF-α/β-actin levels, (F)
relative IL-α/β-actin levels, (G) relative 4-HNE/β-actin levels,
(H) relative GPx4/β-actin levels and (I) relative xCT/β-actin
levels. *P<0.05 vs. control group. ox-LDL, oxidized
low-density lipoprotein; GPx4, glutathione peroxidase 4; xCT,
cystine-glutamate antiporter; 4-HNE, 4-hydroxynonenal; DFOM,
deferoxamine mesylate; HUVEC, human umbilical vein cell. |
To further assess whether ox-LDL-induced endothelial
dysfunction is mediated by ferroptosis, the expression of central
ferroptosis regulators, including GPx4, xCT and 4-HNE, was
evaluated after ox-LDL treatment. ox-LDL markedly reduced the
expression of GPx4 and xCT, while increasing the expression of
4-HNE in human endothelial cells (Fig.
1A and G-I). Subsequently, the
proliferation of human endothelial cells was assessed by MTT assay.
As shown in Fig. 1D, ox-LDL reduced
the proliferation of endothelial cells by 49.3%. DFOM, an iron
chelator, has been shown to reduce unbound iron levels and inhibit
the production of ROS and the occurrence of ferroptosis (11). It was observed that pre-treatment
with DFOM (125 µM) increased the expression of GPx4 and xCT, while
decreasing that of 4-HNE after ox-LDL treatment compared to that
observed in the vehicle control (Fig.
1A and G-I). In addition, the
ox-LDL-mediated inhibition of endothelial cell proliferation was
also rescued by DFOM (Fig. 1D),
indicating that DFOM inhibits ox-LDL-induced ferroptosis in human
endothelial cells. The data further demonstrated that inhibition of
ferroptosis by DFOM reduced ox-LDL-induced pro-inflammatory
cytokine production (Fig. 1A,
E and F) and increased ox-LDL-inhibited
endothelial cell migration (Fig.
1B) and angiogenesis (Fig.
1C).
RNAi-mediated GPx4 and xCT knockdown
(KD) promotes ox-LDL-induced ferroptosis in human endothelial
cells
Previous studies have demonstrated that inhibition
of GPx4 and xCT activity induces ferroptosis in cancer cells
(20,21). The data of the present study
demonstrated that ox-LDL significantly reduced CPx4 and xCT
expression and induced ferroptosis in endothelial cells (Fig. 1A). To further confirm the role of
GPx4 and xCT in the ox-LDL-mediated induction of endothelial cell
ferroptosis, GPx4 and xCT expression was knocked down by siRNA
transfection in human endothelial cells (Fig. 2). The results presented in Fig. 2A show successful Knockdown (KD) of
GPx4 and xCT in human endothelial cells. It was observed that KD of
either GPx4 or xCT increased pro-inflammatory cytokine production
after ox-LDL treatment in endothelial cells compared to that
observed in endothelial cells transfected with scramble control
siRNA (Fig. 2A, E and F).
Cell proliferation was further reduced after KD of either GPx4 or
xCT in ox-LDL-treated endothelial cells (Fig. 2D). Additionally, decreased
endothelial cell migration (Fig.
2B) and tube formation (Fig.
2C) were observed in ox-LDL-treated endothelial cells after KD
of either GPx4 or xCT. Interestingly, the application of DFOM (125
µM) attenuated the increased the production of pro-inflammatory
cytokines (Fig. 2A, E and F)
and decreased cell proliferation (Fig.
2D), migration (Fig. 2B) and
angiogenesis (Fig. 2C) after KD of
GPx4 or xCT in ox-LDL-treated endothelial cells. These data
demonstrate that ox-LDL-induced endothelial cell ferroptosis is
mediated by GPx4 and xCT activity.
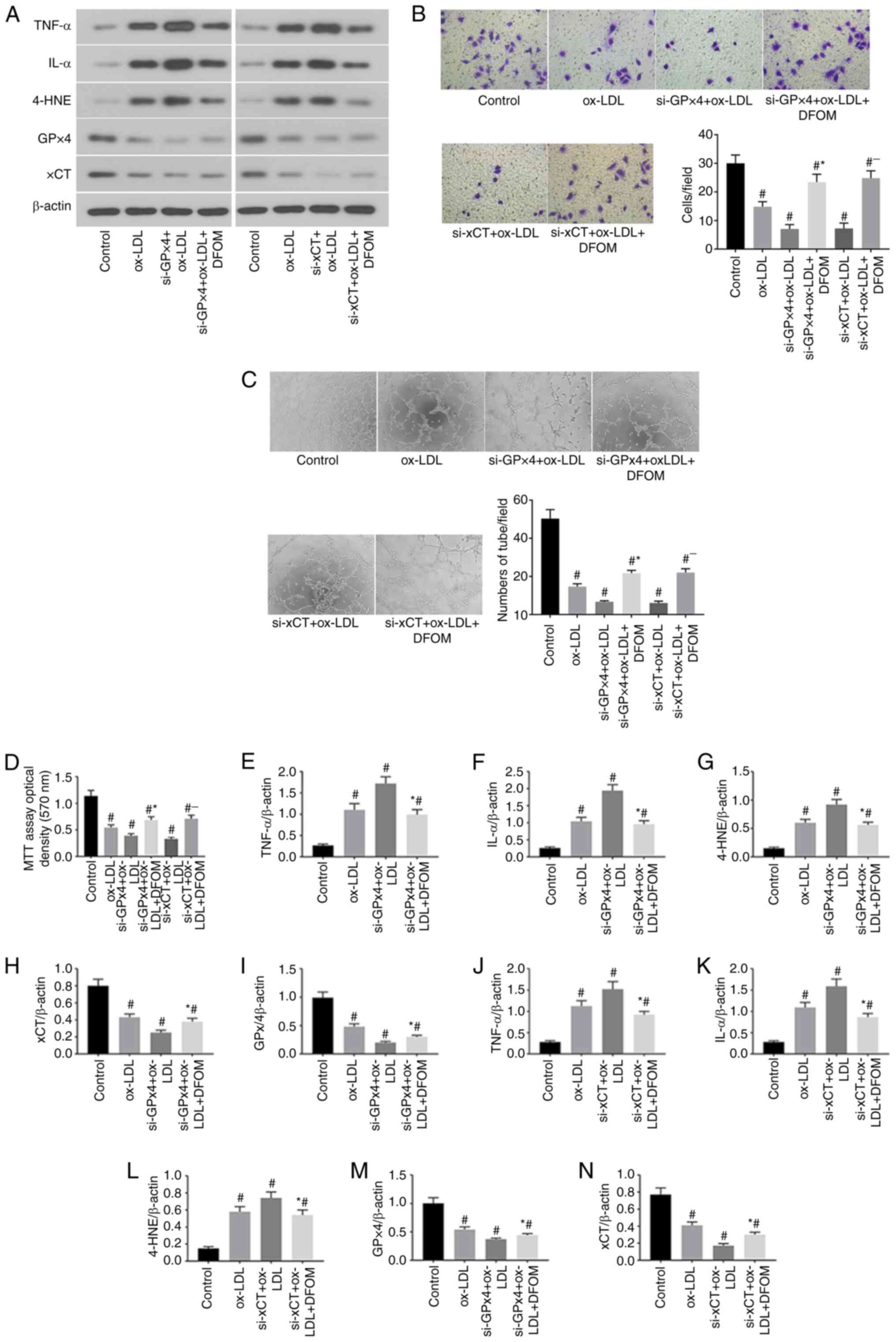 | Figure 2RNAi-mediated GPx4 and xCT knockdown
promotes ox-LDL-induced ferroptosis in human endothelial cells. (A)
