Introduction
The neural L1 cell adhesion molecule (L1CAM) is a member of the immunoglobulin superfamily 1, constituted by six immunoglobulin-like domains, five fibronectin-III-like domains, a single pass transmembrane domain and a short cytoplasmic domain (1). It produces various molecular and cellular effects, which are key to nervous system development and are involved in neural migration and differentiation, as well as memory and learning processes (2-5).
The L1CAM gene is the known causative gene of L1 syndrome. It is close to the telomere on the long arm of the X chromosome and contains 28 exons (6). Mutation of the L1CAM gene may lead to four different X-linked neurological conditions, including hydrocephalus, mental retardation, spasticity of lower limbs and flexion deformity of the thumbs (7,8). The phenotype varies greatly from mild to severe, largely depending on the specific affected domains of the L1CAM protein. In detail, the mutations located on the extracellular domains generally cause severe conditions, while the pathologies associated with dysfunction of the cytoplasmic domains tend to be milder (9-11).
Hydrocephalus is defined as distension of the ventricular system of the brain owing to the inadequate passage of cerebrospinal fluid from the position of production within the cerebral ventricles to its region of absorption into the systemic circulation (12). Congenital hydrocephalus is one of the most common characteristics of L1 syndrome. It is most frequently caused by mutations in the L1CAM gene, which are responsible for 5-10% of male cases of isolated congenital hydrocephalus (13,14). Furthermore, the prognosis for this condition is poor.
