Peutz-Jeghers syndrome (PJS), a rare genodermatosis
with an autosomal dominant inheritance of a high-penetrance
profile, is caused by serine/threonine kinase (STK)11 gene mutation
on chromosome 19p13.3(1). This is a
tumor-suppressor gene, also known as liver kinase B1 (LKB1)
gene, which involves master serine-threonine protein kinase
activity that communicates with different growth and angiogenesis
factors representing tumorigenic pathways of associated neoplasia
(2). STK11/LKB1 is also
involved in signaling pathways involved in the DNA damage response
to sun ultraviolet radiation as a contributor to skin cancer and it
has been connected to other non-PJS-related epithelial cancers
(3,4). In addition, control of certain immune
cells has been reported to be under the influence of the
STK11/LKB1 gene (5).
The syndrome, with a reported incidence of
1/25,000-1/280,00 persons/year, includes multiple gastro-intestinal
mucosal lesions including benign hamartomatouspolyps causing local
bleeding, occlusion and intussusception, post-resection small bowel
syndrome; hyperpigmentation of the skin and mucosa especially at
the oral and lips levels, and a higher risk of developing other
non-gastrointestinal tumors including ovarian/testicular,
pancreatic, breast, and uterine neoplasia (6,7)
(Fig. 1). The cumulative risk of
cancer is higher with 76% in the general population and with
females being more exposed (8).
Cancer of the small intestine usually occurs during the third
decade of life making early assessment and serial follow-up
crucial, once the skin lesions or gene anomalies are identified in
a carrier or a specific family (9).
Esophago-gastro-duodenoscopy screening and further surveillance are
essential (10). Familial genetic
consult is useful since the digestive disease may be asymptomatic
for years (11). Lifelong follow-up
is required (12).
The need for multidisciplinary teams of surveillance
in PJS patients is essential, from dermatology to gastroenterology,
from endocrinology to oncology, in both the pediatric and adult
patients. Muco-cutaneous hyperpigmentation (deposits of melanin in
skin and mucosa) represents an essential dermal clue for assessment
of this multi-system condition (13,14).
This is a systematic, narrative review of the
literature conducted based on two main aspects in PJS: on the one
hand, skin and mucosa anomalies and, on the other hand, endocrine
manifestations. Additional genetic and neoplasia data are provided
in order to integrate the general multidisciplinary, complex image
of this hereditary syndrome.
The research of the review started from the PubMed
database with respect to the following key words: ‘Peutz-Jeghers
syndrome’ and ‘skin’, ‘mucosa’, ‘oral’, ‘endocrine, ‘puberty’,
‘ovary’, ‘testes’, and ‘polyps’. A total of 101 references were
cited between January 2010 and May 2021. Full-length original
papers of different types were exclusively introduced due to the
rarity of the publications in areas of interest. The selection was
based on clinical relevance.
Dermatological findings may be identified before
gastro-intestinal polyps and other PJS-related tumors; skin and
mucosal manifestations being a valuable clue of this hereditary
syndrome (15,16). In some cases, vitiligo (usually
focal depigmentation) may be followed by focal hyperpigmentation in
the skin, oral mucosa, lips, and labia (17,18).
Pigmentation features may mimic xeroderma pigmentosum in the early
stages (xeroderma pigmentosum is an extreme sun
sensitivity-associated high risk of skin cancer) (19). Melanin hyperpigmentation present on
a patient who is not previously known to have the syndrome must be
biopsied in order to obtain a clear diagnostic (20). Previous findings have revealed a
certain genotype-phenotype correlation of the STK11/LKB1
gene involving the fact that more severe dermatological findings
are correlated with a more aggressive profile of gastro-intestinal
hamartomatous polyps (21). A total
of 90% of subjects with positive PJS criteria have
STK11/LKB1 mutations; an oral hyperpigmentation suggestive
for PJS is an indication of STK11 genetic testing and
further familial genetic assessment (22). Abdominal pain in a patient with oral
pigmentations is suggestive of a complication of a digestive PJS
polyp (23). Except for a digestive
field, specific protocols of investigations and follow-up regarding
skin and mucosal PJS lesions remain suboptimal (24).