Immunoblots of TNF-α, IL-α, 4-HNE, GPx4 and xCT in the normal
(control), ox-LDL, si-GPx4 + ox-LDL, si-GPx4 + ox-LDL + DFOM,
si-xCT + ox-LDL and si-xCT + ox-LDL + DFOM treatment groups. (B)
Representative Transwell migration assay images of HUVECs treated
with ox-LDL, si-GPx4 + ox-LDL, si-GPx4 + ox-LDL + DFOM, si-xCT +
ox-LDL and si-xCT + ox-LDL + DFOM (magnification, x10).
#P<0.05 vs. control group; *P<0.05 vs.
si-GPx4 + ox-LDL; -P<0.05 vs. si-xCT + ox-LDL. (C)
Representative images showing HUVEC tube formation in the presence
of PBS (control) or 100 µg/ml ox-LDL,si-GPx4 + 100 µg/ml ox-LDL,
si-GPx4 + 100 µg/ml ox-LDL + 125 µM DFOM, si-xCT + 100 µg/ml ox-LDL
or si-xCT + 100 µg/ml ox-LDL + 125 µM DFOM (all magnifications,
x10). #P<0.05 vs. control group,
*P<0.05 vs. si-GPx4 + ox-LDL, -P<0.05
vs. si-xCT + ox-LDL. (D) Analysis of cell viability using the MTT
assay in HUVECs after ox-LDL, si-GPx4 + ox-LDL, si-GPx4 + ox-LDL +
DFOM, si-xCT + ox-LDL and si-xCT + ox-LDL + DFOM treatments.
Columns, mean of three experiments; bars, SD. #P<0.05
vs. control group, *P<0.05 vs. si-GPx4 + ox-LDL,
-P<0.05 vs. si-xCT + ox-LDL. (E) Histogram of
relative TNF-α/β-actin levels. #P<0.05 vs. control
group, *P<0.05 vs. si-GPx4 + ox-LDL. (F) Histogram of
relative IL-α/β-actin levels. #P<0.05 vs. control
group, *P<0.05 vs. si-GPx4 + ox-LDL. (G) Histogram of
relative 4-HNE/β-actin levels. #P<0.05 vs. control
group, *P<0.05 vs. si-GPx4 + ox-LDL. (H) Histogram of
relative xCT/β-actin levels. #P<0.05 vs. control
group, *P<0.05 vs. si-GPx4 + ox-LDL. (I) Histogram of
relative GPx4/β-actin levels. #P<0.05 vs. control
group, *P<0.05 vs. si-GPx4 + ox-LDL. (J) Histogram of
relative TNF-α/β-actin levels. #P<0.05 vs. control
group, *P<0.05 vs. si-xCT + ox-LDL. (K) Histogram of
relative IL-α/β-actin levels. #P<0.05 vs. control
group, *P<0.05 vs. si-xCT + ox-LDL. (L) Histogram of
relative 4-HNE/β-actin levels. #P<0.05 vs. control
group, *P<0.05 vs. si-xCT + ox-LDL. (M) Histogram of
relative GPx4/β-actin levels. #P<0.05 vs. control
group, *P<0.05 vs. si-xCT + ox-LDL. (N) Histogram of
relative xCT/β-actin levels. #P<0.05 vs. control
group, *P<0.05 vs. si-xCT + ox-LDL. ox-LDL, oxidized
low-density lipoprotein; GPx4, glutathione peroxidase 4; xCT,
cystine-glutamate antiporter; 4-HNE, 4-hydroxynonenal; DFOM,
deferoxamine mesylate; HUVEC, human umbilical vein cell. |
Fluvastatin attenuates ox-LDL-induced
endothelial cell dysfunction and ferroptosis
The results of previous studies revealed that
fluvastatin can reverse endothelial dysfunction in arthritis
(22). To investigate whether
fluvastatin could rescue ox-LDL-induced ferroptosis in endothelial
cells, ox-LDL-induced human endothelial cells were treated with
different concentrations of fluvastatin (Fig. 3). It was observed that fluvastatin
ameliorated the ox-LDL-mediated decrease in GPx4 and xCT expression
and the increased 4-HNE expression in a dose-dependent manner
(Fig. 3A, E, F and
G). Fluvastatin also restored
ox-LDL-inhibited cell proliferation (Fig. 3B) and lowered the ox-LDL-induced
increase in inflammatory cytokine levels (Fig. 3C and D), suggesting that fluvastatin attenuated
ox-LDL-induced endothelial ferroptosis. Of note, fluvastatin
treatment rescued the ox-LDL-mediated inhibition of endothelial
cell migration (Fig. 3H) and
angiogenesis (Fig. 3I). Taken
together, these data indicated that fluvastatin may normalized
ox-LDL-induced endothelial dysfunction via inhibition of
ferroptosis.