The present study reported on a nonsense mutation in the L1CAM gene causing congenital hydrocephaly in a fetus from a Chinese family.
Case report
Case study
A 30-year-old healthy multipara visited The Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University (Wenzhou, China) at 23 weeks of gestation on August 2018. She had already given birth to one healthy female infant and experienced one voluntary pregnancy termination at another hospital due to fetal hydrocephalus and the aborted male fetus was after the first girl was born. As indicated in Fig. 1, the familial pedigree suggested X-linked recessive hydrocephalus because the first girl was healthy, while both of aborted male fetuses presented with congenital hydrocephalus. As presented in Fig. 2, the ultrasound scan indicated progressive lateral ventricular dilatation, indicating severe fetal hydrocephalus. The maternal patient requested to terminate the pregnancy and provided written informed consent.
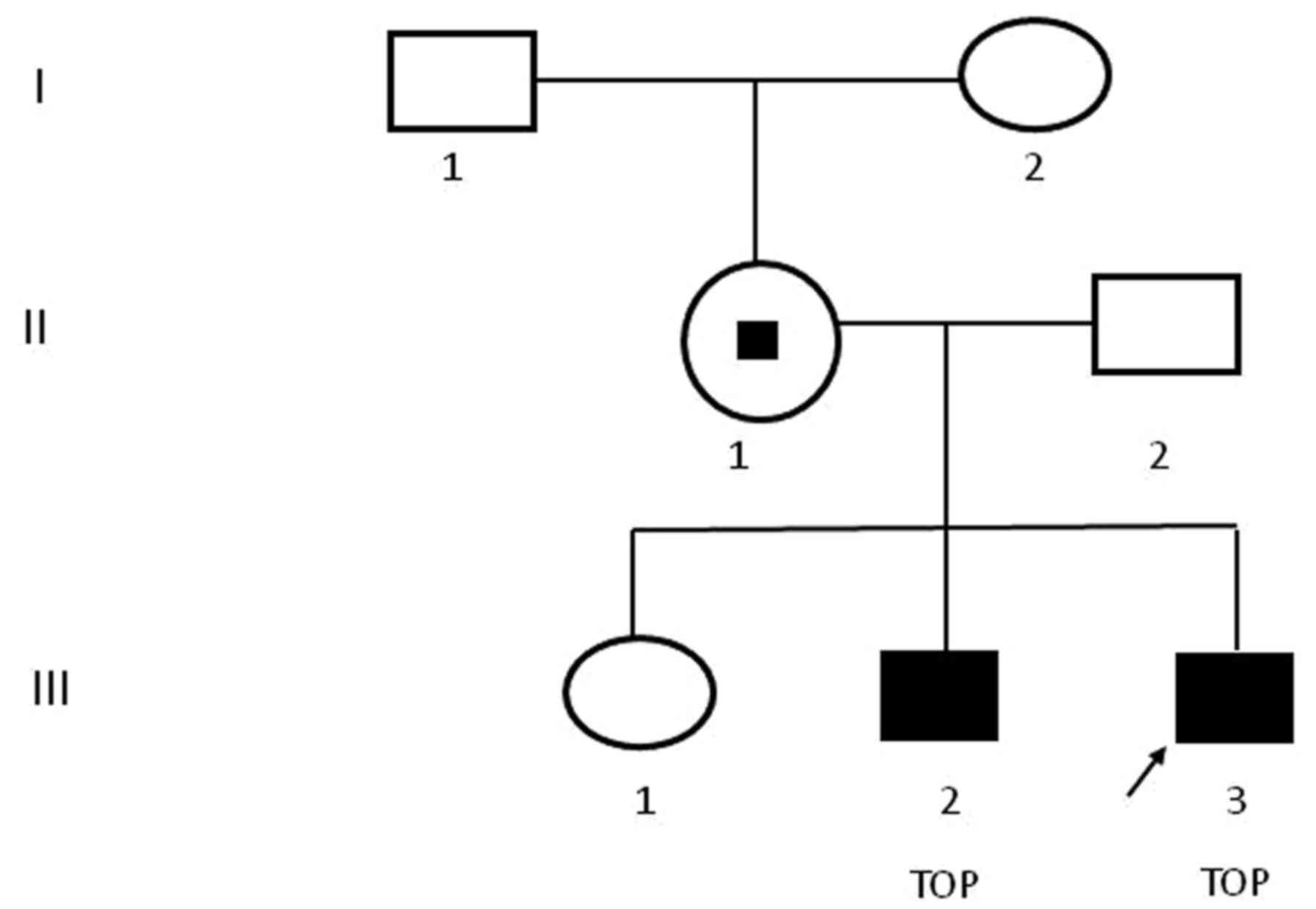 |
Figure 1
Pedigree chart. Females are indicated with circles and males with squares. The proband is indicated by an arrow. Filled squares indicate affected individuals and the small black square indicates mutation carrier status. TOP, termination of pregnancy.
|
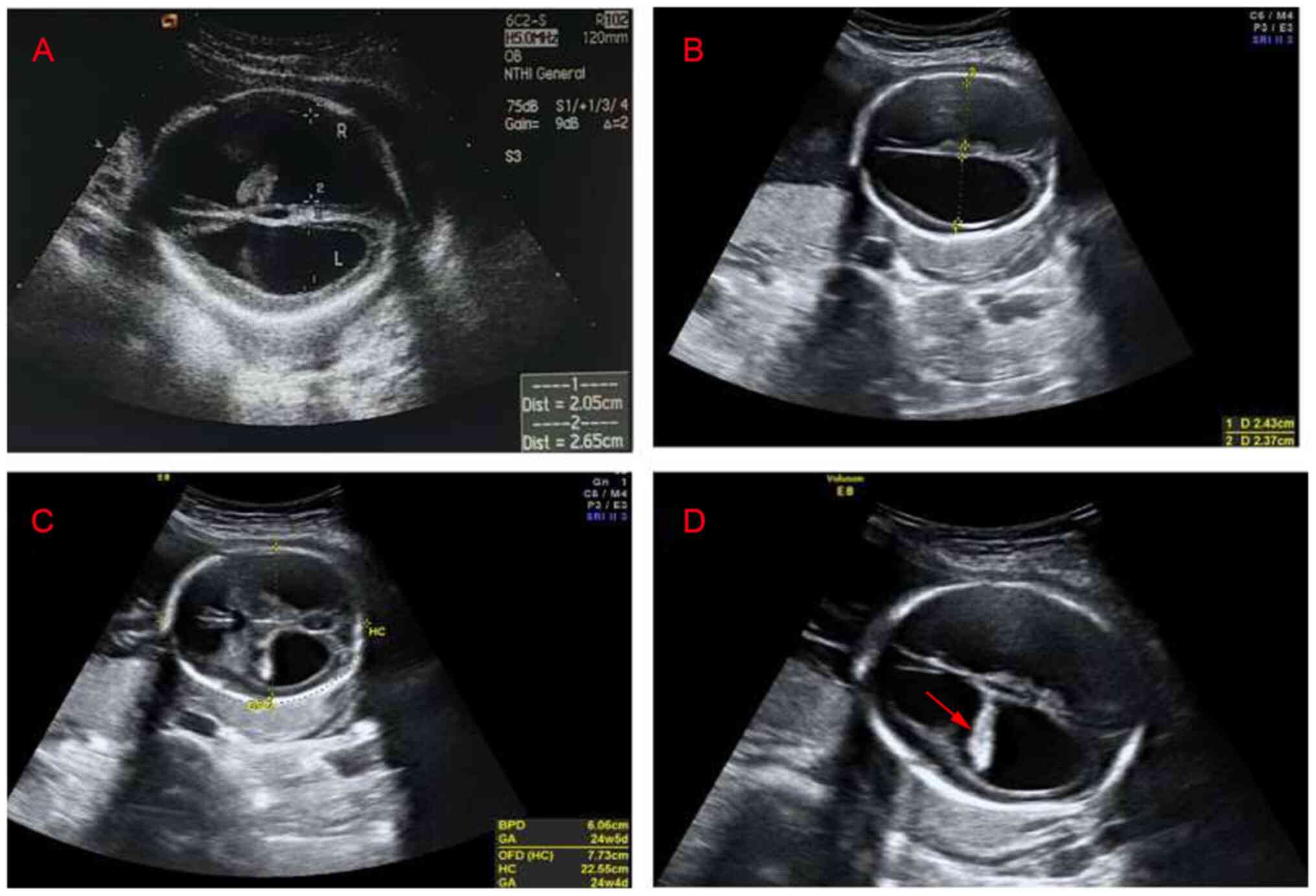 |
Figure 2