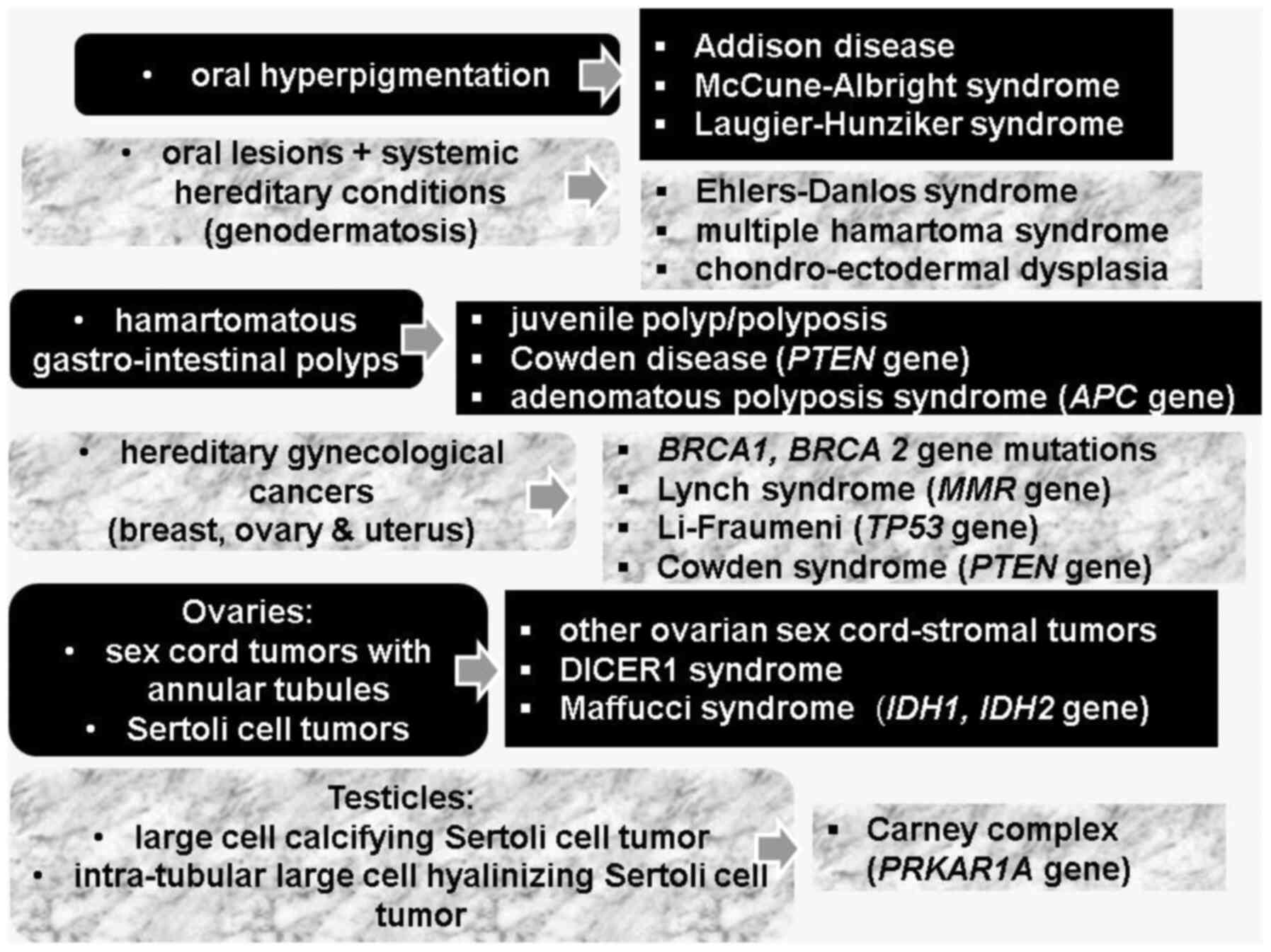
Skin and mucosa hyperpigmentation need to be
differentiated from other conditions that are indicated by
hyperpigmentation: Addison disease (AD), McCune-Albright syndrome
(MAS), and Laugier-Hunziker syndrome (LHS) (25).
AD, a chronic primary adrenal insufficiency,
requiring lifelong glucocorticoid and mineralocorticoid
substitution, is accompanied by hyperpigmentation at general level
or at the level of scars, areolas, and skin-fold, a
hyperpigmentation that is reversible to some extent under adequate
endocrine therapy (26). The areas
that are more exposed to the sun are also more sensitive to
hyperpigmentation (26). The
underlying mechanism involves high adrenocorticotropic hormone
(ACTH) in addition to increased levels of melanocyte-stimulating
hormone (MSH) (26). Concurrent
focal or general depigmentation may also be found since vitiligo is
associated with poly-glandular autoimmune syndrome, especially with
chronic autoimmune thyroiditis (26).
MAS, an underlying endocrine condition usually
accompanied by hyper-function, presents focal hyperpigmentation
including ‘café au lait’ spots, similar to type 1 neurofibromatosis
(27,28).
LHS is an exceptionally rare, acquired condition
(the level of evidence is case reports) associated with idiopathic
hyperpigmentation at the level of the lip, acral area, oral mucosa,
and even nails (29). The skin
color changes are due to diffuse spreading of black macules with
dimensions that vary from 1 to 5 mm (29,30).
Longitudinal melanonychia has been described in both children and
adults (30). To date, the cause is
unknown (31). It seems that skin
anomalies are not associated with a higher risk compared to general
manifestations including endocrine conditions or non-endocrine
tumors/cancers, as observed in PJS (32). The overall prognosis is a favorable
one (33). Melanonychia striata,
caused by higher melanocyte activity and secondary hyperplasia at
the nail level, is also described in constitutional circumstances
(individuals with dark skin), after local traumatisms or
infections, in conditions such as alkaptonuria, hemochromatosis,
porphyria, and LHS (34). Since LHS
is a benign, rather harmless condition when it comes to general
complications, the endocrine assessment is necessary to exclude the
diagnosis of AD and MAS (35).
PJS is a type of genodermatosis representing a
complex cluster of hereditary syndromes with cutaneous-mucosal
manifestations in addition to systemic complications of tumor and
non-tumor type (for instance, Ehlers-Danlos syndrome, multiple
hamartoma syndrome, and chondro-ectodermal dysplasia) (36,37).