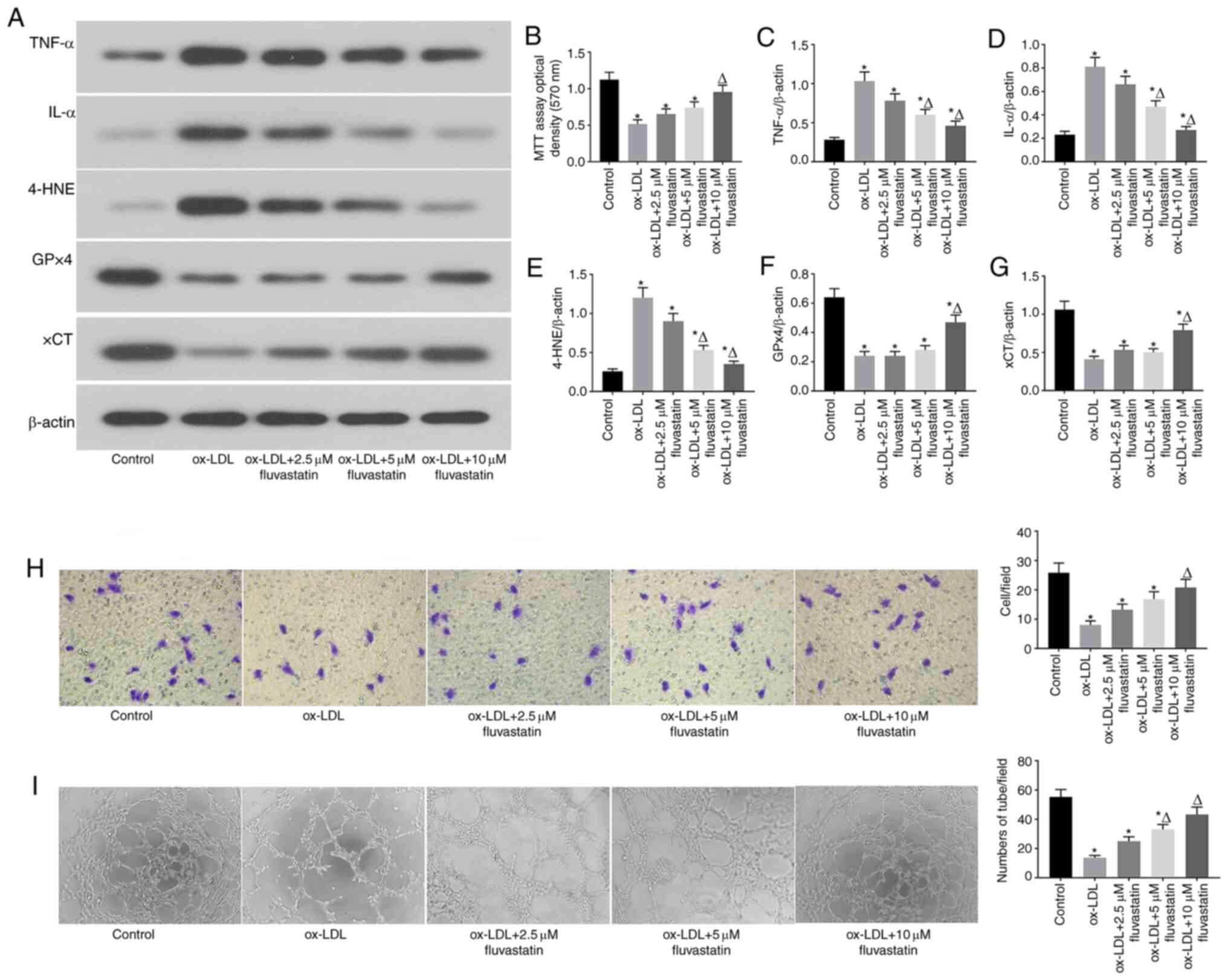 | Figure 3Fluvastatin attenuates ox-LDL-induced
endothelial cell dysfunction and ferroptosis. (A) Immunoblots of
TNF-α, IL-α, 4-HNE, GPx4 and xCT in the normal, ox-LDL, ox-LDL +
2.5 µM fluvastatin, ox-LDL + 5 µM fluvastatin, ox-LDL + 10 µM
fluvastatin treatment groups. (B) Analysis of cell viability using
the MTT assay in HUVECs after ox-LDL, ox-LDL + 2.5 µM fluvastatin,
ox-LDL + 5 µM fluvastatin and ox-LDL + 10 µM fluvastatin
treatments. Columns, means of three experiments; bars, SD.