Transabdominal view of the fetal brain. (A) Ultrasound image of the first hydrocephalus fetus. The width of the left ventricle was 2.05 cm, the width of the right ventricle was 2.65 cm and the cerebral cortex became thinner. (B) Ultrasound image of the second hydrocephalus fetus. The width of the left ventricles was 2.43 cm, the width of the right ventricle was 2.37 cm and the cerebral cortex became thinner. (C) Ultrasound image of the second hydrocephalus fetus. The biparietal diameter of the fetus was 6.06 cm, the occipitofrontal diameter was 7.73 cm and the head circumference was 22.55 cm. (D) Ultrasound image of the second hydrocephalus fetus. Hyperechoic choroid hanging sign (arrow), the bilateral lateral ventricles were expanded and the cerebral cortex became thinner.
|
To identify disease-causing factors, blood samples from the parents and umbilical cord blood of the aborted fetus were collected at 24 weeks of gestation. Preliminary karyotype analysis and single-nucleotide polymorphism array did not provide any evidence (15). Whole-exome sequencing (WES) and in-depth mutation analysis were then used to analyze the samples from the parents and umbilical cord blood (16). All underlying pathogenic mutants were confirmed by Sanger sequencing (6). Potential mutants were called by Genome Analysis Toolkit (GATK v3.7; https://software.broadinstitute.org/gatk/download/) (17) and the variants were filtered by gnomAD database (https://gnomad.broadinstitute.org; Exome Aggregation Consortium; ExAC Version 0.3.1) (17) and annotated by ANNOVAR (https://annovar.openbioinformatics.org/en; 2018 Mar 25 version) (17).