Non-genodermatoses involve lesions such as acanthosis nigricans
associated with insulin resistance, diabetes mellitus, and
polycystic ovary syndrome (38).
Other hereditary syndromes that involve lesions of the oral region
are associated predominantly with cutaneous manifestations
(Brooke-Spiegler syndrome, Muir-Torre syndrome) or predominant
endocrine complications such as multiple endocrine neoplasia (MEN)
type 1 (MEN gene) and type 2 (RET gene) syndrome,
Carney complex (PRKAR1A gene), or head and neck tumors
(Cowden syndrome-PTEN gene) or systemic tumors
(neurofibromatosis type 1) (39,40).
In many cases, the skin lesions are less important in regards to
the overall prognosis even though they represent the obvious mark
of the syndrome or the first step in its identification (41,42).
So-called familial lentiginosis dermato-endocrine syndromes include
PJS, Carney complex, Cowden disease and Noonan syndrome (43). A study published in 2021 that
analyzed the published papers focusing on oral pigmented lesions
(OPL) introduced 9 different syndromes in individuals with a mean
age at diagnosis of 35 years and female predominance (68%)
(44). Multiple lesions were more
frequent than single (73.15% vs. 26.85%); lip followed by buccal
mucosa were the more affected sites, in 75% of cases, OPL preceded
the recognition of the syndrome (44).
Multiple endocrine anomalies have been reported in
PJS as mentioned subsequently. Individuals presenting with PJS have
a lifelong higher risk of gynecological cancers with endocrine
components, so-called hereditary gynecological cancers (breast,
ovarian and uterine), also including those related to Lynch
syndrome (mutations of the MMR gene), Li-Fraumeni (mutations
of the TP53 gene), and Cowden syndrome (45). Similarity with hereditary conditions
with an identical malignancy pattern includes mutations of
BRCA1 and BRCA2 genes, respectively (46). Lynch syndrome is mostly related to
endometrial cancer, while Cowden syndrome and Li-Fraumeni are
related to breast cancer (Li-Fraumeni syndrome is also an important
cause of adrenocortical carcinoma especially in the pediatric
population) while PJS is equally associated with all three
mentioned malignancies (47).
Conditions with benign behavior have been reported
in PJS including breast hyperplasia and ovarian cysts, the
STK11 gene being related to primordial follicles reserve and
female fertility (48). Another
potential link involves STK11 gene polymorphism in
polycystic ovary syndrome that has been reported in PJS subjects,
although an incidental overlap cannot be excluded (49).
PJS constitutes two types of ovarian tumors, namely
sex cord tumors with annular tubules (SCTAT) and pure Sertoli cell
tumors (SCT), both originating from ovarian sex cord-stroma
(50,51). Overall, ovarian sex-cord stromal
neoplasia accounts for 8% of all cases diagnosed with ovarian
cancer (the most frequent cause of ovarian malignancy among
sex-cord stromal tumors is due to granulosa cell tumors) (52). Hereditary syndromes that have been
related to this large group of ovarian sex cord-stromal tumors
includes, besides PJS, DICER1 syndrome, Maffucci syndrome and
Ollier disease (52). The most
important hereditary syndromes in ovarian cancer are PJS, Lynch
syndrome and BRCA1 and BRCA2- related disease, overall accounting
for 1 out of 4-5 females with ovarian cancer (53).
SCTAT, representing 2% of all sex cord-stromal
tumors, are considered very rare neoplasias (54). An increased progesterone production
has been revealed in some cases (55). In non-PJS cases, one in five are
malignant; thus, candidates are subjected to chemotherapy and/or
surgical resection (56). Overall,
the risk of malignancy is higher compared to other sex cord stromal
tumors (55,57). Surgical removal remains the
first-line therapy, regardless of the presence of PJS (58). The presentation of SCTAT-PJS vs.
SCTAT-nonPJS reveals bilateral or multifocal tumors of small size
with a rather benign behavior vs. a unique ovarian tumor usually
with increased diameters and a higher malignant profile, including
the risk of developing distant metastases (59). Previous findings have shown
synchronous detection of SCTAT and SCT in the same PJS patient
(60). Other data suggest a poorer
prognosis of SCTAT-PJS because of the associated higher risk of
non-ovarian malignancies (61).
SCTAT has also been associated with co-presence of dysgerminomas
and gonadoblastomas or with Turner syndrome or endometriotic cysts,
most probably incidental-based (62). The association of PJS with
Sertoli-Leydig cell ovarian tumor has been reported in a very
limited number of cases and it seems atypical (63,64).
The first recurrent Sertoli-Leydig cell tumor of the ovary was
reported in 2016 in an African-American female with PJS diagnosed
at the age of 3 years due to precocious puberty followed by
recurrence at age of 17 years (65).
The specific manifestation of PJS at the digestive
system level is the presence of hamartomatous polyps, which
otherwise are described as single lesion (outside PJS), juvenile
polyp/polyposis or as clinical manifestation of Cowden disease
(78,79). Solitary Peutz-Jeghers polyp is a
distinct entity and it does not underline PJS; similarly,
LKB1/STK11 mutations (80,81).