*P<0.05 vs. control group, ΔP<0.05 vs.
ox-LDL group. (C) Histogram of relative TNF-α/β-actin levels.
*P<0.05 vs. control group, ΔP<0.05 vs.
ox-LDL group. (D) Histogram of relative IL-α/β-actin levels.
*P<0.05 vs. control group, ΔP<0.05 vs.
ox-LDL group. (E) Histogram of relative 4-HNE/β-actin levels.
*P<0.05 vs. control group, ΔP<0.05 vs.
ox-LDL group. (F) Histogram of relative GPx4/β-actin levels.
*P<0.05 vs. control group, ΔP<0.05 vs.
ox-LDL group. (G) Histogram of relative xCT/β-actin
levels.*P<0.05 vs. control group,
ΔP<0.05 vs. ox-LDL group. (H) Representative
Transwell migration assay images of HUVECs treated with ox-LDL,
ox-LDL + 2.5 µM fluvastatin, ox-LDL + 5 µM fluvastatin, or ox-LDL +
10 µM fluvastatin. *P<0.05 vs. control group,
ΔP<0.05 vs. ox-LDL group (magnification, 100x). (I)
Representative tube formation assay images of HUVEC in the presence
of PBS (control) or with 100 µg/ml ox-LDL, 100 µg/ml ox-LDL + 2.5
µM fluvastatin, 100 µg/ml ox-LDL + 5 µM fluvastatin, or 100 µg/ml
ox-LDL + 10 µM fluvastatin (magnification, 100x).
*P<0.05 vs. control group, ΔP<0.05 vs.
ox-LDL group. ox-LDL, oxidized low-density lipoprotein; GPx4,
glutathione peroxidase 4; xCT, cystine-glutamate antiporter; 4-HNE,
4-hydroxynonenal; HUVEC, human umbilical vein cell. |
GPx4 and xCT are required for the
fluvastatin-mediated attenuation of ferroptosis in human
endothelial cells
Subsequently, it was assessed whether the
fluvastatin-mediated attenuation of ferroptosis in ox-LDL-treated
endothelial cells requires GPx4 and xCT (Fig. 4). To this end, GPx4 and xCT were
knocked down in endothelial cells by transfection with specific
siRNA (cells transfected with scramble siRNA were used as
controls). It was observed that KD of either GPx4 or xCT partially
blocked the fluvastatin-mediated attenuation of pro-inflammatory
cytokine production in ox-LDL-treated endothelial cells (Fig. 4A, C
and D). The KD of endothelial GPx4
or xCT also attenuated the protective effects of fluvastatin on
cell proliferation (Fig. 4B),
migration (Fig. 4H) and
angiogenesis (Fig. 4I).
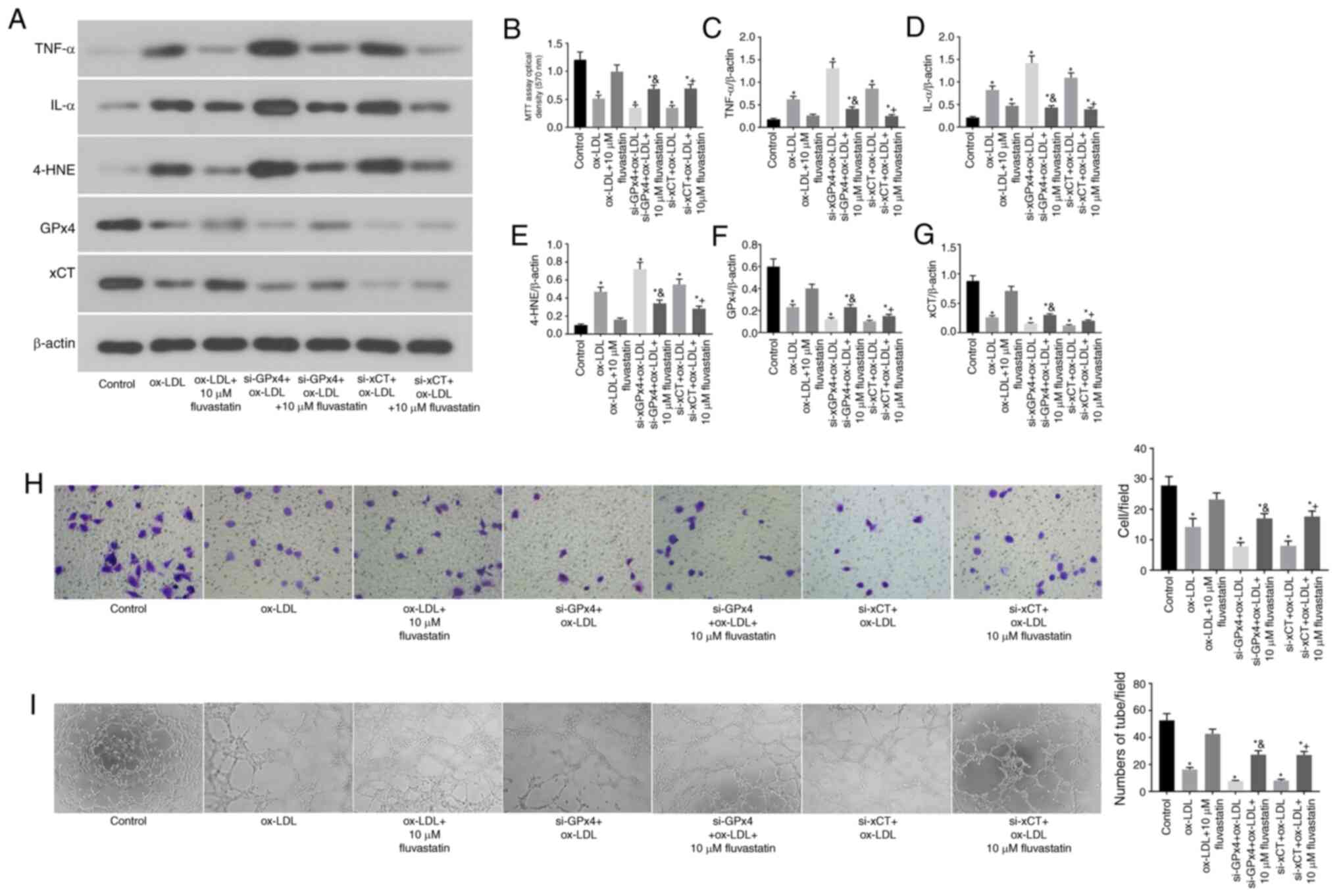 | Figure 4GPx4 and xCT are required for the
fluvastatin-mediated attenuation of ferroptosis in human
endothelial cells. (A) Immunoblots of TNF-α, IL-α, 4-HNE, GPx4 and
xCT in the normal, ox-LDL, ox-LDL + 10 µM fluvastatin, si-GPx4 +