Literature search
Using PubMed (https://pubmed.ncbi.nlm.nih.gov/) as the primary search database, the literature was examined for nonsense mutations in the L1CAM gene. PubMed searches of the references from database were performed using the key terms ‘prenatal’, ‘hydrocephalus’ and ‘nonsense mutation’. Inclusion criteria were as follows: Fetal ventriculomegaly >15 mm; singleton pregnancies; normal karyotype and microarray; fetal ultrasound excluding additional central nervous system (CNS); or extra-CNS malformations. Cases with aqueductal stenosis or progressive enlargement of ventricles were excluded. A total of 11 publications related to the topic published between 1989 and October 25, 2020 were retrieved and their features are summarized in Table I. To date, 30 different nonsense mutations in the L1CAM gene have been reported (7,8,16,18-27).
 |
Table I
Nonsense mutations in the L1 cell adhesion molecule gene.
|
Table I
Nonsense mutations in the L1 cell adhesion molecule gene.
| Author (year) |
Position in the gene (exon no.) |
Nucleotide change |
Consequence |
Country |
(Refs.) |
| Gu et al, 1996 |
08 |
c.828G>A |
p.Trp276Ter |
Germany |
(21) |
| Gu et al, 1997 |
11 |
c.1318C>T |
p.Gln440Ter |
Germany |
(22) |
| |
23 |
c.3124C>T |
p.Gln1042Ter |
|
|
| Finckh et al, 2000 |
04 |
c.316C>T |
p.Q106X |
Germany |
(7) |
| |
11 |
c.1267C>T |
p.Q423X |
|
|
| |
26 |
c. 3496C>T |
p.R1166X |
|
|
| |
02 |
c.79G>T |
p.E27X |
|
|
| |
21 |
c.2746G>T |
p.G916X |
|
|
| |
06 |
c.614C>A c.615C>G |
p.S205X |
|
|
| |
13 |
c.1672C>T |
p.R558X |
|
|
| |
14 |
c.1786G>T |
p.E597X |
|
|
| Okamoto et al, 2004 |
22 |
c.2974C>T |
p.Gln992Ter |
Japan |
(8) |
| Kanemura et al, 2006 |
26 |
c.3496C>T |
p.R1166X |
Japan |
(23) |
| Jackson et al, 2009 |
13 |
c.1674C>T |
p.R558X |
USA |
(24) |
| Takenouchi et al, 2012 |
01 |
c.61C>T |
p.Q21X |
Japan |
(25) |
| Adle-Biassette et al, 2013 |
06 |
c.621C>A |
p.Tyr207X |
France |
(26) |
| |
07 |
c.772C>T |
p.gln258X |
|
|
| |
08 |
c.871C>T |
p.gln291X |
|
|
| |
08 |
c.892C>T |
p.gln298X |
|
|
| |
12 |
c.1380G>A |
p.Trp460X |
|
|
| |
12 |
c.1453C>T |
p.Arg485X |
|
|
| |
12 |
c.1453C>T |
p.Arg485X |
|
|
| |
15 |
c.1882C>T |
p.gln628X |
|
|
| |
16 |
c.1989C>G |
p.Tyr663X |
|
|
| |
21 |
c.2785C>T |
p.gln929X |
|
|
| |
23 |
c.3163G>T |
p.gly1055X |
|
|
| |
23 |
c.3107G>A |
p.Trp1036X |
|
|
| Marín et al, 2015 |
26 |
c.3478C>T |
P.Gln1160X |
Spain |
(27) |
| Ochando et al, 2016 |
18 |
c.2239C>T |
P.Gln747X |
Spain |
(18) |
| Guo et al, 2020 |
21 |
c.2865C>A |
p.Y955X |
China |
(16) |
Results. The proband fetus was confirmed to be hemizygous for a mutant in the L1CAM gene, NM_001278116.1:c.1267C>T:p.(Q423X), whilst the mother was identified to be a heterozygote and the father was healthy (Fig. 3). The nonsense mutation is located on exon 11 of the L1CAM gene [chromosome (chr)X:153134975] and computational analysis [Sorting Intolerant From Tolerant (SIFT) (https://genetics.bwh.harvard.edu/pph2) (28) and PolyPhen-2 (https://sift.jcvi.org/) (29)] revealed that exon 11 is involved in encoding the fibronectin type-III of L1CAM protein. According to the gnomAD database (https://gnomad.broadinstitute.org; Exome Aggregation Consortium; ExAC Version 0.3.1) (17), this mutation had not been previously reported among Asian populations. Based on the Online Mendelian Inheritance in Man database (OMIM; https://www.omim.org), disease-associated mutations in the L1CAM gene were determined to be associated with X-linked hydrocephalus and the characteristics matched those of the proband. The identified mutation was X-linked recessive inheritance and pathogenic.