Their evaluations combine family medical history, endoscopy
findings, histological and immunohistochemistry report (after
biopsy or after resection) and serial endoscopic follow-up because
of post-polypectomy recurrence and increased malignancy risk
(78,82). The clue at first endoscopic
evaluation is the number of polyps; multiple polyps generally have
a genetic background and require further dermatological and
endocrine workup if PJS is suspected (78). A polyp takes time to grow, thus its
detection is less likely to occur during the first decade of life
(83).
The most frequent sites are the small intestine as
well as the colon and stomach, with the duodenum and appendix being
rare sites (84). Recent findings
indicate the duodenum as the most frequent location of complicated
polyps (85). PJS-related polyps at
the level of the small bowel need to be differentiated from other
benign tumors including lipomas, leiomyomas, neurofibromas or
syndromic circumstances such as adenomatous polyposis syndromes
(caused by APC gene mutations) (86,87).
The PJS-related polyp histological profile includes peripheral
edema, mucin-filled, dilated cystic glands (88).
Not all PJS polyps have the same malignant potential
of transformation because it seems that an underlying cell
population is not homogenous and the exact mechanisms and
prevalence of PJS polyps are not fully known at present (89). Recent findings show that
hypo-methylation of the LKB1/STK11 promoter is correlated
with a more aggressive profile (90). STK11 genotype-phenotype
correlations regarding malignant potential remain a subject of
discussion (91). Other authors
suggest that sporadic PJS polyps are less malignant than familial
cases (92). Non-STK11 gene
mutations have been found in hamartomatous PJS polyps such as the
MMR (DNA mismatch repair) gene (93).
Specific multidisciplinary guidelines and protocols
for the dermatological manifestations in addition to
gastro-intestinal, endocrine and oncologic complications in PJS are
sub-optimal. The skin and mucosal lesions are useful markers of the
syndrome, assisting in early identification of hamartomatous polyps
and serial surveillance concerning the associated higher risk of
breast, uterine, ovarian, and pancreatic neoplasia.
Not applicable.
No funding was received.
All data generated or analyzed during this study are
included in this published article.
FS drafted the manuscript and critically revised the
final form, AP researched the literature and generated the figure,
MCD drafted the manuscript, RCP researched the literature, MC
drafted the manuscript and approved the final form. All authors
read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Nevozinskaya Z, Korsunskaya I, Sakaniya L,
Perlamutrov Y and Sobolev V: Peutz-Jeghers syndrome in dermatology.
Acta Dermatovenerol Alp Pannonica Adriat. 28:135–137.
2019.PubMed/NCBI
|
|
2
|
Zhang W, Ding Y, Zhang C, Lu Q, Liu Z,
Coughlan K, Okon I and Zou MH: Deletion of endothelial
cell-specific liver kinase B1 increases angiogenesis and tumor
growth via vascular endothelial growth factor. Oncogene.
36:4277–4287. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Esteve-Puig R, Gil R, González-Sánchez E,
Bech-Serra JJ, Grueso J, Hernández-Losa J, Moliné T, Canals F,
Ferrer B, Cortés J, et al: A mouse model uncovers LKB1 as an
UVB-induced DNA damage sensor mediating CDKN1A (p21WAF1/CIP1)
degradation. PLoS Genet. 10(e1004721)2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Herrmann JL, Byekova Y, Elmets CA and
Athar M: Liver kinase B1 (LKB1) in the pathogenesis of epithelial
cancers. Cancer Lett. 306:1–9. 2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zhang Y, Meng Q, Sun Q, Xu ZX, Zhou H and
Wang Y: LKB1 deficiency-induced metabolic reprogramming in
tumorigenesis and non-neoplastic diseases. Mol Metab.
44(101131)2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Syarifuddin E, Masadah R, Lusikooy RE,
Warsinggih Uwuratuw JA and Faruk M: Peutz-Jeghers syndrome in a
woman presenting as intussusception: A case report. Int J Surg Case
Rep. 79:286–290. 2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Shrivastava A, Gupta A, Gupta A and
Shrivastava J: Unusual presentation of intussusception of the small
bowel with peutz jeghers syndrome: Report of a case. J Clin Diagn
Res. 7:2296–2297. 2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Intratubular Armijo B, Bocklage T and
Heideman R: Large cell hyalinizing sertoli cell tumor of the testes
in a 4-year-old male with peutz-jeghers syndrome. J Pediatr Hematol
Oncol. 37:e184–e187. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Khanna K, Khanna V and Bhatnagar V:
Peutz-Jeghers syndrome: Need for early screening. BMJ Case Rep.
11(e225076)2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Tomas C, Soyer P, Dohan A, Dray X, Boudiaf
M and Hoeffel C: Update on imaging of Peutz-Jeghers syndrome. World
J Gastroenterol. 20:10864–10875. 2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ben Hammouda S, Njima M, Ben Abdeljelil N,
Bellalah A, Njim L and Zakhama A: An unusual presentation revealing
Peutz-Jeghers syndrome in adult. Ann Med Surg (Lond). 58:87–90.
2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Dutta A, Ghosh SK and Kundu SK: Peutz
Jegher syndrome. Indian Pediatr. 52:176–177. 2015.PubMed/NCBI
|
|
13
|
Tacheci I, Kopacova M and Bures J:
Peutz-Jeghers syndrome. Curr Opin Gastroenterol. 37:245–254.