ox-LDL, si-GPx4 + ox-LDL + 10 µM fluvastatin, si-xCT + ox-LDL and
si-xCT + ox-LDL + 10 µM fluvastatin treatment groups. (B) Analysis
of cell viability using the MTT assay in HUVECs after ox-LDL,
ox-LDL + 10 µM fluvastatin, si-GPx4 + ox-LDL, si-GPx4 + ox-LDL + 10
µM fluvastatin, si-xCT + ox-LDL or si-xCT + ox-LDL + 10 µM
fluvastatin treatments. Columns, means of three experiments; bars,
SD. *P<0.05 vs. control group,
&P<0.05 vs. si-GPx4 + ox-LDL,
+P<0.05 vs. si-xCT + ox-LDL. (C) Histogram of
relative TNF-α/β-actin levels. *P<0.05 vs. control
group, &P<0.05 vs. si-GPx4 + ox-LDL,
+P<0.05 vs. si-xCT + ox-LDL. (D) Histogram of
relative IL-α/β-actin levels. *P<0.05 vs. control
group, &P<0.05 vs. si-GPx4 + ox-LDL,
+P<0.05 vs. si-xCT + ox-LDL. (E) Histogram of
relative 4-HNE/β-actin levels. *P<0.05 vs. control
group, &P<0.05 vs. si-GPx4 + ox-LDL,
+P<0.05 vs. si-xCT + ox-LDL. (F) Histogram of
relative GPx4/β-actin levels. *P<0.05 vs. control
group, &P<0.05 vs. si-GPx4 + ox-LDL,
+P<0.05 vs. si-xCT + ox-LDL. (G) Histogram of
relative xCT/β-actin levels. *P<0.05 vs. control
group, &P<0.05 vs. si-GPx4 + ox-LDL,
+P<0.05 vs. si-xCT + ox-LDL. (H) Representative
Transwell migration assay images of HUVECs treated with ox-LDL,
ox-LDL + 10 µM fluvastatin, si-GPx4 + ox-LDL, si-GPx4 + ox-LDL + 10
µM fluvastatin, si-xCT + ox-LDL and si-xCT + ox-LDL + 10 µM
fluvastatin. *P<0.05 vs. control group,
&P<0.05 vs. si-GPx4 + ox-LDL,
+P<0.05 vs. si-xCT + ox-LDL (magnification, 100x).
(I) Representative tube formation assay images of HUVECs in the
presence of PBS or with 100 µg/ml ox-LDL, 100 µg/ml ox-LDL + 10 µM
fluvastatin, si-GPx4 + 100 µg/ml ox-LDL, si-GPx4 + 100 µg/ml ox-LDL
+ 10 µM fluvastatin, si-xCT + 100 µg/ml ox-LDL or si-xCT + 100
µg/ml ox-LDL + 10 µM fluvastatin (magnification, 100x).
*P<0.05 vs. control group, &P<0.05
vs. si-GPx4 + ox-LDL, +P<0.05 vs. si-xCT + ox-LDL.
ox-LDL, oxidized low-density lipoprotein; GPx4, glutathione
peroxidase 4; xCT, cystine-glutamate antiporter; 4-HNE,
4-hydroxynonenal; HUVEC, human umbilical vein cell. |
Discussion
The endothelium is one of the largest organs in the
body by area (23). Furthermore,
the endothelium interacts with nearly every organ and system in the
body, and dysregulation of endothelial function is implicated in a
diverse range of diseases, including cancer, stroke and
cardiovascular diseases (24-26).