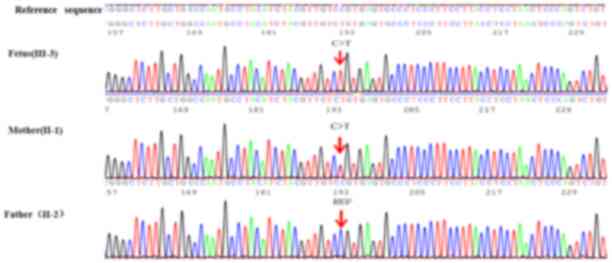 |
Figure 3
Gene sequencing results in L1 cell adhesion molecule for the fetus and the parents. The ref sequence was cited from Human GRCh37/hg19 (https://ncbiinsights.ncbi.nlm.nih.gov/2020/11/12/human-grch37-hg19-refseq-annotation-update/). The mutation site is indicated by arrows.
|
Discussion
Hydrocephalus, the abnormal accumulation of intracranial cerebrospinal fluid, is a common congenital malformation in fetuses. It occurs in 0.4-0.8% of newborns (14). The specific causes of this disease have remained to be fully identified. However, a number of ideas have been put forward. In a certain proportion of patients with genetic disorders, congenital hydrocephalus manifests (13). It has been reported that X-linked hydrocephalus (XLH), which is caused by mutations in the L1CAM gene, contributes to 5% of all cases of congenital hydrocephalus (10,14).
To date, >270 different pathogenetic mutations in the L1CAM gene have been identified, including frameshifts, nonsense, missense mutations and alterations in splice-site junction (13). Certain studies have explored the associations between specific mutations in the L1CAM gene and the severity of syndromes. Mutations leading to extracellular protein truncation may produce severe pathogeneses, whereas mutations in cytoplasmic domains tend to lead to milder manifestations (7,9-11).
The criteria for identifying a high-risk mutation in the L1CAM gene are family history and the clinical L1 syndromes, including stenosis of the aqueduct of Sylvius, mental retardation, aphasia, spastic paraplegia, adducted thumbs, X-linked complicated hereditary spastic paraplegia type 1 and X-linked agenesis of the corpus callosum (7,8,10,13). In the present study, in view of that the patient had experienced a previous pregnancy with fetal hydrocephalus and the current fetal ultrasound revealed hydrocephalus, genetic causes were suspected. A nonsense mutation, NM_001278116.1:c.1267C>T:p.(Q423X) in exon 11 of the L1CAM gene (chrX:153134975) was identified in the Chinese family. The mutant converts a glutamine into a premature stop codon on amino acid position 423, which in turn leads to extracellular protein truncation that abrogates the actions of L1CAM in its intracellular signaling pathways (7,18,19). However, there was no history of hydrocephalus in the mother's family. Nonsense-mediated mRNA decay (NMD) is an RNA surveillance mechanism, which may be used to detect mRNAs that harbor premature termination codons and initiate the degradation of the truncated or potentially harmful proteins. In the present case, it may have been suitable to perform NMD analysis of the nonsense mutation. However, due to improper preservation of specimens, this was not performed. In a future study, it may even be possible to trigger the RNA surveillance mechanism artificially to observe whether the harmful effects resulting from the nonsense mutation may be alleviated by NMD.
Since only one case has so far been reported, it is impossible to clarify the exact mechanisms of how a mutation at this site causes disease in individuals. This is a limitation of this study. However, it is possible to estimate the pathogenicity of a mutation with some available online tools, e.g., MutationTaster. Specifically, the pathogenesis observed to result from nonsense mutations on other sites in the L1CAM gene may provide clues. To date, 30 different nonsense mutations in the L1CAM gene have been reported (7,18,21-27). All of the mutations were unique to each family, with the exception that one mutation appeared in two different families (c.3496C>T) (7,23) and two different mutations were detected in one family (c.614C>A, c.615C>G) (7). All of these 30 mutations are considered capable of causing disease.