2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kidambi TD, Kohli DR, Samadder NJ and
Singh A: Hereditary polyposis syndromes. Curr Treat Options
Gastroenterol. 17:650–665. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Shah KR, Boland CR, Patel M, Thrash B and
Menter A: Cutaneous manifestations of gastrointestinal disease:
Part I. J Am Acad Dermatol. 68:211.e1–e33. 2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Shen Z, Hoffman JD, Hao F and Pier E: More
than just skin deep: Faciocutaneous clues to genetic syndromes with
malignancies. Oncologist. 17:930–936. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Patel LM, Lambert PJ, Gagna CE, Maghari A
and Lambert WC: Cutaneous signs of systemic disease. Clin Dermatol.
29:511–522. 2011.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Duan N, Zhang YH, Wang WM and Wang X:
Mystery behind labial and oral melanotic macules: Clinical,
dermoscopic and pathological aspects of Laugier-Hunziker syndrome.
World J Clin Cases. 6:322–334. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Lehmann AR, McGibbon D and Stefanini M:
Xeroderma pigmentosum. Orphanet J Rare Dis. 6(70)2011.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Vageli DP, Doukas SG and Markou A:
Mismatch DNA repair mRNA expression profiles in oral melanin
pigmentation lesion and hamartomatous polyp of a child with
Peutz-Jeghers syndrome. Pediatr Blood Cancer. 60:E116–E117.
2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhang Y, Ke Y, Zheng X, Liu Q and Duan X:
Correlation between genotype and phenotype in three families with
Peutz-Jeghers Syndrome. Exp Ther Med. 13:507–514. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Duong BT and Winship I: The role of STK 11
gene testing in individuals with oral pigmentation. Australas J
Dermatol. 58:135–138. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chan TC and Sirlin C: Abdominal pain in a
young man with oral pigmentations. J Emerg Med. 50:335–336.
2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wagner A, Aretz S, Auranen A, Bruno MJ,
Cavestro GM, Crosbie EJ, Goverde A, Jelsig AM, Latchford A, Leerdam
MEV, et al: The Management of peutz-jeghers syndrome: European
hereditary tumour group (EHTG) guideline. J Clin Med.
10(473)2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Sputa-Grzegrzolka P, Wozniak Z, Akutko K,
Pytrus T, Baran W, Calik J, Glatzel-Plucinska N, Domagala Z,
Podhorska-Okolow M, Stawarski A and Dziegiel P: Laugier-Hunziker
syndrome: A case report of the pediatric patient and review of the
literature. Int J Dermatol. 59:1513–1519. 2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Lause M, Kamboj A and Fernandez Faith E:
Dermatologic manifestations of endocrine disorders. Transl Pediatr.
6:300–312. 2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Legrand MA, Raverot G, Nicolino M and
Chapurlat R: GNAS mutated thyroid carcinoma in a patient with Mc
Cune Albright syndrome. Bone Rep. 13(100299)2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Sandru F, Carsote M, Valea A, Albu SE,
Petca RC and Dumitrascu MC: Somatostatinoma: Beyond
neurofibromatosis type 1 (Review). Exp Ther Med. 20:3383–3388.
2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wang WM, Wang X, Duan N, Jiang HL and
Huang XF: Laugier-Hunziker syndrome: A report of three cases and
literature review. Int J Oral Sci. 4:226–230. 2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Lalosevic J, Zivanovic D, Skiljevic D and
Medenica L: Laugier-Hunziker syndrome-Case report. An Bras
Dermatol. 90 (3 Suppl 1):S223–S225. 2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Wei Z, Li GY, Ruan HH, Zhang L, Wang WM
and Wang X: Laugier-Hunziker syndrome: A case report. J Stomatol
Oral Maxillofac Surg. 119:158–160. 2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Cusick EH, Marghoob AA and Braun RP:
Laugier-Hunziker syndrome: A case of asymptomatic mucosal and acral
hyperpigmentation. Dermatol Pract Concept. 7:27–30. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Rangwala S, Doherty CB and Katta R:
Laugier-Hunziker syndrome: A case report and review of the
literature. Dermatol Online J. 16(9)2010.PubMed/NCBI
|
|
34
|
Leung AKC, Lam JM, Leong KF and Sergi CM:
Melanonychia striata: Clarifying behind the Black Curtain. A review
on clinical evaluation and management of the 21st century. Int J
Dermatol. 58:1239–1245. 2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Miličević T, Žaja I, Tešanović D and
Radman M: Laugier-Hunziker syndrome in endocrine clinical practice.
Endocrinol Diabetes Metab Case Rep:. 2018:18–0025. 2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wilder EG, Frieder J, Sulhan S, Michel P,
Cizenski JD, Wright JM and Menter MA: Spectrum of orocutaneous
disease associations: Genodermatoses and inflammatory conditions. J
Am Acad Dermatol. 77:809–830. 2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Babu NA, Rajesh E, Krupaa J and
Gnananandar G: Genodermatoses. J Pharm Bioallied Sci. 7 (Suppl
1):S203–S206. 2015.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Pinna R, Cocco F, Campus G, Conti G, Milia
E, Sardella A and Cagetti MG: Genetic and developmental disorders
of the oral mucosa: Epidemiology; molecular mechanisms; diagnostic
criteria; management. Periodontol 2000. 80:12–27. 2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Kennedy RA, Thavaraj S and Diaz-Cano S: An
overview of autosomal dominant tumour syndromes with prominent
features in the oral and maxillofacial region. Head Neck Pathol.