Moreover, the endothelium functions as one of the first lines of
defense of the immune system by combating the invasion of microbes
and endogenous substances (27). In
the context of inflammatory diseases, such as the development of
atherosclerotic lesions, endothelial cells are activated by ox-LDL
and immune cell-derived cytokines, which is characterized by an
increased expression of intercellular adhesion molecule-1,
chemokines, cytokines and vascular cell adhesion molecule-1
(28,29). These changes in endothelial cells
further promote the formation of atherosclerotic lesions. In the
present study, it was observed that treatment with ox-LDL (100
µg/ml) significantly induced the production of pro-inflammatory
cytokines, including TNF-α and IL-1α. The results of a previous
study by Valente et al (7)
suggested that ox-LDL may induce endothelial cell dysfunction and
apoptosis via activation of TRAF3 interacting protein 2 (TRAF3IP2).
It was observed that ox-LDL reduced the migration and angiogenesis
of endothelial cells, indicating that ox-LDL caused endothelial
injury. Additionally, MTT assays demonstrated that ox-LDL
significantly inhibited endothelial cell proliferation, prompting
further investigation of whether ox-LDL may promote endothelial
cell death.
Ferroptosis, an iron-dependent form of regulated
cell death, is distinct from apoptosis, and accumulating evidence
indicates that ferroptosis is a significant type of cell death
observed in various cell populations, including cardiomyocytes
(18,30). In the present study, it was observed
that ox-LDL significantly inhibited GPx4 expression in human
endothelial cells. Inhibited GPx4 activity decreases the reduction
of lipid peroxides, which is one of the major pathways that lead to
ferroptosis (31). The expression
of xCT, a specific cys2/glutamate antiporter that plays a role in
negatively regulating ferroptosis, is also inhibited by ox-LDL
(32). These findings suggest that
ox-LDL-induced endothelial dysfunction occurs by promoting
ferroptosis. To further confirm these results, ferroptosis in
ox-LDL-treated endothelial cells was inhibited using the
ferroptosis inhibitor DFOM, which blunted the ox-LDL-mediated
decrease in GPx4 and xCT expression in endothelial cells. In
addition, DFOM attenuated the ox-LDL-mediated induction of cytokine
production and rescued the decreased proliferation, migration and
angiogenesis of endothelial cells treated with ox-LDL. Moreover, KD
of GPx4 and xCT by transfecting endothelial cells with specific
siRNAs aggravated the ox-LDL-induced ferroptosis and dysfunction of
endothelial cells.
Fluvastatin has been used as a drug to lower serum
cholesterol levels (33). Haruna
et al (22) showed that
fluvastatin exerted protective effects against endothelial
dysfunction by reducing the levels of p22phox mRNA. To investigate
whether fluvastatin protects against endothelial ferroptosis,
endothelial cells were treated with low doses of fluvastatin (2.5,
5 and 10 µM) in the presence of ox-LDL. The data demonstrated that
10 µM fluvastatin significantly blunted ox-LDL-induced endothelial
cell dysfunction. Importantly, fluvastatin also reversed the
ox-LDL-mediated decrease in GPx4 and xCT expression. Combined with
its protective effect on cell proliferation, fluvastatin inhibited
ox-LDL-induced ferroptosis in human endothelial cells. The
expression of GPx4 and xCT was then knocked down in endothelial
cells by siRNA transfection. It was observed that the suppression
of GPx4 and xCT partly blocked the protective effects of
fluvastatin against the ox-LDL-mediated decrease in endothelial
cell proliferation, migration and angiogenesis, which further
confirmed that fluvastatin functions by regulating GPx4 and xCT to
inhibit endothelial cell dysfunction and ferroptosis.
Bioccaa et al (10) demonstrated that, in endothelial
cells, ox-LDL functions through lectin-like ox-LDL receptor 1
(LOX-1), and all tested statins (atorvastatin, fluvastatin,
lovastatin, pitavastatin and pravastatin) were able to displace the
binding of ox-LDL to LOX-1 by directly interacting with LOX-1,
leading to a significant loss of ox-LDL-induced LOX-1 function,
including ox-LDL-induced ferroptosis. Based on the results of this
previous study, it was inferred that the underlying mechanism
through which fluvastatin alleviates endothelial cell ferroptosis
involves counteracting the effects of ox-LDL by disrupting the
interaction of ox-LDL with the C-type lectin-like recognition
domain of LOX-1(10). As all
statins can disrupt the binding of ox-LDL to the C-type lectin-like
recognition domain of LOX-1, they all have the same effects of
alleviating ox-LDL-induced endothelial ferroptosis (10). Previous studies have demonstrated
that ox-LDL does not interact with xCT and GPx4 directly, but that
it rather acts via LOX-1 (34-36).
Similarly, all tested statins (atorvastatin, fluvastatin,
lovastatin, pitavastatin and pravastatin) did not directly interact
with xCT and GPx4 to improve ferroptosis (10), but protected endothelial cells from
ferroptosis by disrupting the interaction of ox-LDL with LOX-1.
Therefore, it was observed in the present study that fluvastatin
sequentially activated GPx4 and xCT indirectly.
Recently, a number of studies reported various
mechanisms through which statins modulate ox-LDL toxicity (10). For example, in endothelial cells,
ox-LDL functions via binding LOX-1, which upregulates the
expression of the cytoplasmic adaptor protein TRAF3IP2, further
activating the downstream signaling pathway of IKK/NF-κB
p65(10), resulting in endothelial
dysfunction and endothelial cell death, as well as impaired
vasorelaxation (7,9,10).