The characteristics of XLH include the enlargement of both third and lateral ventricles, and agenesis of the corpus callosum (7,8). Previous data revealed that 41% of patients with congenital hydrocephalus carried pathogenic mutations in the L1CAM gene (26). Furthermore, the prognosis of these carriers is exclusively worse. In certain cases, hydrocephaly in fetuses may cause stillbirth, neurodevelopmental delay in survivors or death in early infancy (13). Facing such conditions, most parents choose to terminate their pregnancies. However, in other cases, terminating pregnancy was not a legal option given the gestational age. Intrauterine ventriculo-amniotic shunting is an option for managing such cases. Theoretically, such therapy is beneficial for preventing amniotic fluid reflux and cerebral injury. The efficacy of this treatment has been proven in animal experiments (30). However, attempts in the 1980s were generally abandoned due to the high prevalence of severe neurodevelopmental delay among survivors (31). Recently, a retrospective study including 44 fetuses with severe bilateral ventriculomegaly treated with ventriculo-amniotic shunting demonstrated the feasibility of this treatment for severe fetal cerebral ventriculomegaly. However, repeated shunting was required in nearly half of the cases and a high proportion of survivors have neurodevelopmental delay. It has been proposed that the possible beneficial role of ventriculo-amniotic shunting requires to be confirmed by randomized studies with a large sample size (32).
There are currently no effective therapies for curing genetic disabilities. The only available method is to prevent the birth of fetuses with diagnosed hydrocephalus and the reoccurrence of genetic hydrocephalus in future pregnancies. Comprehensive prenatal diagnostic measures have considerable significance in assisting clinicians in making informed decisions and providing guidance for families with L1CAM gene mutants.
In addition, prenatal ultrasound may detect the presence of hydrocephalus, but its dependability is not optimal. Kanemura et al (23) demonstrated only 21.7% of cases of XLH were identifiable by ultrasound screening prior to 22 weeks of gestation. Of note, enlargement of the ventricles usually does not begin until 20 weeks of gestation and certain individuals may only develop this condition after live birth or not at all (33). On one hand, prenatal diagnosis of hydrocephalus cannot always be made as early as possible. On the other hand, false-negative results undoubtedly increase the clinical variability for diagnosing L1-associated disease (33). Accordingly, using DNA sequencing to identify the genetic causes of hydrocephalus is particularly important in families carrying L1CAM gene mutations. It has been reported that the detection rate of L1AM gene mutations in patients without a family history of L1 syndrome is 9-50%, while among those with a family history of L1 syndrome, the probability of L1CAM gene mutations is up to 50-80% (7,10,34).
In conclusion, the present study reported on a nonsense mutation, NM_001278116.1:c.1267C>T:p.(Q423X), in the L1CAM gene as the probable cause of XLH in a Chinese family. According to the diagnostic experience in this case, prenatal ultrasound and DNA screening should be offered to high-risk families to assist them in deciding whether to continue a pregnancy (14). Determining which mutations are associated with fetal malformations may also improve counseling for future pregnancies.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from Zhejiang Provincial Natural Science Foundation (grant nos. LY19H040006 and LQ15H040006), the Research Fund for Lin He's Academician Workstation of New Medicine and Clinical Translation (grant no. 18331209) and the Medical and Scientific Research Foundation of Zhejiang Province (grant no. 2020KY641).
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in the LOVD database repository (https://databases.lovd.nl/shared/variants/0000796593#00025606).
Authors' contributions
RW, HC, and XWu performed genetic diagnoses and drafted and critically revised the manuscript. RW and XWu Authenticated the raw data in this manuscript. RW, XWa, SH, and AX clinically diagnosed patients, collected and analysed samples and performed imaging tests. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
This study has been approved by the Ethics Committee/Institutional Review Board of the Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University (Wenzhou, China). Written informed consent, according to the Declaration of Helsinki, were acquired from both parents of the fetus.