11:364–376. 2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Sandru F, Carsote M, Albu SE, Valea A,
Petca A and Dumitrascu MC: Glucagonoma: From skin lesions to the
neuroendocrine component (Review). Exp Ther Med. 20:3389–3393.
2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Stratakis CA: Hereditary syndromes
predisposing to endocrine tumors and their skin manifestations. Rev
Endocr Metab Disord. 17:381–388. 2016.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Ponti G, Tomasi A, Manfredini M and
Pellacani G: Oral mucosal stigmata in hereditary-cancer syndromes:
From germline mutations to distinctive clinical phenotypes and
tailored therapies. Gene. 582:23–32. 2016.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Lodish MB and Stratakis CA: The
differential diagnosis of familial lentiginosis syndromes. Fam
Cancer. 10:481–490. 2011.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Ferreira LDS, Calderipe CB, Maass JB,
Carrard VC, Martins MD, Abreu LG, Shuch LF and Uchoa Vasconcelos
AC: Oral pigmented lesions in syndromic individuals: A systematic
review. Oral Dis: Jan 4, 2021 (Epub ahead of print).
|
|
45
|
Ueki A and Hirasawa A: Molecular features
and clinical management of hereditary gynecological cancers. Int J
Mol Sci. 21(9504)2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Pietragalla A, Arcieri M, Marchetti C,
Scambia G and Fagotti A: Ovarian cancer predisposition beyond BRCA1
and BRCA2 genes. Int J Gynecol Cancer. 30:1803–1810.
2020.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Chung SH, Woldenberg N, Roth AR, Masamed
R, Conlon W, Cohen JG, Joines MM and Patel MK: BRCA and Beyond:
Comprehensive image-rich review of hereditary breast and
gynecologic cancer syndromes. Radiographics. 40:306–325.
2020.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Jiang ZZ, Hu MW, Ma XS, Schatten H, Fan
HY, Wang ZB and Sun QY: LKB1 acts as a critical gatekeeper of
ovarian primordial follicle pool. Oncotarget. 7:5738–5753.
2016.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Smith KJ and Germain M: Polycystic ovary
syndrome (PCOS) with melanocytic mucosal macules: The role of STK11
gene polymorphisms in PCOS and Peutz-Jeghers syndrome. Int J
Dermatol. 55:177–180. 2016.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Gheorghisan-Galateanu AA, Carsote M,
Terzea D, Valea A and Ghemigian A: Ovarian Sertoli cell tumours:
Practical points. J Pak Med Assoc. 70:129–133. 2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Garg K, Karnezis AN and Rabban JT:
Uncommon hereditary gynaecological tumour syndromes: Pathological
features in tumours that may predict risk for a germline mutation.
Pathology. 50:238–256. 2018.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Fuller PJ, Leung D and Chu S: Genetics and
genomics of ovarian sex cord-stromal tumors. Clin Genet.
91:285–291. 2017.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Weissman SM, Weiss SM and Newlin AC:
Genetic testing by cancer site: Ovary. Cancer J. 18:320–327.
2012.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Jaegle WT, Keyser EA, Messersmith L, Brady
RO and Miller C: Extraovarian sex cord tumor with annular tubules
discovered arising from a leiomyoma. Gynecol Oncol Rep. 26:17–20.
2018.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Young RH: Ovarian sex cord-stromal tumours
and their mimics. Pathology. 50:5–15. 2018.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Plevová P and Geržová H: Genetic causes of
rare pediatric ovarian tumors. Klin Onkol. 32 (Suppl 2):S79–S91.
2019.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Yahaya JJ, Mshana D and Mremi A: Ovarian
sex cord tumour with annular tubules in a 13-year-old female: A
case report. Oxf Med Case Reports. 2020(omaa024)2020.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Choudhary F, Tanveer N, Mangla G and
Gayatree A: Non-syndromic sex cord tumor with annular tubules: A
rare diagnosis. Indian J Surg Oncol. 11:313–315. 2020.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Han Y, Li S, Wu L, Zhang X and Cao D:
Non-Peutz-Jeghers syndrome-associated ovarian sex cord tumor with
annular tubules: Report of a malignant case. J Obstet Gynaecol Res.
42:224–227. 2016.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Ravishankar S, Mangray S, Kurkchubasche A,
Yakirevich E and Young RH: Unusual sertoli cell tumor associated
with sex cord tumor with annular tubules in peutz-jeghers syndrome:
Report of a case and review of the literature on ovarian tumors in
peutz-jeghers syndrome. Int J Surg Pathol. 24:269–273.
2016.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Kwon SY, Choe MS, Lee HW, Lee HJ, Shin SJ
and Cho CH: Minimal deviation adenocarcinoma of the cervix and
tumorlets of sex-cord stromal tumor with annular tubules of the
ovary in Peutz-Jeghers syndrome. J Gynecol Oncol. 24:92–95.
2013.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Singh M, Mandal S and Majumdar K: Sex cord
tumor with annular tubules: An incidental finding in an
endometriotic cyst-the first known co-occurrence. Biomed Res Int.