Fluvastatin can inhibit this ox-LDL-mediated toxicity by displacing
the binding of ox-LDL to LOX-1(10). Previous studies have also
demonstrated that statins target the 3-hydroxy-3-methylglutaryl
(HMG)-CoA/mevalonate/isopentenyl-pp/TRIT1 pathways to alleviate
ferroptosis by inhibiting HMG-CoA reductase (37). In addition, fluvastatin can inhibit
ferroptosis induced by erastin and RSL3 by inhibiting LOX-1
(10,38).
The present study further demonstrated that ox-LDL
promotes endothelial cell ferroptosis by inhibiting GPx4 and xCT,
and that fluvastatin may protect endothelial cells from
ox-LDL-induced endothelial ferroptosis by counteracting the
ox-LDL-induced inhibition of GPx4 and xCT expression. These
findings revealed a new mechanism by which statins modulate ox-LDL
toxicity in addition to other well-known mechanisms. Furthermore,
Yu et al (32) and Friedmann
Angeli et al (37) reported
that statins inhibited HMG-CoA reductase, thereby regulating the
activity of GPX4. Sui et al (31) found that inhibiting GPX4 may lead to
ferroptosis by decreasing the reduction of lipid peroxide levels.
Therefore, fluvastatin can alleviate ferroptosis via inhibiting
HMG-CoA reductase to regulate GPX4, the function of which is also
indirect.
The results of the present study demonstrated that
fluvastatin exerts potent protective effects against ox-LDL-induced
endothelial cell dysfunction through regulation of GPx4 and xCT,
providing a scientific rationale for the clinical use of
fluvastatin in atherosclerosis. Importantly, our data indicate that
statins may have clinical benefits in patients beyond their lipid
level-lowering properties, by improving endothelial cell function.
However, supporting data from pre-clinical and clinical studies are
required to confirm these hypotheses.
A limitation of the present study was that it
primarily focused on the mechanisms through which fluvastatin
regulates HUVC ferroptosis in vitro. However, in vivo
experiments could further elucidate the function of fluvastatin in
endothelial ferroptosis.
In the future, in vivo studies will aim to
use Cre-LoxP technology to generate transgenic mice with vascular
endothelial-specific knockout of GPx4/xCT and establish an ox-LDL
overexpression animal model through high-fat diet.
Acknowledgements
Not applicable.
Funding
Funding: The present study was funded by Shaanxi Social
Development Funding (grant no. 2017SF-134) and Shaanxi Nature
Science Funding (grant no. 2020JQ-553).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JD, QL, CL, EX, YT and ZC designed the experiments;
analyzed and interpreted the data; and drafted the manuscript. JD,
QL, CL, LD, EX, YT, ZC and NY were involved in the data acquisition
and analysis. All authors revised the manuscript critically for
important intellectual content and approved the final version to be
published. JD is responsible for the integrity of the work as a
whole. JD, QL, CL, LD, EX, YT and ZC confirm the authenticity of
all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Khaddaj Mallat R, Mathew John CM, Kendrick
DJ and Braun AP: The vascular endothelium: A regulator of arterial
tone and interface for the immune system. Crit Rev Clin Lab Sci.
54:458–470. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Rajendran P, Rengarajan T, Thangavel J,
Nishigaki Y, Sakthisekaran D, Sethi G and Nishigaki I: The vascular
endothelium and human diseases. Int J Biol Sci. 9:1057–1069.
2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Beerepoot LV, Mehra N, Vermaat JS,
Zonnenberg BA, Gebbink MF and Voest EE: Increased levels of viable
circulating endothelial cells are an indicator of progressive
disease in cancer patients. Ann Oncol. 15:139–145. 2004.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Furstenberger G, von Moos R, Lucas R,
Thürlimann B, Senn HJ, Hamacher J and Boneberg EM: Circulating
endothelial cells and angiogenic serum factors during neoadjuvant
chemotherapy of primary breast cancer. Br J Cancer. 94:524–531.
2006.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Park KH and Park WJ: Endothelial
dysfunction: Clinical implications in cardiovascular disease and
therapeutic approaches. J Korean Med Sci. 30:1213–1225.
2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Li TB, Zhang YZ, Liu WQ, Zhang JJ, Peng J,
Luo XJ and Ma QL: Correlation between NADPH oxidase-mediated
oxidative stress and dysfunction of endothelial progenitor cell in
hyperlipidemic patients. Korean J Intern Med. 33:313–322.
2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Valente AJ, Irimpen AM, Siebenlist U and
Chandrasekar B: OxLDL induces endothelial dysfunction and death via
TRAF3IP2: Inhibition by HDL3 and AMPK activators. Free Radic Biol
Med. 70:117–128. 2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Morawietz H: LOX-1 receptor as a novel
target in endothelial dysfunction and atherosclerosis. Dtsch Med
Wochenschr. 135:308–312. 2010.PubMed/NCBI View Article : Google Scholar : (In German).
|
|
9
|
Wahyudi S and Sargowo D: Green tea
polyphenols inhibit oxidized LDL-induced NF-KB activation in human
umbilical vein endothelial cells. Acta Med Indones. 39:66–70.
2007.PubMed/NCBI
|
|
10
|
Biocca S, Iacovelli F, Matarazzo S,
Vindigni G, Oteri F, Desideri A and Falconi M: Molecular mechanism
of statin-mediated LOX-1 inhibition. Cell Cycle. 14:1583–1595.