Patient consent for publication
All probands provided informed consent for genetic testing and publication of their data in this study.
Competing interests
The authors declare that they have no competing interests.
References
|
1
|
Moos M, Tacke R, Scherer H, Teplow D, Früh K and Schachner M: Neural adhesion molecule L1 as a member of the immunoglobulinsuperfamily with binding domains similar to fibronectin. Nature. 334:701–703. 1988.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Basel-Vanagaite L, Straussberg R, Friez MJ, Inbar D, Korenreich L, Shohat M and Schwartz CE: Expanding the phenotypic spectrum of L1CAM-associated disease. Clin Genet. 69:414–419. 2006.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Patzke C, Acuna C, Giam LR, Wernig M and Sudhof TC: Conditional deletion of L1CAM in human neurons impairs both axonal and dendritic arborization and action potential generation. J Exp Med. 213:499–515. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Chang S, Rathjen FG and Raper JA: Extension of neuritis on axons is impaired by antibodies against specific neural cell surface glycoproteins. J Cell Biol. 104:355–362. 1987.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Rathjen FG and Schachner M: Immunocytological and biochemical characterization of a new neuronal cell surface component (L1 antigen) which is involved in cell adhesion. EMBO J. 3:1–10. 1984.PubMed/NCBI
|
|
6
|
Ferese R, Zampatti S, Griguoli AMP, Fornai F, Giardina E, Barrano G, Albano V, Campopiano R, Scala S, Novelli G and Gambardella S: A new splicing mutation in the L1CAM gene responsible for X-linked hydrocephalus (HSAS). J Mol Neurosci. 59:376–381. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Finckh U, Schröder J, Ressler B, Veske A and Gal A: Spectrum and detection rate of L1CAM mutations in isolated and familial cases with clinically suspected L1-disease. Am J Med Genet. 92:40–46. 2000.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Okamoto N, Del Maestro R, Valero R, Monros E, Poo P, Kanemura Y and Yamasaki M: Hydrocephalus and Hirschsprung's disease with a mutation of L1CAM. J Hum Genet. 49:334–337. 2004.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Fransen E, Van Camp G, D'Hooge R, Vits L and Willems PJ: Genotype-phenotype correlation in L1 associated diseases. J Med Genet. 35:399–404. 1998.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Vos YJ, de Walle HE, Bos KK, Stegeman JA, Ten Berge AM, Bruining M, van Maarle MC, Elting MW, den Hollander NS, Hamel B, et al: Genotype-phenotype correlationsin L1 syndrome: A guide for genetic counselling and mutation analysis. J Med Genet. 47:169–175. 2010.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Yamasaki M, Thompson P and Lemmon V: CRASH syndrome: Mutations in L1CAM correlate with severity of the disease. Neuropediatrics. 28:175–178. 1997.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Rekate HL: The definition and classification of hydrocephalus: A personal recommendation to stimulate debate. Cerebrospinal Fluid Res. 5(2)2008.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Vos YJ and Hofstra RM: An updated and upgraded L1CAM mutation database. Hum Mutat. 31:E1102–E1109. 2010.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Schrander-Stumpel C and Fryns JP: Congenital hydrocephalus: Nosology and guidelines for clinical approach and genetic counselling. Eur J Pediatr. 157:355–362. 1998.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Noronha TR, Rohr SS and Chauffaille MLLF: Identifying the similarities and differences between single nucleotide polymorphism array (SNPa) analysis and karyotyping in acute myeloid leukemia and myelodysplastic syndromes. Rev Bras Hematol Hemoter. 37:48–54. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Guo D, Shi Y, Jian W, Fu Y, Yang H, Guo M, Yong W, Chen G, Deng H, Qin Y, et al: A novel nonsense mutation in the L1CAM gene responsible for X-linked congenital hydrocephalus. J Gene Med. 22(e3180)2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kong W, Wang X, Zhao J, Kang M, Xi N and Li S: A new frameshift mutation in L1CAM producing X-linked hydrocephalus. Mol Genet Genomic Med. 8(e1031)2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Ochando I, Vidal V, Gascón J, Acién M, Urbano A and Rueda J: Prenatal diagnosis of X-linked hydrocephalus in a family with a novel mutation in L1CAM gene. J Obstet Gynaecol. 36:403–405. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Isik E, Onay H, Atik T, Akgun B, Cogulu O and Ozkinay F: Clinical and genetic features of L1 syndrome patients: Definition of two novel mutations. Clin Neurol Neurosurg. 172:20–23. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Vinci M, Falco M, Castiglia L, Grillo L, Spalletta A, Sturnio M, Galesi O, Salemi M, Gloria A, Amata S, et al: Identification of novel mutations in L1CAM gene by a DHPLC-based assay. Genes Genomics. 38:1159–1164. 2016.