2014(970243)2014.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Howell L, Bader A, Mullassery D, Losty P,
Auth M and Kokai G: Sertoli Leydig cell ovarian tumour and gastric
polyps as presenting features of Peutz-Jeghers syndrome. Pediatr
Blood Cancer. 55:206–207. 2010.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Poiana C, Virtej I, Carsote M, Banceanu G,
Sajin M, Stanescu B, Ioachim D, Hortopan D and Coculescu M:
Virilising Sertoli-Leydig cell tumour associated with thyroid
papillary carcinoma: Case report and general considerations.
Gynecol Endocrinol. 26:617–622. 2010.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Bellfield EJ and Alemzadeh R: Recurrent
ovarian Sertoli-Leydig cell tumor in a child with Peutz-Jeghers
syndrome. Oxf Med Case Reports. 2016(omw048)2016.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Kong F, Wang M, Huang X, Yue Q, Wei X, Dou
X, Peng X, Jia Y, Zheng K, Wu T, et al: Differential regulation of
spermatogenic process by Lkb1 isoforms in mouse testis. Cell Death
Dis. 8(e3121)2017.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Valeri C, Lovaisa MM, Racine C, Edelsztein
NY, Riggio M, Giulianelli S, Venara M, Bedecarrás P, Ballerini MG,
di Clemente N, et al: Molecular mechanisms underlying AMH elevation
in hyperoestrogenic states in males. Sci Rep.
10(15062)2020.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Zhang LJ, Su Z, Liu X, Wang L and Zhang Q:
Peutz-Jeghers syndrome with early onset of pre-adolescent
gynecomastia: A predigree case report and clinical and molecular
genetic analysis. Am J Transl Res. 9:2639–2644. 2017.PubMed/NCBI
|
|
69
|
Renes JS, Knijnenburg J,
Chitoe-Ramawadhdoebe S, Gille JJP, de Bruin C and Barge-Schaapveld
DQCM: Possible hints and pitfalls in diagnosing Peutz-Jeghers
syndrome. J Pediatr Endocrinol Metab. 31:1381–1386. 2018.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Kaluzny A, Matuszewski M, Wojtylak S,
Krajka K, Cichy W, Plawski A, Gintowt A and Lipska BS:
Organ-sparing surgery of the bilateral testicular large cell
calcifying Sertoli cell tumor in patient with atypical
Peutz-Jeghers syndrome. Int Urol Nephrol. 44:1045–1048.
2012.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Tracey AJ and Cerwinka WH: Benign
large-cell calcifying sertoli tumor of the testis in a 13-year-old
male patient treated with partial orchiectomy. Urology.
107:226–228. 2017.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Simões-Pereira J, Santos F, Lopes L and
Limbert C: Prepubertal gynaecomastia in a boy with Peutz-Jeghers
syndrome: Managing the aromatase overexpression. J Pediatr
Endocrinol Metab. 31:1149–1154. 2018.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Gourgari E, Saloustros E and Stratakis CA:
Large-cell calcifying Sertoli cell tumors of the testes in
pediatrics. Curr Opin Pediatr. 24:518–522. 2012.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Pelit ES, Erol B, Zenginkinet T and
Çaşkurlu T: Testis-sparing surgery of unilateral testicular
large-cell calcifying Sertoli cell tumor: A sporadic case. Turk J
Urol. 44:370–372. 2018.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Koç Yekedüz M, Şıklar Z, Burgu B, Kuloğlu
Z, Kocaay P, Çamtosun E, İsakoca M, Kansu A, Soygür T and
Berberoğlu M: Response to anastrozole treatment in a case with
peutz-jeghers syndrome and a large cell calcifying Sertoli cell
tumor. Clin Res Pediatr Endocrinol. 9:168–171. 2017.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Crocker MK, Gourgari E, Lodish M and
Stratakis CA: Use of aromatase inhibitors in large cell calcifying
sertoli cell tumors: Effects on gynecomastia, growth velocity, and
bone age. J Clin Endocrinol Metab. 99:E2673–E2680. 2014.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Idrees MT, Ulbright TM, Oliva E, Young RH,
Montironi R, Egevad L, Berney D, Srigley JR, Epstein JI and Tickoo
SK: Members of the international society of urological pathology
testicular tumour panel. The World Health Organization 2016
classification of testicular non-germ cell tumours: A review and
update from the International Society of Urological Pathology
Testis Consultation Panel. Histopathology. 70:513–521.
2017.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Cauchin E, Touchefeu Y and Matysiak-Budnik
T: Hamartomatous tumors in the gastrointestinal tract. Gastrointest
Tumors. 2:65–74. 2015.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Rosty C: The Role of the surgical
pathologist in the diagnosis of gastrointestinal polyposis
syndromes. Adv Anat Pathol. 25:1–13. 2018.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Suzuki K, Higuchi H, Shimizu S, Nakano M,
Serizawa H and Morinaga S: Endoscopic snare papillectomy for a
solitary Peutz-Jeghers-type polyp in the duodenum with ingrowth
into the common bile duct: Case report. World J Gastroenterol.
21:8215–8220. 2015.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Endo K, Kawamura K, Murakami K, Murakami
K, Nagao M, Satoh T, Takasu A, Kogure T, Hirota M, Meguro T, et al:
A case of jejunal solitary Peutz-Jeghers polyp with intussusception
identified by double-balloon enteroscopy. Clin J Gastroenterol.