2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao
N, Sun B and Wang G: Ferroptosis: Past, present and future. Cell
Death Dis. 11(88)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Maiorino M, Conrad M and Ursini F: GPx4,
lipid peroxidation, and cell death: Discoveries, rediscoveries, and
open issues. Antioxid Redox Signal. 29:61–74. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wortmann M, Schneider M, Pircher J,
Hellfritsch J, Aichler M, Vegi N, Kölle P, Kuhlencordt P, Walch A,
Pohl U, et al: Combined deficiency in glutathione peroxidase 4 and
vitamin E causes multiorgan thrombus formation and early death in
mice. Circ Res. 113:408–417. 2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ridker PM, Mora S, Rose L and Group JTS:
Percent reduction in LDL cholesterol following high-intensity
statin therapy: Potential implications for guidelines and for the
prescription of emerging lipid-lowering agents. Eur Heart J.
37:1373–1379. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Correction to: Pleiotropic effects of
statins on the cardiovascular system. Circ Res. 123(e20)2018.
|
|
16
|
Wang J, Tokoro T, Matsui K, Higa S and
Kitajima I: Pitavastatin at low dose activates endothelial nitric
oxide synthase through PI3K-AKT pathway in endothelial cells. Life
Sci. 76:2257–2268. 2005.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Weis M, Heeschen C, Glassford AJ and Cooke
JP: Statins have biphasic effects on angiogenesis. Circulation.
105:739–745. 2002.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285.
2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Di Pietro N, Formoso G and Pandolfi A:
Physiology and pathophysiology of oxLDL uptake by vascular wall
cells in atherosclerosis. Vascul Pharmacol. 84:1–7. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wang Z, Ding Y, Wang X, Lu S, Wang C, He
C, Wang L, Piao M, Chi G, Luo Y and Ge P: Pseudolaric acid B
triggers ferroptosis in glioma cells via activation of Nox4 and
inhibition of xCT. Cancer Lett. 428:21–33. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Haruna Y, Morita Y, Yada T, Satoh M, Fox
DA and Kashihara N: Fluvastatin reverses endothelial dysfunction
and increased vascular oxidative stress in rat adjuvant-induced
arthritis. Arthritis Rheum. 56:1827–1835. 2007.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ait-Oufella H, Maury E, Lehoux S, Guidet B
and Offenstadt G: The endothelium: Physiological functions and role
in microcirculatory failure during severe sepsis. Intensive Care
Med. 36:1286–1298. 2010.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Cosentino F, Rubattu S, Savoia C,
Venturelli V, Pagannonne E and Volpe M: Endothelial dysfunction and
stroke. J Cardiovasc Pharmacol. 38 (Suppl 2):S75–S78.
2001.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Franses JW, Drosu NC, Gibson WJ, Chitalia
VC and Edelman ER: Dysfunctional endothelial cells directly
stimulate cancer inflammation and metastasis. Int J Cancer.
133:1334–1344. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Widmer RJ and Lerman A: Endothelial
dysfunction and cardiovascular disease. Glob Cardiol Sci Pract.
2014:291–308. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Young MR: Endothelial cells in the eyes of
an immunologist. Cancer Immunol Immunother. 61:1609–1616.
2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Stroka KM, Levitan I and Aranda-Espinoza
H: OxLDL and substrate stiffness promote neutrophil transmigration
by enhanced endothelial cell contractility and ICAM-1. J Biomech.
45:1828–1834. 2012.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Feng Y, Cai ZR, Tang Y, Hu G, Lu J, He D
and Wang S: TLR4/NF-κB signaling pathway-mediated and oxLDL-induced
up-regulation of LOX-1, MCP-1, and VCAM-1 expressions in human
umbilical vein endothelial cells. Genet Mol Res. 13:680–695.
2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Fang X, Wang H, Han D, Xie E, Yang X, Wei
J, Gu S, Gao F, Zhu N, Yin X, et al: Ferroptosis as a target for
protection against cardiomyopathy. Proc Natl Acad Sci USA.
116:2672–2680. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Sui X, Zhang R, Liu S, Duan T, Zhai L,
Zhang M, Han X, Xiang Y, Huang X, Lin H and Xie T: RSL3 drives
ferroptosis through GPX4 inactivation and ROS production in
colorectal cancer. Front Pharmacol. 9(1371)2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yu H, Guo P, Xie X, Wang Y and Chen G:
Ferroptosis, a new form of cell death, and its relationships with
tumourous diseases. J Cell Mol Med. 21:648–657. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Adams SP, Sekhon SS, Tsang M and Wright
JM: Fluvastatin for lowering lipids. Cochrane Database Syst Rev.
3(CD012282)2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Lubrano V and Balzan S: Role of oxidative
stress-related biomarkers in heart failure: Galectin 3,
α1-antitrypsin and LOX-1: New therapeutic perspective? Mol Cell
Biochem. 464:143–152. 2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Singh S and Gautam AS: Upregulated LOX-1
receptor: Key player of the pathogenesis of atherosclerosis. Curr
Atheroscler Rep. 21(38)2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zeya B and Chandra NC: LOX-1: Its
cytotopographical variance and disease stress. J Biochem Mol
Toxicol. 33(e22375)2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Friedmann Angeli JP and Conrad M: Selenium
and GPX4, a vital symbiosis. Free Radic Biol Med. 127:153–159.
2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Shintoku R, Takigawa Y, Yamada K, Kubota
C, Yoshimoto Y, Takeuchi T, Koshiishi I and Torii S:
Lipoxygenase-mediated generation of lipid peroxides enhances
ferroptosis induced by erastin and RSL3. Cancer Sci. 108:2187–2194.
2017.PubMed/NCBI View Article : Google Scholar
|