|
|
21
|
Gu SM, Orth U, Vske A, Enders H, Klunder K, Schlosser M, Engel W, Schwinger E and Gal A: Five novel mutations in the L1CAM gene in families with X linked hydrocephalus. J Med Genet. 33:103–106. 1996.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Gu SM, Orth U, Zankl M, Schröder J and Gal A: Molecular analysis of the L1CAM gene in patients with X-linked hydrocephalus demonstrates eight novel mutations and suggests non-allelic heterogeneity of the trait. Am J Med Genet. 71:336–340. 1997.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Kanemura Y, Okamoto N, Sakamoto H, Shofuda T, Kamiguchi H and Yamasaki M: Molecular mechanisms and neuroimaging criteria for severe L1 syndrome with X-linked hydrocephalus. J Neurosurg. 105 (Suppl 5):S403–S412. 2006.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Jackson SR, Guner YS, Woo R, Randolph LM, Ford H and Shin CE: L1CAM mutation in association with X-linked hydrocephalus and Hirschsprung's disease. Pediatr Surg Int. 25:823–825. 2009.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Takenouchi T, Nakazawa M, Kanemura Y, Shimozato S, Yamasaki M, Takahashi T and Kosaki K: Hydrocephalus with Hirschsprung disease: Severe end of X-linked hydrocephalus spectrum. Am J Med Genet A. 158:812–815. 2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Adle-Biassette H, Saugier-Veber P, Fallet-Bianco C, Delezoide AL, Razavi F, Drouot N, Bazin A, Beaufrère AM, Bessières B, Blesson S, et al: Neuropathological review of 138 cases genetically tested for X-linked hydrocephalus: Evidence for closely related clinical entities of unknown molecular bases. Acta Neuropathol. 126:427–442. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Marín R, Ley-Martos M, Gutiérrez G, Rodríguez-Sánchez F, Arroyo D and Mora-López F: Three cases with L1 syndrome and two novel mutations in the L1CAM gene. Eur J Pediatr. 174:1541–1544. 2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kumar P, Henikoff S and Ng PC: Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A method and server for predicting damaging missense mutations. Nat Methods. 7:248–249. 2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Peiro JL and Fabbro MD: Fetal therapy for congenital hydrocephalus-where we came from and where we are going. Childs Nerv Syst. 36:1697–1712. 2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Manning FA, Harrison MR and Rodeck C: Catheter shunts for fetal hydronephrosis and hydrocephalus. Report of the international fetal surgery registry. N Engl J Med. 315:336–340. 1986.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Litwinska M, Litwinska E, Czaj M, Polis B, Polis L and Szaflik K: Ventriculo-amniotic shunting for severe fetal ventriculomegaly. Acta Obstet Gynecol Scand. 98:1172–1177. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Serikawa T, Nishiyama K, Tohyama J, Tazawa R, Goto K, Kuriyama Y, Haino K, Kanemura Y, Yamasaki M, Nakata K, et al: Prenatal molecular diagnosis of X-linked hydrocephalus via a silent C924T mutation in the L1CAM gene. Congenit Anom (Kyoto). 54:243–245. 2014.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Weller S and Gärtner J: Genetic and clinical aspects of X-linked hydrocephalus (L1 disease): Mutations in the L1CAM gene. Hum Mutat. 18:1–12. 2001.PubMed/NCBI View Article : Google Scholar
|