13:1129–1135. 2020.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Kumar S, Arora P and Goswami P: Recurrent
intestinal obstruction in a patient of Peutz-Jeghers syndrome. J
Cancer Res Ther. 15:252–254. 2019.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Mărginean CO, Meliţ LE, Patraulea F,
Iunius S and Mărginean MO: Early onset Peutz-Jeghers syndrome, the
importance of appropriate diagnosis and follow-up: A case report.
Medicine (Baltimore). 98(e16381)2019.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Duan SX, Wang GH, Zhong J, Ou WH, Fu MX,
Wang FS, Ma SH and Li JH: Peutz-Jeghers syndrome with intermittent
upper intestinal obstruction: A case report and review of the
literature. Medicine (Baltimore). 96(e6538)2017.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Rodríguez Lagos FA, Sorlí Guerola JV,
Romero Martínez IM and Codoñer Franch P: Register and clinical
follow-up of patients with Peutz-Jeghers syndrome in Valencia. Rev
Gastroenterol Mex. 85:123–139. 2020.PubMed/NCBI View Article : Google Scholar
|
|
86
|
De Latour RA, Kilaru SM and Gross SA:
Management of small bowel polyps: A literature review. Best Pract
Res Clin Gastroenterol. 31:401–408. 2017.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Achatz MI, Porter CC, Brugières L, Druker
H, Frebourg T, Foulkes WD, Kratz CP, Kuiper RP, Hansford JR,
Hernandez HS, et al: Cancer screening recommendations and clinical
management of inherited gastrointestinal cancer syndromes in
childhood. Clin Cancer Res. 23:e107–e114. 2017.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Shaco-Levy R, Jasperson KW, Martin K,
Samadder NJ, Burt RW, Ying J and Bronner MP: Morphologic
characterization of hamartomatous gastrointestinal polyps in Cowden
syndrome, Peutz-Jeghers syndrome, and juvenile polyposis syndrome.
Hum Pathol. 49:39–48. 2016.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Linhart H, Bormann F, Hutter B, Brors B
and Lyko F: Genetic and epigenetic profiling of a solitary
Peutz-Jeghers colon polyp. Cold Spring Harb Mol Case Stud.
3(a001610)2017.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Li T, Lin W, Zhao Y, Zhu J, Sun T and Ren
L: Distinct promoter methylation patterns of LKB1 in the
hamartomatous polyps of Peutz-Jeghers syndrome and its potential in
gastrointestinal malignancy prediction. Orphanet J Rare Dis.
15(208)2020.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Wang Z, Wu B, Mosig RA, Chen Y, Ye F,
Zhang Y, Gong W, Gong L, Huang F, Wang X, et al: STK11 domain XI
mutations: Candidate genetic drivers leading to the development of
dysplastic polyps in Peutz-Jeghers syndrome. Hum Mutat. 35:851–858.
2014.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Jedrzkiewicz J, Quencer K, Matynia AP,
Morrow E, Pletneva M and Barraza G: Peutz-jeghers type polyp of the
appendix with review of literature. Case Rep Pathol.
2019(7584070)2019.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Zhang Z, Duan FX, Gu GL and Yu PF:
Mutation analysis of related genes in hamartoma polyp tissue of
Peutz-Jeghers syndrome. World J Gastroenterol. 26:1926–1937.
2020.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Jung I, Gurzu S and Turdean GS: Current
status of familial gastrointestinal polyposis syndromes. World J
Gastrointest Oncol. 7:347–355. 2015.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Yu Q, Subedi S, Tong Y, Wei Q, Xu H, Wang
Y, Gong Y and Shi Y: Scalp metastases as first presentation of
pulmonary adenocarcinomas: A case report. Onco Targets Ther.
11:6147–6151. 2018.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Zaba O, Holbe D, Aretz S and Grohé C: LKB1
mutant in a KRAS activated adenocarcinoma of the lung associated
with Peutz-Jeghers syndrome: A case report. Lung Cancer.
82:368–369. 2013.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Osoegawa A, Kometani T, Nosaki K, Ondo K,
Hamatake M, Hirai F, Seto T, Sugio K and Ichinose Y: LKB1 mutations
frequently detected in mucinous bronchioloalveolar carcinoma. Jpn J
Clin Oncol. 41:1132–1137. 2011.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Kojima Y, Ohtsuka K, Ishii S, Aso N, Ohki
A, Hashimoto Y, Takeuchi H, Ohnishi H and Abe N: STK11. p. F354L
germline mutation in a case of multiple gastrointestinal tumors.
Case Rep Gastroenterol. 14:547–553. 2020.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Boda D: Cellomics as integrative omics for
cancer. Curr Proteomics. 10:237–245. 2013.
|
|
100
|
Neagu M, Constantin C, Tanase C and Boda
D: Patented biomarker panels in early detection of cancer. Recent
Patents Biomarkers. 1:10–14. 2011.
|
|
101
|
Lupu M, Caruntu A, Caruntu C, Papagheorghe
LML, Ilie MA, Voiculescu V, Boda D, Constantin C, Tanase C, Sifaki
M, et al: Neuroendocrine factors: The missing link in non melanoma
skin cancer. Oncol Rep. 38:1327–1340. 2017.PubMed/NCBI View Article : Google Scholar
|